
Abstract
S100B, a calcium-binding protein produced by astroglial cells, mediates paracrine and autocrine effects on neurons and glial cells. It regulates the balance between proliferation and differentiation in neurons and glial cells by affecting protective and apoptotic mechanisms. Post-mortem studies have demonstrated a deficit in synapses and dendrites in brains of schizophrenics. Recent studies have shown increased S100B levels in medicated acutely psychotic schizophrenic patients as well as unmedicated or drug naive schizophrenics. One study reported a positive correlation between negative symptoms and S100B. S100B serum levels (quantitative immunoassay) and psychopathology (Positive and Negative Syndrome Scale, PANSS) were examined upon study admission and after 12 and 24 weeks of standardized treatment in 98 chronic schizophrenic patients with primarily negative symptoms. Compared to age- and sex-matched healthy controls, the schizophrenic patients showed significantly increased S100B concentrations upon admission and after 12 and 24 weeks of treatment. High PANSS negative scores were correlated with high S100B levels. Regression analysis comparing psychopathology subscales and S100B identified negative symptomatology as the predicting factor for S100B. S100B is not just elevated during acute stages of disease since it remains elevated for at least 6 months following an acute exacerbation. With regard to psychopathology, negative symptomatology appears to be the predicting factor for the absolute S100B concentration. This might indicate that S100B in schizophrenic patients either promotes apoptotic mechanisms by itself or is released from astrocytes as part of an attempt to repair a degenerative or destructive process.
Similar content being viewed by others

INTRODUCTION
Although enormous efforts have been made to understand the pathogenesis of schizophrenia, research is still arrested at the stage of forwarding and corroborating hypotheses. Findings from post-mortem brain morphology, immunohistochemistry, neurochemistry, structural, and functional brain imaging have contributed towards a gradual comprehension of the underlying pathogenetic mechanisms of schizophrenia. Recently, several authors endeavored to integrate the neurodevelopmental and neurodegenerative aspects of this disease spectrum into a comprehensive pathogenetic hypothesis of schizophrenia (eg Bartzokis, 2002; Henn and Braus, 1999; Lieberman et al, 1997; Mirnics et al, 2001; Weinberger and McClure, 2002; Woods, 1998).
One integrative approach based on microarray findings considers schizophrenia as a disease of the synapse (Mirnics et al, 2001). The authors hypothesize that defects in the gene expression of presynaptic secretory pathway elements (PSYN group and G-protein regulators) might affect the extended postnatal developmental process of synapse formation and pruning. Impaired synaptic transmission in schizophrenic patients most likely occurs in a subset of synapses from early life, but the exuberant synaptic connections that form during the third trimester of gestation and early childhood might compensate for the curtailing of synaptic function. The exuberant pruning of synapses by late adolescence might then expose any existing synaptic impairment. Inadequate synaptic activity might then lead to overpruning and manifestation of the symptoms of schizophrenia. The recent finding that neuregulin, a peptide involved in the regulation of proliferation and differentiation in neurons and glia, was identified as a candidate gene for schizophrenia (Stefansson et al, 2002, 2003), provides additional support for the synapse hypothesis.
Regarding the neurodegenerative aspect there is convincing evidence that progression of cerebral volume reduction occurs during the course of disease in at least a subgroup of schizophrenics (Gur et al, 1998; DeLisi, 1999; Pearlson and Marsh, 1999; Shenton et al, 2001). A classical neurodegenerative mechanism involving loss of neurons and development of gliosis, however, is unlikely to be relevant for schizophrenia. Brains from patients having suffered schizophrenia usually show unchanged numbers of neurons and a reduction in neuronal cell size and neuropil (Harrison, 1999; Powers, 1999; Selemon and Goldman-Rakic, 1999; McGlashan and Hoffman, 2000). A reduction of neuropil entails compromised cell structure and impoverishment of neuronal connectivity so that a loss in functional communication between neurons is presumed to occur. Several studies reported changes in synaptic proteins and their gene expression (Kung et al, 1998; Karson et al, 1999; Glantz and Lewis, 2000; Harrison and Eastwood, 2001; Mirnics et al, 2001). These findings support a diminishment or dysfunction of dendrites, neurites, and synapses in schizophrenia.
Considering these facts it would be useful to have access to a method that provides data on ongoing synaptic pathology at different stages of disease, and at different time points. There is now evidence that the calcium-binding astrocytic protein S100B might serve as a marker for such a procedure.
S100B is a dimeric protein with a molecular weight of 21 kDa that exerts paracrine and autocrine effects on neurons and glia (Beaudeux et al, 1999; Griffin et al, 1998). In adult brains it plays a role in maintaining neuronal plasticity and allowing long-term potentiation. From in vitro and in vivo animal experiments it is known that S100B plays important roles regarding cell proliferation and differentiation, cellular energy metabolism, and cytoskeletal modification. Those effects appear to be mediated primarily via binding to key synaptic proteins and inhibition of their phosphorylation. S100B in particular has an important role in regulating the protein kinase C phosphorylation of GAP-43, a growth-associated protein that is involved in axonal growth and synaptogenesis during development, synaptic remodeling, and long-term potentiation. S100B interacts with and stabilizes microtubule-associated proteins (MAPs), such as tau and MAP-2. Increased S100B and MAP-2 promote dendritic maturation and induce loss of dendrites (Whitaker-Azmitia et al, 1997). Lower concentrations of extracellular S100B act on glial and neuronal cells as a growth differentiating factor, while higher concentrations induce apoptosis (Fano et al, 1995). Physiologically or pathologically (ie excessively) released S100B reaches the extracellular compartment and cerebrospinal fluid before entering the bloodstream (Beaudeux et al, 1999). In animal studies, changes in the cerebral concentration of S100B cause behavioral disturbances and cognitive deficits (for a review see, Donato, 2001; Heizmann et al, 2002; Rothermundt et al, 2003, 2004).
In humans, increased S100B has been detected in various clinical conditions. Brain trauma and ischemia is associated with increased S100B concentrations, probably due to the destruction of astrocytes. In neurodegenerative and neuroinflammatory diseases, increased S100B levels may result from S100B secretion or release from damaged astrocytes. In schizophrenia, several studies have shown increased S100B serum levels in the acute stage of disease (Wiesmann et al, 1999; Lara et al, 2001; Rothermundt et al, 2001; Schroeter et al, 2003). Lara et al (2001) reported higher levels in patients with recent onset of disease compared to chronic patients while Rothermundt et al (2001) demonstrated that continuously increased S100B levels after 6 weeks are associated with negative symptomatology. Schroeter et al (2003) found increased S100B levels in patients with deficit schizophrenia compared to nondeficit patients.
These earlier studies provide evidence that S100B might be relevant for the pathophysiology of schizophrenia. However, several essential aspects have not been addressed until now, and for this reason these have been tackled in the study presented here:
-
1)
In most earlier studies the schizophrenic patients were examined just once. In the only longitudinal study the observation period was 6 weeks (Rothermundt et al, 2001). Since in most patients symptoms of schizophrenia, especially negative symptoms, improve slowly and the majority of patients have not reached a stable state after 6 weeks, our present study covers a treatment period of 24 weeks.
-
2)
So far none of the investigations were performed with standardized medication. In our study, patients received either flupenthixol or risperidone (randomly assigned) over the whole study period and no other antipsychotic medication was allowed.
-
3)
Earlier studies featured small sample sizes of n=30 at the most. We recruited a larger sample of patients (n=98).
-
4)
Since associations between negative symptoms or deficit schizophrenia and S100B concentrations have been reported (Rothermundt et al, 2001; Schroeter et al, 2003), we focused on the long-term course of negative symptoms.
METHODS
Patient and Control Samples
In this multicenter study, designed according to Good Clinical Practice Guidelines (GCP) of the ICH, 98 patients (56 males, 42 females) were recruited in 31 psychiatric treatment centers throughout Germany and Austria. All patients were diagnosed to be suffering from schizophrenia according to ICD-10 criteria (ICD-10: F20.0–F20.3, F20.5–F20.9) by two psychiatrists independently. They showed considerable negative symptoms with ⩾4 points in at least three items of the Positive and Negative Syndrome Scale (PANSS) negative subscale and the duration of illness exceeded 2 years. Exclusion criteria included substance dependency, internal or neurological disorders, or pregnancy.
After written informed consent according to the standards of the Declaration of Helsinki was acquired, former medication was discontinued for 1 week prior to the investigation time point T0. The vast majority of patients (70.4%) had received typical antipsychotic drugs, 19.4% atypical antipsychotics, and 10.2% had been without medication. Patients were (random and double-blind) assigned to treatment with either risperidone (n=53) or flupenthixol (n=45). They were started on 2 mg of risperidone or 4 mg of flupenthixol. In the following weeks dosages ranged from 2 to 6 mg of risperidone or 4 to 12 mg of flupenthixol. Throughout the whole study period only comedication with benzodiazepines, anticholinergic drugs, or nonbenzodiazepine-hypnotics were allowed.
Psychopathology was assessed using the PANSS (Kay et al, 1987). In order to ensure inter-rater reliability, all raters participated in a comprehensive rater training. None of the raters knew the medication status at any point of investigation. All patients were examined at intake (T0), after 12 (T12), and after 24 (T24) weeks of treatment. After 12 weeks of treatment 73 patients and after 24 weeks 59 patients remained in the study. The dropout rates were 25 and 40%, respectively. The reasons for discontinuation from the study (T24) were withdrawal of consent (11.2%), lack of efficacy (9.2%), adverse event (8.2%), patient decision (7.2%), poor compliance (2%), satisfactory response (1.1%), and death (1.1%). There was no significant difference between the dropout rates of the two treatment groups (χ2=0.018, df=1, p=0.89).
The mean age of the patients was 42.1 years (SD 11.1), ranging from 23 to 64 years. Precisely age- and sex-matched healthy blood donors (mean age 42.1 years, SD 11.1) were recruited for comparison of S100B concentrations. The controls had no lifetime history of any psychiatric disorder. Systematic diseases (neoplasms, autoimmune diseases, infectious diseases, neurological, and cardiovascular diseases), brain injury, and substance abuse were excluded by taking a detailed history and performing a physical examination.
S100B Measurements
Blood samples were centrifuged within 4 h, aliquoted, and sent to a central laboratory where they were frozen at −80°C until analysis. S100B concentrations were determined by applying the LIAISON Sangtec 100 assay (AB Sangtec Medical, Bromma, Sweden), a quantitative automated luminometric immunoassay, according to the manufacturer's instructions. Briefly, the two-site sandwich assay is based on paramagnetic particles coated with two monoclonal antibodies and a monoclonal tracer antibody labeled with an isoluminol derivative. The magnetic particles, assay buffer and sample are first incubated together before unbound material is removed by a wash cycle. Tracer is added, and after a second incubation, unbound tracer is removed by a second washing cycle. Subsequently, the starter reagents are added, and the S100B concentration is determined from the chemiluminescence reaction induced. The light signal measured in relative light units (RLUs) is directly proportional to the amount of S100B in the sample. The antibody is highly specific for the S100β monomer; two S100β monomers form the S100B dimer. The assay's lower detection limit for S100B is 0.02 μg/l. The intra-assay (within-run) imprecision (CVs) is 6.4% at 0.11 μg/l, 3.1% at 0.31 μg/l, 2.8% at 1.6 μg/l, 3.9% at 2.6 μg/l, 2.8% at 9.9 μg/l, and 3.6% at 18.4 μg/l. The interassay variation (CVs) is 10.7% at 0.11 μg/l, 8.2% at 0.3 μg/l, 3.7% at 1.6 μg/l, 2.2% at 2.6 μg/l, 6.3% at 9.9 μg/l, and 3.2% at 18.4 μg/l. Analytical recovery ranges between 91 and 100%.
Statistics
Owing to the non-Gaussian distribution of our data, nonparametric tests were employed for statistical evaluation. The Wilcoxon's matched-pairs signed-ranks test, the Mann–Whitney U-test, the Spearman's correlation coefficient, the Friedman test, and the stepwise multiple regression analysis were used as provided by the SPSS 10.0 program.
RESULTS
The psychopathology measurements (Table 1) show that the patients suffered from moderate to severe symptoms at intake with predominantly negative symptoms. Treatment with risperidone or flupentixole resulted in a significant improvement of the PANSS total score (χ2=69.5, p⩽0.0001, df=2) and all subscores between T0, T12, and T24. The groups treated with risperidone and flupenthixol (Table 2) did not differ psychopathologically from each other at any time point.
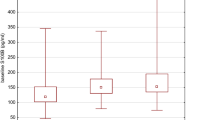
The mean S100B serum concentration of the schizophrenic patients at intake (T0) was 0.073±0.032 μg/l. After 12 (T12) and 24 weeks (T24) of treatment the concentrations were 0.070±0.026 or 0.078±0.041 μg/l, respectively. No difference between the three study time points could be observed (χ2=0.12, p=0.94, df=2), but the patients’ S100B concentration at all study time points was significantly higher than that of the matched healthy controls (0.044±0.015 μg/l, χ2=47.03, p⩽0.0001, df=3; Figure 1). There was no difference between the two treatment groups (risperidone or flupenthixol) at any time point (T0: Z=−0.043, p=0.966; T12: Z=−0.312, p=0.755; T24: Z=−1.077, p=0.282; controls: Z=0.403, p=0.687). In all, 38.8% of the patients at T0, 38.4% at T12, and 40.7% at T24 showed S100B serum concentrations that laid above the mean plus two standard deviations (SDs) (0.074 μg/l) of the healthy controls.

S100B in patients with schizophrenia at intake (T0), after 12 (T12) and after 24 weeks (T24) of treatment and healthy controls.
A significant correlation between S100B serum levels and the PANSS negative subscale score at T0 (ρ=−0.17, p=0.048) could be found while the PANSS positive score (ρ=−0.13, p=0.10), the PANSS general psychopathology score (ρ=−0.06, p=0.29), and the PANSS total score (ρ=−0.13, p=0.11) were not significantly correlated with S100B.
A stepwise multiple regression analysis including PANSS psychopathology subscales and S100B (as dependent variable) identified the PANSS negative subscale as the predicting factor for S100B. This also held true if other factor analytic approaches for subclassifying the PANSS such as the five-factor models of Lindenmayer et al (1994) and Marder et al (1997) or the eight-factor model of Peralta and Cuesta (1994) were applied (Table 3).
In order to assess the changes in negative psychopathology between T0 and T12 or T24, we calculated the relative response measure ΔRes (%) from the difference between the PANSS negative subscale score at T0 and the PANSS negative subscale score at T12 or T24, and divided the result by the PANSS negative subscale score at T0 before this quotient was multiplied by 100. Between T0 and T12 the median ΔRes was 28.6% (range: −40 to 73.5%) and between T0 and T24 42.4% (range: 35.7–74.3%). We then divided the patient sample into two groups. One group contained those patients who experienced a psychopathological change above the median (PP+ group), while the other consisted of patients who showed a psychopathological change below the median (PP−). Schizophrenics who demonstrated little or no improvement in the PANSS negative subscale score (PP−) at T12 had significantly higher S100B serum levels at T0 (Z=−2.45, p=0.014) and T12 (Z=−3.11, p=0.002) compared with the group of patients who showed a ΔRes of more than 28.6%. At T24, no difference was observed any more (Z=−0.43, p=0.67). Concerning the PANSS negative score changes after 24 weeks of treatment (T24), the S100B serum concentrations were significantly higher in the low improvement group (PP−) at T12 (Z=−2.52, p=0.012) while at T0 (Z=−0.59, p=0.56) and T24 (Z=−0.57, p=0.57) they did not differ from the S100B levels of the high improvement group (PP+). No differences between the risperidone and the flupenthixol treatment groups could be observed concerning the relative response measure ΔRes (data not shown). Furthermore, no sex or age effects could be detected (data not shown).
In a second approach we divided the whole group according to the S100B serum concentrations at T0. Patients who had S100B levels higher than the mean plus two SDs of the control group (0.074 μg/l) were assigned to group H (‘high’; n=38), while group N (‘normal’; n=60) consisted of individuals whose S100B concentration was lower than 0.074 μg/l. Groups H and N did not differ regarding PANSS total score (Z=−1.21, p=0.23), PANSS positive subscale score (Z=−1.08, p=0.28), and PANSS general psychopathology subscale score (Z=−0.46, p=0.64) at T0. The score of the PANSS negative subscale at T0 was higher in group N than it was in group H (Z=−2.17, p=0.03). After 12 and 24 weeks of treatment, no differences in psychopathology between groups H and N could be detected any more. Both groups showed significant improvement of psychopathology in all subscales between T0 and T12, or T0 and T24. However, there was a major difference between groups H and N regarding the changes in psychopathology between T12 and T24. In group N the psychopathological improvement occurred within the first 12 weeks of treatment, while during the second 12 weeks period no more changes could be observed. In group H, less psychopathological improvement occurred within the first 12 weeks of treatment, but the progress continued during the second 12 weeks treatment period so that a comparable psychopathology was reached as could be seen in group N (Figures 2 and 3).

Improvement of psychopathology in schizophrenic patients with increased S100B serum levels at intake (group H).

Improvement of psychopathology in schizophrenic patients with normal S100B serum levels at intake (group N).
In case of a Gaussian distribution of the data a repeated measure ANOVA would be a relevant analysis for examination of differential longitudinal effects of S100B levels. There would be no significance concerning the main effect of time (F=1.4, p=0.26) but the interaction effect (F=10.5, p⩽0.0001) and the main effect of group (F=29.0, p⩽0.0001) would be significant. These results support the findings demonstrated in Figures 2 and 3.
DISCUSSION
To our knowledge, this is the first published longitudinal study investigating S100B in schizophrenic patients covering a 24-week treatment period from an acute stage of disease to recovery or a stable residual state. Regarding the entire patient sample, S100B serum concentrations were increased at intake, after 12 weeks and after 24 weeks of treatment compared to age- and sex-matched healthy controls. About 40% of the schizophrenics showed S100B levels higher than the mean plus two SDs of the healthy control group at all investigation time points. Comparable numbers were reported earlier for an acute stage of disease (Lara et al, 2001; Rothermundt et al, 2001; Schroeter et al, 2003; Wiesmann et al, 1999). It appears that a subgroup of schizophrenic patients (about 40%) exhibits increased S100B levels that are relatively stable over time and which do not normalize after recovery from an acute episode.
The increased S100B concentrations were associated with high scores of negative symptoms as have been reported in two earlier studies (Rothermundt et al, 2001; Schroeter et al, 2003). This association holds true not only for acute stages of disease and short observation periods, but remains stable for at least 6 months following an acute episode.
Negative symptoms measured by the PANSS appear to predict the S100B serum concentration. Positive symptoms or other psychopathological factors seem to be independent of S100B levels. The strong association between negative symptoms and S100B was stable even if different factor analytic models for grouping the negative parameters of the PANSS were applied. Kay et al (1987) presented a model of negative symptoms containing seven parameters: blunted affect (N1), emotional withdrawal (N2), poor rapport (N3), passive/apathetic withdrawal (N4), difficulty in abstract thinking (N5), lack of spontaneity (N6), and stereotyped thinking (N7). The negative component assembled by Lindenmayer et al (1994) consisted of the parameters N1, N2, N3, N4, N6, and G16 (active social avoidance). Marder et al (1997) expanded Lindenmayer's negative component by including G7 (motor retardation). Peralta and Cuesta's (1994) negative component is composed of 10 items including the seven parameters used by Marder as well as the items G7, G13 (disturbance of volition), and G16. No matter which factor analytic model was applied, the negative component of each model predicted the S100B levels. Common to all models are the six items: blunted affect, emotional withdrawal, poor rapport, passive/apathetic withdrawal, lack of spontaneity, and active social avoidance, that is, those factors building the negative component of Lindenmayer et al's (1994) model. Patients with increased S100B appear to have problems in emotional expression, contact with others, spontaneity and taking initiatives. Animal experiments studying the behavior of transgenic S100B mice expressing multiple genetic copies of S100B revealed a decreased spontaneous alternation rate (Gerlai et al, 1994) among other symptoms such as impaired memory and learning deficits (Gerlai and Roder, 1996; Whitaker-Azmitia et al, 1997; Winocur et al, 2001). The decreased spontaneous alternation rate was interpreted as a lack of motivation to explore novelty, impaired ability to attend to stimuli, or a failure to remember acquired information (Gerlai et al, 1994). Although it is difficult to draw conclusions from animal studies on complex human diseases such as schizophrenia, it raises the question whether changes in the concentration of S100B might be involved in the pathogenesis of this disorder.
To take a closer look at the changes in negative symptoms throughout the treatment course we calculated the relative response rates between T0 and T12 or T0 and T24. Patients with an improvement below the median of the total group showed significantly higher S100B levels than those patients who experienced a greater improvement. These results are in line with the earlier findings of sustained negative symptoms in those patients who showed high S100B serum levels (Rothermundt et al, 2001).
Division of the whole group according to S100B levels into patients with highly elevated S100B concentrations and those with close to normal levels confirmed the association between S100B and negative symptomatology. Patients with high S100B levels (group H) showed more and more persistent negative symptoms compared to those of group N who had close to normal S100B levels. Interestingly, group N showed faster improvement of psychopathology: There was a pronounced decrease of PANSS scores between T0 and T12 with hardly any further change between T12 and T24. Group H finally reached comparable PANSS scores after 24 weeks. The improvement, however, was much slower and more prolonged.
These findings suggest that S100B might play a role in the pathogenesis of schizophrenia. S100B displays neuroprotective or neurodegenerative abilities depending on its concentration. In cell culture and animal experiments, micromolar concentrations of S100B stopped proliferation and induced differentiation of neurons leading to aging and finally apoptosis of cells, synapses or dendrites. Nanomolar concentrations of S100B, however, appear to boost proliferation of neurons, dendrites, and neurites, thus inducing an increase in synaptic contacts between the cells. Since a healthy brain requires the potential to implement proliferative as well as differentiating mechanisms according to varying requirements, S100B should be present in balanced amounts (Donato, 2001; Rothermundt et al, 2003). We still do not know what causes the loss of dendrites and synapses in brains of schizophrenic patients (Harrison, 1999; Powers, 1999; Selemon and Goldman-Rakic, 1999; McGlashan and Hoffman, 2000), but from our knowledge concerning the mechanisms of regeneration and degeneration it appears likely that S100B might be involved in this process (Donato, 2001; Rothermundt et al, 2003). The increased S100B concentrations in patients with schizophrenia might cause a shift in the regeneration–degeneration balance towards degeneration so that a rarification of synapses and dendrites might result. In the long run these morphological changes could lead to clinically overt symptoms such as negative symptoms. Another and probably more likely model views the increased S100B concentration as an attempt of the brain to counter an ongoing destructive process. In this model S100B would then be released as an attempt to repair and regenerate the damage. A prolonged increase of S100B would then point towards an ongoing destructive process.
At the beginning of the study all patients were randomly assigned to receive either flupenthixol or risperidone as a single antipsychotic drug. This standardized medication regime was maintained throughout the whole study period. In this group of patients the S100B serum concentrations did not change according to medication status. Patients showed increased S100B concentrations in an unmedicated stage as well as after 12 or 24 weeks of treatment. No differences between the treatment with flupenthixol or risperidone could be observed. The dropout rates of patients during the study of 25% (T12) or 40% (T24), respectively, were within the expected range of such long-term trials. In two double-blind multicenter studies with schizophrenic patients over 28 weeks dropout rates of 40 or 55%, respectively, were reported (Tran et al, 1997; Hirsch et al, 2002). The improvement of psychopathology, especially the PANSS negative score during the trial, is remarkable but not completely unusual. Similar longitudinal investigations reported comparable findings (Tran et al, 1997; Peuskens et al, 1999). Others, however, showed much smaller changes in stable outpatients or first-episode patients (eg Hirsch et al, 2002; Lieberman et al, 2003). The selection of patients with leading negative symptoms and the fact that 80% were untreated or receiving typical antipsychotic drugs prior to the study might account for these changes.
In earlier cross-sectional studies either unmedicated schizophrenic patients (Lara et al, 2001; Schroeter et al, 2003) or patients treated with various antipsychotic drugs (Wiesmann et al, 1999; Gattaz et al, 2000; Schroeter et al, 2003) were investigated. In the only longitudinal study published so far, patients were studied initially in an unmedicated state and then after 6 weeks of neuroleptic treatment with various antipsychotic drugs (Rothermundt et al, 2001). In that study the S100B levels of the total group decreased after 6 weeks of treatment but in the subgroup of patients with persistent negative symptoms S100B concentrations remained high. This was interpreted to be linked to the psychopathology rather than to be caused by the antipsychotic medication. Schroeter and co-workers reported increased S100B serum concentrations in patients receiving antipsychotic medication for 3 weeks on the average compared to unmedicated patients. They discussed whether antipsychotic medication might cause an increase in S100B serum levels during the early treatment phase before a decrease is observed after 6 weeks (as reported by Rothermundt et al, 2001) or long-term treatment (as reported by Gattaz et al, 2000). However, Schroeter and co-workers investigated their patients only once, either in an unmedicated or medicated state, enabling only interpersonal and not intrapersonal comparisons. The findings of Gattaz and co-workers showing decreased S100B levels in chronic schizophrenics should be interpreted with caution. In that study the S100B concentrations measured in the citrate plasma of healthy controls (0.55 μg/l) were almost 10 times higher than the S100B concentrations measured in serum (0.066 μg/l) using the same assay (Lara et al, 2001). This questions the appropriateness of the chosen assay for detecting S100B in plasma. Additional animal and clinical studies are clearly called for to better understand the influence of antipsychotic drugs on S100B serum concentration.
S100B concentrations were measured in the serum of patients and controls. Since schizophrenia is a brain disorder, it has to be considered how reliably S100B serum measures reflect processes going on in the brains of the individual patients. S100B is produced by astrocytes and as a rather small protein of only 21 kDa in its dimeric form it can easily traverse the blood–brain barrier (BBB). In healthy individuals as well as patients with various neurological diseases, serum measures have been proven to reflect the S100B concentration in the CSF (Reiber, 2001). Moreover, S100B serum levels might in fact reflect brain metabolism more reliably than CSF levels since, due to natural CSF flow, lumbar CSF only reflects defects in brain tissue that are less than 30 mm from the lateral ventricles and the third ventricle (Reiber, 2001). For small proteins such as S100B, the short transit from the extracellular space through the wall of the blood vessel to the blood might be accomplished more easily than the potentially long passage from the extracellular space to the ventricles. However, the relationship between S100B in the CSF and serum of schizophrenic patients has not yet been published and requires further study.
Even though the S100B serum concentrations in schizophrenic patients were significantly increased compared to healthy controls the absolute values were within a range that has been considered normal in studies designed to discover brain damage (for a review see, Rothermundt et al, 2003). In brain damage, S100B levels are measured to detect a vast destruction of astrocytes. In schizophrenia, however, much more subtle neuroplastic processes are to be expected. Therefore, it appears necessary to concomitantly investigate the S100B concentration of matched healthy controls. Repeated measurements at various time points are helpful to ensure the stability of values.
S100B has a very high brain specificity. The only known condition when serum S100B is not necessarily originated from the brain is a major soft tissue damage such as after polytrauma or cardiac surgery. The S100B released from the soft tissue is most probably not originated from S100ββ homodimers but rather from other S100 species that contain the β-subunit in a heterodimer. Unfortunately, all available assays to measure S100B contain antibodies which are specific for the monomer and not for the functionally relevant dimer. However, patients with recent soft tissue damage were excluded from the study.
In order to reinforce the biological relevance of the S100B findings for schizophrenia, we should not confine our studies to psychopathological evaluations. Investigations combining S100B measurements with established parameters such as neurocognitive functions, evoked potentials or structural and functional imaging should be undertaken. Furthermore, additional cell culture experiments are required to further our knowledge on the molecular pathways of S100B in neurons and glial cells. These investigations offer the chance to improve our understanding of the pathogenetic mechanisms involved in the development of schizophrenia. Finally these findings could open up new therapeutic strategies since the release of S100B is modulated via 5HT1A receptors (Rothermundt et al, 2004).
References
Bartzokis G (2002). Schizophrenia: breakdown in the well-regulated lifelong process of brain development and maturation. Neuropsychopharmacology 27: 672–683.
Beaudeux JL, Dequen L, Foglietti MJ (1999). La protéine S-100β: un nouveau marquer biologique de pathologie cérébrale. Ann Biol Clin 57: 261–272.
DeLisi LE (1999). Defining the course of brain structual change and plasticity in schizophrenia. Psychiatry Res Neuroimaging 92: 1–9.
Donato R (2001). S100: a multigenic family of calcium-modulated proteins of the EF-hand type with intracellular and extracellular functional roles. Int J Biochem Cell Biol 33: 637–668.
Fano G, Biocca S, Fulle S, Mariggio MA, Belia S, Calissano P (1995). The S-100: a protein family in search of a function. Prog Neurobiol 46: 71–82.
Gattaz WF, Lara DR, Elkis H, Portel LV, Goncalves CA, Tort AB et al (2000). Decreased S100-beta protein in schizophrenia: preliminary evidence. Schizophr Res 43: 91–95.
Gerlai R, Marks A, Roder J (1994). T-maze spontaneous alternation rate is decreased in S100β transgenic mice. Behav Neurosci 108: 100–106.
Gerlai R, Roder J (1996). Spatial and nonspatial learning in mice: effects of S100β overexpression and Age. Neurobiol Learn Mem 66: 143–154.
Glantz LA, Lewis DA (2000). Decreased dendritic spine density on prefrontal cortical pyramidal neurons in schizophrenia. Arch Gen Psychiatry 57: 65–73.
Griffin WST, Sheng JG, Mrak RE (1998). Senescence-accelerated overexpression of S100β in brain of SAMP6 mice. Neurobiol Aging 19: 71–76.
Gur RE, Cowell P, Turetsky BI, Gallacher F, Cannon T, Bilker W et al (1998). A follow-up magnetic resonance imaging study of schizophrenia. Arch Gen Psychiatry 55: 145–152.
Harrison PJ (1999). The neuropathology of schizophrenia. A critical review of the data and their interpretation. Brain 122: 593–624.
Harrison PJ, Eastwood SL (2001). Neuropathological studies of synaptic connectivity in the hippocampal formation in schizophrenia. Hippocampus 11: 508–519.
Heizmann CW, Fritz G, Schäfer BW (2002). S100 proteins: structure, functions and pathology. Front Biosci 7: 1356–1368.
Henn FA, Braus DF (1999). Structural neuroimaging in schizophrenia. An integrative view of neuromorphology. Eur Arch Psychiatry Clin Neurosci 249(Suppl 4): IV/48–IV/56.
Hirsch SR, Kissling W, Bauml J, Power A, O’Connor R (2002). A 28-week comparison of ziprasidone and haloperidole in outpatients with stable schizophrenia. J Clin Psychiatry 63: 516–523.
Karson CN, Mrak RE, Schluterman KO, Sturner WQ, Sheng JG, Griffin WST (1999). Alterations in synaptic proteins and their encoding mRNAs in prefrontal cortex in schizophrenia: a possible neurochemical basis for ‘hypofrontality’. Mol Psychiatry 4: 39–45.
Kay SR, Fiszbein A, Opler LA (1987). The positive and negative syndrome scale (PANSS) for schizophrenia. Schizophr Bull 13: 261–276.
Kung L, Conley R, Chute DJ, Smialik J, Roberts RC (1998). Synaptic changes in the striatum of schizophrenic cases: a controlled postmortem ultrastructural study. Synapse 28: 125–139.
Lara DR, Gama CS, Belmonte-de-Abreu P, Portela LVC, Goncalves CA, Fonseca M et al (2001). Increased serum S100B protein in schizophrenia: a study in medication-free patients. J Psychiatr Res 35: 11–14.
Lieberman JA, Sheitman BB, Kinon BJ (1997). Neurochemical sensitization in the pathophysiology of schizophrenia: deficits and dysfunction in neuronal regulation and plasticity. Neuropsychopharmacology 17: 205–229.
Lieberman JA, Tollefson G, Tohen M, Green AI, Gur RE, Kahn R et al (2003). Comparative efficacy and safety of atypical and conventional antipsychotic drugs in first-episode psychosis: a randomized, double-blind trial of alonazapine versus haloperidol. Am J Psychiatry 160: 1396–1404.
Lindenmayer JP, Bernstein-Hyman R, Grochowski S (1994). Five-factor model of schizophrenia. J Nerv Ment Dis 182: 631–638.
Marder SR, Davis JM, Chouinard G (1997). The effects of risperidone on the five dimensions of schizophrenia derived by factor analysis: combined results of the North American Trials. J Clin Psychiatry 58: 538–546.
McGlashan TH, Hoffman RE (2000). Schizophrenia as a disorder of developmentally reduced synaptic connectivity. Arch Gen Psychiatry 57: 637–648.
Mirnics K, Middleton FA, Lewis DA, Levitt P (2001). Analysis of complex brain disorders with gene expression microarrays: schizophrenia as a disease of the synapse. Trends Neurosci 24: 479–486.
Pearlson GD, Marsh L (1999). Structural brain imaging in schizophrenia: a selective review. Biol Psychiatry 46: 627–649.
Peralta V, Cuesta MJ (1994). Psychometric properties of the positive and negative syndrome scale (PANSS) in schizophrenia. Psychiatry Res 53: 31–40.
Peuskens J, Bech P, Möller HJ, Bale R, Fleurot O, Rein W (1999). Amisulpride vs Risperidone in the treatment of acute exacerbations of schizophrenia. Psychiatry Res 88: 107–117.
Powers RE (1999). The neuropathology of schizophrenia. J Neuropathol Exp Neurol 58: 679–690.
Reiber H (2001). Dynamics of brain-derived proteins in cerebrospinal fluid. Clin Chim Acta 310: 173–186.
Rothermundt M, Missler U, Arolt V, Peters M, Leadbeater J, Wiesmann M et al (2001). Increased S100B blood levels in unmedicated and treated schizophrenic patients are correlated with negative symptomatology. Mol Psychiatry 6: 445–449.
Rothermundt M, Peters M, Prehn JHM, Arolt V (2003). S100B in brain damage and neurodegeneration. Microsc Res Technol 60: 614–632.
Rothermundt M, Ponath G, Arolt V (2004). S100B in schizophrenic psychosis. Int Rev Neurobiol 59: 445–470.
Schroeter ML, Abdul-Khaliq H, Frühauf S, Höhne R, Schick G, Diefenbacher A et al (2003). Serum S100B is increased during early treatment with antipsychotics and in deficit schizophrenia. Schizophr Res 62: 231–236.
Selemon LD, Goldman-Rakic PS (1999). The reduced neuropil hypothesis: a circuit based model of schizophrenia. Biol Psychiatry 45: 17–25.
Shenton ME, Dickey CC, Frumin M, McCarley RW (2001). A review of MRI findings in schizophrenia. Schizophr Res 49: 1–52.
Stefansson H, Sarginson J, Kong A, Yates P, Steinthorsdottir V, Gudfinnsson E et al (2003). Association of neuregulin 1 with schizophrenia confirmed in a Scottish population. Am J Hum Genet 72: 83–87.
Stefansson H, Sigurdsson E, Steinthorsdottir V, Bjornsdottir S, Sigmundsson T, Ghosh S et al (2002). Neuregulin 1 and susceptibility to schizophrenia. Am J Hum Genet 71: 877–892.
Tran PV, Hamilton SH, Kuntz AJ, Potvin JH, Andersen SW, Beasley C et al (1997). Double-blind comparison of olanzapine versus risperidone in the treatment of schizophrenia and other psychotic disorders. J Clin Psychopharmacol 17: 407–418.
Weinberger DR, McClure RK (2002). Neurotoxicity, neuroplasticity, and magnetic resonance imaging morphometry. Arch Gen Psychiatry 59: 553–558.
Whitaker-Azmitia PM, Wingate M, Borella A, Gerlai R, Roder J, Azmitia EC (1997). Transgenic mice overexpressing the neurotrophic factor S-100β show neuronal cytoskeletal and behavioral signs of altered aging processes: implications for Alzheimer's disease and Down's syndrome. Brain Res 776: 51–60.
Wiesmann M, Wandinger KP, Missler U, Eckhoff D, Rothermundt M, Arolt V et al (1999). Elevated plasma levels of S-100b protein in schizophrenic patients. Biol Psychiatry 45: 1508–1511.
Winocur G, Roder J, Lobaugh N (2001). Learning and memory in S100-β transgenic mice: an analysis of impaired and preserved function. Neurobiol Learn Mem 75: 230–243.
Woods BT (1998). Is schizophrenia a progressive neurodevelopmental disorder? Toward a unitary pathogenetic mechanism. Am J Psychiatry 155: 1661–1670.
Acknowledgements
We thank Dr Thomas Suslow for statistical advice and Dr Julian Patrick Keogh for editorial support. The financial support of Bayer Vital Pharmaceuticals for laboratory reagents and assays is appreciated.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Rothermundt, M., Ponath, G., Glaser, T. et al. S100B Serum Levels and Long-Term Improvement of Negative Symptoms in Patients with Schizophrenia. Neuropsychopharmacol 29, 1004–1011 (2004). https://doi.org/10.1038/sj.npp.1300403
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.npp.1300403
Keywords
This article is cited by
-
The relationship between inflammatory markers, clinical characteristics, and cognitive performance in drug-naïve patients with schizophrenia
European Archives of Psychiatry and Clinical Neuroscience (2023)
-
Linking Inflammation, Aberrant Glutamate-Dopamine Interaction, and Post-synaptic Changes: Translational Relevance for Schizophrenia and Antipsychotic Treatment: a Systematic Review
Molecular Neurobiology (2022)
-
Blood-brain barrier associated tight junction disruption is a hallmark feature of major psychiatric disorders
Translational Psychiatry (2020)
-
S100B polymorphisms are associated with age of onset of Parkinson’s disease
BMC Medical Genetics (2018)
-
Elevated Plasma S100B, Psychotic Symptoms, and Cognition in Schizophrenia
Psychiatric Quarterly (2018)
