
Abstract
Bipolar disorder (BPD) has traditionally been conceptualized as a neurochemical disorder, but there is mounting evidence for impairments of cellular plasticity and resilience. Here, we review and synthesize the evidence that critical aspects of mitochondrial function may play an integral role in the pathophysiology and treatment of BPD. Retrospective database searches were performed, including MEDLINE, abstract booklets, and conference proceedings. Articles were also obtained from references therein and personal communications, including original scientific work, reviews, and meta-analyses of the literature. Material regarding the potential role of mitochondrial function included genetic studies, microarray studies, studies of intracellular calcium regulation, neuroimaging studies, postmortem brain studies, and preclinical and clinical studies of cellular plasticity and resilience. We review these data and discuss their implications not only in the context of changing existing conceptualizations regarding the pathophysiology of BPD, but also for the strategic development of improved therapeutics. We have focused on specific aspects of mitochondrial dysfunction that may have major relevance for the pathophysiology and treatment of BPD. Notably, we discuss calcium dysregulation, oxidative phosphorylation abnormalities, and abnormalities in cellular resilience and synaptic plasticity. Accumulating evidence from microarray studies, biochemical studies, neuroimaging, and postmortem brain studies all support the role of mitochondrial dysfunction in the pathophysiology of BPD. We propose that although BPD is not a classic mitochondrial disease, subtle deficits in mitochondrial function likely play an important role in various facets of BPD, and that enhancing mitochondrial function may represent a critical component for the optimal long-term treatment of the disorder.
Similar content being viewed by others


INTRODUCTION
Despite the fact that bipolar disorder (BPD) is a common, severe, often life-threatening illness, the biochemical abnormalities underlying the predisposition to, and the pathophysiology of, this complex and intriguing neuropsychiatric disorder have yet to be fully elucidated (Goodwin and Jamison, 2007). The brain systems that have heretofore received the greatest attention in neurobiological studies of BPD have been the monoaminergic neurotransmitter systems, which are extensively distributed throughout the network of limbic, striatal, and prefrontal cortical neuronal circuits thought to support the behavioral and visceral manifestations of the disease (Drevets, 2000). Neurobiological studies of mood disorders over the last 40 years have primarily focused on abnormalities of these systems, on characterizing alterations of individual neurotransmitters in disease states, and on assessing response to mood stabilizer and antidepressant medications. Studies of cerebrospinal fluid chemistry, neuroendocrine responses to pharmacological challenge, and neuroreceptor and transporter binding have demonstrated a number of abnormalities in monoaminergic neurotransmitter and neuropeptide systems in mood disorders (Goodwin and Jamison, 2007).
Unfortunately, these observations have not yet greatly advanced our understanding of the underlying biology of recurrent mood disorders, which must include an explanation for the predilection to episodic, and often profound, mood disturbance that can become progressive over time. BPD likely arises from the complex interaction of multiple susceptibility (and protective) genes and environmental factors, and the phenotypic expression of the disease includes not only mood disturbance, but also a constellation of cognitive, motor, autonomic, endocrine, and sleep/wake abnormalities. Furthermore, while most antidepressants exert their initial effects by increasing intrasynaptic levels of serotonin and/or norepinephrine, their clinical antidepressant effects are observed only after chronic administration (over days to weeks), suggesting that a cascade of downstream events is ultimately responsible for their therapeutic effects. These observations have led to the idea that while dysfunction within the monoaminergic neurotransmitter systems is likely to play an important role in mediating some facets of the pathophysiology of BPD, it likely represents the downstream effects of other, more primary abnormalities in signaling pathways.
Plasticity, the ability to undergo and sustain change, is essential for the proper functioning of our nervous system. This capacity for change allows organisms to adapt to complex alterations in both their internal and external environments, a feature fundamentally important for survival and reproduction. The biological basis of this capacity to adapt encompasses a diverse set of cellular and molecular mechanisms that fall under the general term ‘neuroplasticity’; in this paper, we make the distinction between synaptic plasticity and neuroplasticity.
Synaptic plasticity refers to the cellular process that results in lasting changes in the efficacy of neurotransmission. More specifically, the term synaptic plasticity refers to the variability of the strength of a signal transmitted through a synapse. The regulation of transmission at the synapse may be mediated by changes in neurotransmitter levels, receptor subunit phosphorylation, surface/cellular levels of receptors, and conductance changes, among others.
Neuroplasticity is a broader term that encapsulates changes in intracellular signaling cascades and gene regulation, modifications of synaptic number and strength, variations in neurotransmitter release, modeling of axonal and dendritic architecture and, in some areas of the CNS, the generation of new neurons. Modifications arising from neuroplastic mechanisms can be of short duration or long lasting, and this is determined by the qualitative, quantitative, and temporal characteristics of the precipitating stimuli.
Research on the biological underpinnings of mood disorders has therefore moved away from focusing on absolute changes in neurochemicals such as monoamines and neuropeptides, and instead has begun highlighting the role of neural circuits and synapses, and the plastic processes controlling their function. Thus, these illnesses can best be conceptualized as genetically influenced disorders of synapses and circuits rather than simply as deficits or excesses in individual neurotransmitters (Bachmann et al, 2005; Schloesser et al, 2007). Most germane to the present discussion is the fact that it is now clear that mitochondria regulate not only long-term cell survival/cell death, but also immediate synaptic function—both of which are clearly very relevant for BPD. Indeed, Kato and co-workers had anticipated some of the recent developments in the field when they first proposed that mitochondrial dysfunction might play an important role in the pathophysiology of BPD (Kato and Kato, 2000; Kato et al, 2001; Murashita et al, 2000).
It is important to emphasize at the outset that it is not our contention that BPD is necessarily a classic mitochondrial disorder. Indeed, the vast majority of BPD patients do not show the symptoms of classic mitochondrial disorders (eg, optic and retinal atrophy, seizures, dementia, ataxia, myopathy, exercise intolerance, cardiac conduction defects, diabetes, and lactic acidosis; Fadic and Johns, 1996). Instead, emerging data suggest that upstream abnormalities (likely encoded in the nucleus) converge on mitochondrial function, leading to altered synaptic plasticity and impaired cellular resilience. In this paper, we synthesize and focus on the emerging data that support the contention that mitochondrial dysfunction may play a role in the impairments of cellular plasticity and resilience manifest in the context of BPD. It should be noted that it is beyond the scope of this paper to discuss in detail the myriad functions performed by mitochondria. Thus, we limit ourselves to a discussion of those facets most likely to play a role in the pathophysiology and treatment of BPD, namely intracellular calcium regulation, cytoprotection, and synaptic plasticity.
MITOCHONDRIA PLAY CRITICAL ROLES IN INTRACELLULAR CA2+ REGULATION, CYTOPROTECTION, AND REMODELING NEUROPLASTICITY
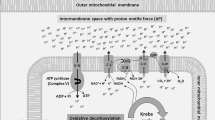
Mitochondrial physiology has the well-known function of energy production through the Krebs tricarboxylic acid cycle and oxidative phosphorylation. One byproduct of oxidative phosphorylation is the production of reactive oxidative species (ROS) that are capable of reacting with a wide variety of biological substrates, including protein thiol groups, membrane lipids, and nucleic acids, leading to cell damage and mutations.
However, mitochondria have additional important roles in the regulation of intracellular calcium (Ca2+), cytoprotection, and synaptic plasticity. Mitochondrial Ca2+ uptake from and release into the cytosol has important consequences for neuronal and glial activity, modulating both physiological and pathophysiological intracellular responses (Simpson and Russell, 1998). Calcium ions influence the synthesis and release of neurotransmitters, receptor signaling, the action potential, and neuronal periodicity (Kandel et al, 2000; Torok, 1989; Wolff et al, 1977). The diffusion of free Ca2+ ion in subcellular regions is normally discrete and short-lived (it is estimated to be free for ∼50 ms before encountering a Ca2+-binding protein); it is then sequestered in the mitochondria and endoplasmic reticulum (ER).
A large movement of positively charged Ca2+ into the mitochondrion will exert a depolarizing effect; most importantly for the present discussion, this increase can surpass mitochondrial capacity to export protons (as well as other cations), and it has the potential to lead to the cessation of ATP synthesis and the initiation of the apoptotic (programmed cell death) process (see below). On N-methyl-D-aspartate (NMDA) glutamate receptor activation, channel opening allows a rapid influx of Ca2+ into the cytosol and mitochondria that are able to rapidly buffer this load (Nicholls and Ward, 2000; Stout et al, 1998). Ca2+ uptake into mitochondria may activate the permeability transition pore (PTP, a channel crossing the outer and inner mitochondrial membranes) independently of ROS production (Chalmers and Nicholls, 2003; Figure 1).

Intracellular signaling pathways relevant to the pathophysiology of BPD and its role in synaptic and neuronal apoptosis. Several neurotransmitters exert their action through G protein-coupled receptors associated with PLC, which is involved in the PIP2 intracellular pathway; IP3 and DAG acting on the endoplasmic reticulum modify the intracellular concentration of Ca2+. Glutamate activation of NMDA receptors also induces a rapid influx of Ca2+ into the cytosol and subsequently, mitochondria. A large movement of positively charged Ca2+ into the mitochondrion will exert a depolarizing effect, surpassing mitochondrial capacity to export protons (as well as other cations), potentially leading to the cessation of ATP synthesis and the activation of the permeability transition pore—independently of ROS production—initiating apoptotic processes. The figure also depicts the neuroprotective role of the activation of G protein-coupled receptors associated with the activation of the PKA intracellular pathway, which mediates the phosphorylation and activation of CREB and upregulates bcl-2—an antiapoptotic protein that acts by increasing the stability of the PTP. Similar upregulation of bcl-2 occurs through the activation of Trk-B receptor by BDNF, through the MAP/ERK pathway, in addition to the interference against propapototic mitochondrial proteins (such as Bad). AC, adenylyl cyclase; AIF, apoptosis-inducing factor; Akt, protein kinase that inactivate GSK; ATP, adenosine triphosphate; Bad, pro-apoptotic protein regulated by RSK; Bag-1, bcl-2-binding antiapoptotic protein; bcl-2, antiapoptotic protein B-cell leukemia/lymphoma; BDNF, brain-derived neurotrophic factor; Ca2+, calcium; cAMP, cyclic adenosine monophosphate; CREB, cAMP response element-binding protein; Cyt c, cytochrome C; DAG, diacylglycerol; E Ret, endoplasmic reticulum; GC, glucocorticoid; Glu, glutamate; GR, glucocorticoid receptor; Gs, protein G stimulatory of adenylyl cyclase; GSK, glycogen synthase kinase; Gq, protein G stimulatory of phospholipase C; IP3, inositol 4,5-trisphosphate; IP3r, inositol triphosphate receptor; MAP/ERK, mitogen-activated protein kinase (MAP) pathway also referred to as extracellular signal-regulated kinase (ERK) pathway; NMDAr, N-methyl-D-aspartate receptor; NT, neurotransmitter and its G protein-coupled receptor; P, phosphate group; PIP2, phosphatidylinositol biphosphate; PI-3K, phosphatidylinositol 3-kinase; PKA, protein kinase A; PLC, phospholipase C; PTP, permeability transition pore; ROS, reactive oxidative species; RSK, kinase of ERK-MAP kinase cascade that downregulates Bad; TCF/LEF, transcription factors for specific genes; Trk-B, tyrosine kinase receptor.
The opening of the PTP has a number of important consequences including not only contributions to learning and synaptic plasticity (Weeber et al, 2002), but also cell death (Bernardi et al, 1998). Mitochondria immediately depolarize (stopping or reversing ATP synthesis) and a number of proteins are released from the intermembrane space. These include the proteins cytochrome c and apoptosis-inducing factor (AIF), which are known to lead to the activation in the cytosol of proteases (caspases). It is believed that this release is the first irreversible step of apoptosis, after which the cell is committed to undergo programmed cell death. Intriguingly, mounting evidence suggests that activation of mitochondrial apoptotic cascades may lead to a process of ‘synaptic apoptosis’ activated in a highly localized manner (Culmsee and Mattson, 2005). Subsequently, individual synapses or neurites may selectively undergo atrophy and provide a mechanism for synapse loss in both physiological and pathophysiological processes. Apoptotic signaling in the synaptic compartment appears to have some synapse-specific effects, such as the degradation of certain glutamate receptors (Glazner et al, 2000).
Importantly, a growing body of evidence suggests mitochondria may be integrally involved in the general processes of synaptic plasticity (Yang et al, 2003). The depolarization of presynaptic mitochondria has been shown to impair neurotransmitter release following tetanic stimulation (likely through the disruption of intracellular Ca2+ buffering; Billups and Forsythe, 2002). In addition, increased synaptic activity has been shown to induce the expression of mitochondrial-encoded genes, suggesting that a long-lasting upregulation of energy production may be triggered by synaptic activity itself, thereby playing a role in the long-term regulation of synaptic strength (Williams et al, 1998; for a review see Mattson and Liu, 2003; Figure 2). Thus, long-term mitochondrial dysfunction is associated with cell death, whereas more subtle mitochondrial dysfunction is associated with ‘here and now’ synaptic dysfunction.

The putative role of mitochondria in synaptic plasticity and possible consequences of mitochondrial pathology and/or treatment. Under healthy conditions (left side), the mitochondrion exerts several effects on presynaptic neurotransmitter release. The surge in intracellular calcium during an individual action potential is rapidly buffered by MMP-driven mitochondrial calcium uptake (1). The slow release of calcium back into the cytoplasm is thought to allow for post-tetanic potentiation (2). The continuous production of ATP and removal of ADP is essential for the energy demanding processes of vesicle docking, fusion, and endocytosis (3). Under pathological conditions (right side), many of these processes may be disrupted. Failure of the MMP reduces the calcium uptake affinity, thereby resulting in more pronounced calcium spikes and chaotic, desynchronized release (4). The return of mitochondrial calcium to the cytoplasm is subsequently impaired, possibly resulting in deficient post-tetantic potentiation (5). Failure of the MMP would also lead to a reduction in ATP production, resulting in more rapid fatigue of energy-dependant processes (6). Finally, abnormally elevated cytosolic calcium may inhibit the efficiency of endocytosis, further impairing subsequent release (7). ANT, adenine nucleotide translocase; ADP/ATP, adenosine di-/tri-phosphate; CU, calcium uniporter; MMP, mitochondrial membrane potential; VDCC, voltage-dependant calcium channel.
THE ROLE OF BCL-2 PROTEINS IN THE MITOCHONDRIA: CRITICAL ARBITERS OF CELLULAR PLASTICITY AND RESILIENCE
As we discuss in more detail later, the bcl-2 family of proteins are major targets for the actions of mood stabilizers; here, we provide a brief overview of their roles in regulating mitochondrial function. The bcl-2 family of proteins consists of both pro- and antiapoptotic proteins embedded in the inner mitochondrial membrane. They may also be present in nuclear membranes and in the ER. Thus, the expression and/or activation of proapoptotic bcl-2 family members (eg, bad and bax) increase mitochondrial membrane permeability, whereas the antiapoptotic members (eg, bcl-2 and bcl-xl) have the opposite effect. The most commonly observed effects of these antiapoptotic proteins are reductions in ROS production, the prevention of PTP opening, and, thus, mitochondrial depolarization (Zamzami et al, 1998). Notably, studies of isolated mitochondria have shown that bcl-2 overexpression increases mitochondria Ca2+ uptake capacity (Murphy et al, 1996), making the cells appear particularly resistant to the destructive influence of elevated intracellular Ca2+. Thus, bcl-2 may exert significant effects on cellular Ca2+ buffering, under the extreme conditions of Ca2+-induced apoptosis and potentially even during normal synaptic activity.
MITOCHONDRIAL GENETICS
Mitochondria are unique in that they are the only organelle to contain nonnuclear genetic information. Mitochondrial DNA (mtDNA) encodes 13 proteins, all components of the electron transport chain, as well as transfer and ribosomal RNAs, which suggests that mitochondria synthesize proteins independently of the mechanism used for nuclear genes; it should be noted, however, that the vast majority of proteins present in the mitochondrion are coded in the nucleus. Mitochondria have the unique properties of maternal inheritance and heteroplasmy. Heteroplasmy refers to the fact that different cells contain different numbers/types of mitochondria; thus, mitochondrial dysfunction may manifest in a very regional-specific manner. Partly because the female gamete contains vastly more cytoplasm (and therefore mitochondria) than the male gamete, the mtDNA of offspring is almost exclusively maternal in origin. Pertinent to the present discussion, it is noteworthy that some previous studies have noted a parent-of-origin effect in BPD (a pattern of maternal transmission; McMahon et al, 1995; Stine et al, 1995). It is also worth noting that mtDNA mutations occur at a much higher frequency than nuclear DNA mutations, likely owing to a combination of closer proximity to free radical production and less efficient DNA repair mechanisms (Richter et al, 1988). We now turn to a discussion of those aspects of mitochondrial function that have most relevance to the pathophysiology and treatment of BPD.
CALCIUM DYSREGULATION IN BPD: POTENTIAL ROLE FOR MITOCHONDRIAL CALCIUM-SEQUESTRATION MACHINERY
Impaired regulation of Ca2+ cascades is one of the most reproducible biological abnormalities described in BPD research (Tables 1 and 2). Studies have consistently revealed elevations in basal intracellular Ca2+ levels in platelets, lymphocytes, or neutrophils of patients with BPD. Higher platelet intracellular Ca2+ elevations have also been found in BPD patients in response to stimulation with thrombin, platelet activator factor (PAF), serotonin, dopamine, and thapsigargin when compared with healthy volunteers or unipolar depressive patients (Table 1). Similarly, higher elevations have been observed in lymphocytes when the cells were stimulated with phytohemagglutinin, concavalin A, thrombin, and, as in platelets, with thapsigargin and serotonin (Table 2). The fact that Ca2+ dysregulation is observed in response to such diverse stimuli suggests that the abnormality is not simply due to the activity of specific receptors, but rather due to a ‘downstream’ function. These observations strongly suggest that calcium release or sequestration (eg, by mitochondria) may represent a site of dysfunction in BPD. However, it is conceivable that the above-mentioned anomalies in peripheral cells are simply the consequence of the myriad other abnormalities of circulating factors (eg, catecholamines or glucocorticoids).
To address this potential confound, Warsh and co-workers used Epstein–Barr-virus-immortalized B lymphoblasts (BLCL) that they had grown in culture for weeks, thus elegantly removing the patients' confounding circulating environment (Yoon et al, 2001). They found that even in the immortalized lymphoblasts, BPD patients showed elevated basal Ca2+ concentration compared with healthy subjects or patients with other psychiatric disorders. In an extension of these studies, they investigated the components of the storage-operated Ca2+ entry (SOCE), and found a reduction in the mRNA expression of the TRPC7 (TRPM2) gene (whose gene product is implicated in SOCE functioning) in BLCLs from a subgroup of BPD I patients (Yoon et al, 2001). This refined work suggests that some calcium abnormalities in BPD are state-independent. As a corollary, it is now important to further delineate the specific nature of these abnormalities, their relationship to illness-state and their pathophysiological significance.
Most recently, Kato and associates (2003) investigated cytosolic and mitochondrial Ca2+ responses in lymphoblastoid cells from BPD subjects. They found that the thapsigargin-induced cytosolic Ca2+ response was significantly higher in patients with BPD (thapsigargin facilitates release of intracellular calcium, thereby bypassing specific receptors). Furthermore, using a mitochondrial uncoupler that abolishes mitochondrial Ca2+ uptake, they observed that responses differed significantly between mitochondrial DNA haplotypes reported to be associated with BPD (Kato et al, 2003). Together, these results clearly suggest that mitochondrial calcium regulation contributes to the Ca2+ abnormalities seen in BPD.
In addition to the mitochondrial contribution to intracellular Ca2+ regulation, Kato and co-workers identified XBP1, a pivotal gene in ER stress response, as contributing to the genetic risk factor for BPD (Kakiuchi et al, 2003). They demonstrated the impaired XBP1 expression after inducing ER stress associated with the polymorphism; these effects were countered by the mood stabilizer valproate (VPA). Finally, they observed that the XBP1C/G genotype was significantly associated with higher stress response in lymphoblastoid cells lines (Kakiuchi et al, 2003). Interestingly, another study that used a similar approach also noted decreased expression levels of NDUFV2 gene (a nuclear-encoded mitochondrial complex I subunit gene) in lymphoblastoid cells from BPD patients (Washizuka et al, 2003). Although these findings await independent replication, they are important in view of the growing body of evidence demonstrating that subcellular compartmentalization of Ca2+ and its source may be large determinants of neural ‘toxicity’ (Mattson et al, 2001; Sapolsky, 2000; Figure 1). It should be emphasized that here we refer not to the neural ‘life and death’ observed in classical neurodegenerative diseases, but to the more subtle atrophic changes observed in BPD, such as those associated with Ca2+ regulation phenomena (discussed in greater detail below).
COULD IMPAIRMENTS OF CELLULAR PLASTICITY AND RESILIENCE BE ASSOCIATED WITH MITOCHONDRIAL DYSFUNCTION IN BPD?
Although BPD is not a classical neurodegenerative disorder, structural neuroimaging studies have demonstrated regional volumetric reductions; these include reduced gray matter volumes in areas of the orbital and medial prefrontal cortex (PFC), temporal lobe, and enlargement of the third ventricle (reviewed by Manji et al, 2003). Recent postmortem neuropathological studies are complementary, showing reductions in cortex volume, region- and layer-specific reductions in number, density, and/or size of neurons and glial cells in the subgenual PFC, orbital cortex, dorsal anterolateral PFC, amygdala, and in basal ganglia and dorsal raphe nuclei in individuals with BPD and other severe mood disorders compared with controls (Manji et al, 2003). It is not currently known whether these alterations constitute developmental abnormalities conferring vulnerability to severe mood episodes, compensatory changes to other pathogenic processes, or the sequelae of recurrent affective episodes. However, a recent report showed that individuals at high risk for developing mood disorders exhibited reduced subgenual prefrontal cortical volumes, raising the possibility that this endophenotype may constitute a heritable vulnerability factor in these patients (Drevets et al, 2004). Overall, the reviewed data clearly show that BPD, undoubtedly a neurochemical illness, is also a disorder associated with impairments of cellular plasticity.
Magnetic resonance spectroscopy (MRS) has increasingly been used in the study of neuropsychiatric disorders. N-Acetyl-aspartate (NAA) is a predominant neurochemical compound that can be quantitatively assessed by MRS of the human brain. Pertinent to the present discussion is the fact that NAA is localized to mature neurons and synthesized within mitochondria. Interestingly, inhibitors of the mitochondrial respiratory chain decrease NAA concentrations, effects that correlate with reductions in ATP and oxygen consumption (Bates et al, 1996).
In BPD, decreased levels of NAA have been found in limbic and frontal cortex; these NAA reductions have been described (i) in hippocampus independent of mood state (Bertolino et al, 2003); (ii) in euthymic and medicated familial BPD I patients (Deicken et al, 2003); (iii) in the dorsolateral prefrontal cortex of euthymic and unmedicated adult BPD I and II patients (Winsberg et al, 2000); (iv) in orbitofrontal cortex of manic/mixed patients (Cecil et al, 2002); and (v) in dorsolateral prefrontal cortex of juvenile BPD patients (Chang et al, 2003). In addition, increased choline/NAA ratios were also found in the basal ganglia in both the depressive and euthymic states (Hamakawa et al, 1998). As discussed above, these findings may be the expression of underlying changes in ATP expenditure and availability, oxygen consumption, and/or glutamatergic activity in BPD. Similar changes in NAA have also been described in schizophrenia (mainly in prefrontal cortex) (Pae et al, 2004; Rowland et al, 2001; Theberge et al, 2004), indicating that some common pathophysiological mechanisms (either as cause or sequelae), may be present in these conditions. However, follow-up studies observing NAA changes after mood stabilizer treatment (described below) may indicate the particular relevance of this metabolite variation in BPD.
Furthermore, although there is some overlap in the MRS results between unipolar depression and BPD, higher specificity has been reported among bipolar populations. Decreased phosphocreatine levels and decreased baseline levels of beta- and total-nucleoside triphosphate have been reported in unipolar depressed subjects (reviewed by Iosifescu and Renshaw, 2003). In addition, Phosphorous-31 magnetic resonance spectroscopy (31P MRS) studies that permit the determination of high energy phosphate metabolism inside the brain have reported normal (Kato et al, 1992) and decreased (Volz et al, 1998) phosphomonoester levels in subjects with unipolar depression compared with controls. Interestingly, independent literature reviews have also suggested that unipolar depression and BPD may possess different pathophysiological bases due to the different direction of the changes reported in H-MRS studies (Yildiz-Yesiloglu and Ankerst, 2006). In fact, a significant role for the phosphoinositol cycle/myoinositol was reported in the pathophysiology of BPD, whereas there was less evidence for a similar role in any other psychiatric condition (Kim et al, 2005).
Additional 31P MRS studies have shown a decrease in phosphocreatine (PCr) and/or ATP levels in patients with BPD, and in one report that studied medicated patients with unipolar depression (Deicken et al, 1995; Kato et al, 1995; Volz et al, 1998). Kato and co-workers have conducted the most extensive series of studies investigating possible abnormalities in brain energy regulation in mood disorders. Consistent with the decreased PCr and ATP levels discussed above, this research group has also found low pH levels in patients with BPD compared with normal controls in the frontal cortex, in basal ganglia, and in the whole brain (Hamakawa et al, 2004; Kato et al, 1998, 2003). Indeed, these observations were among the first to lead to the postulation that BPD may be associated with mitochondrial dysfunction.
Findings of both decreased pH and increased lactate in BPD led to the very recent proposal that BPD subjects exhibit a shift away from oxidative phosphorylation towards glycolysis, thus reducing efficiency and total energy output (Stork and Renshaw, 2005). This suggests that impairment in mitochondrial functioning, which is normally responsible for the oxidative phosphorylation process, might cause the shift changes observed in BPD. In addition, if the respiratory chain of cellular metabolism were less available, the energy production would be displaced towards anaerobic glycolysis, consequently increasing lactate production (Stork and Renshaw, 2005). Notably, mitochondrial dysfunction is associated with reduced pH and increases in lactate. Thus, the seemingly distinct neuroimaging findings in BPD (reduced pH, increased lactate, reduced high energy phosphates, reduced NAA, and regional volumetric reductions) may all be linked by mitochondrial dysfunction as the more proximal cause (Stork and Renshaw, 2005).
ABNORMAL GENE EXPRESSION OF KEY MITOCHONDRIAL PROTEINS IN BPD
Additional evidence of dysregulated mitochondrial processes in BPD comes from an elegant series of postmortem brain microarray studies by Konradi et al (2004) (Table 3). They used gene arrays to analyze 12 558 nuclear genes in hippocampi from three different groups (healthy controls, BPD patients, and schizophrenia patients). Employing stringent statistical analyses, they found that the expression of only 43 genes was decreased in BPD compared with schizophrenia. Notably, 42% of the genes that were reduced in BPD brains coded for mitochondrial proteins and are involved in regulating oxidative phosphorylation in the mitochondrial inner membrane (including subunits of complexes I (NADH dehydrogenase in one gene), IV (cytochrome c oxidase in one gene), and V (ATP synthase in five genes) (Konradi et al, 2004)). These findings, as well as observations of decreased expression of the enzyme glutamic acid decarboxylase 67 and somatostatin (Heckers et al, 2002), indicate abnormal functioning of a subset of hippocampal interneurons in BPD; the possibility that dysfunction of these hippocampal interneurons involves abnormal mitochondrial energy metabolism is an intriguing hypothesis. Benes et al (2006) recently showed a marked downregulation of antioxidant genes in BPD (Table 3). These authors suggested that accumulation of free radicals might then occur in the setting of a previously reported decrease of the electron transport chain. In addition, a large number of apoptotic genes were reported to be upregulated in the hippocampus of BPD subjects (Benes et al, 2006).
Exciting findings pointing in this direction were also reported by Young and co-workers (Sun et al, 2006; Table 3). They showed that genes differentially expressed in subjects with BPD included genes in the mitochondrial electron transport chain. These genes included those responsible for the downregulation of NADH-ubiquinone oxidoreductase (20- and 23-kDa subunits, complex I), cytochrome c oxidase polypeptides (complex IV), and ATP synthase lipid-binding protein and gamma chain (complex V), all confirmed by PCR. Notably, expression of the NADH-ubiquinone oxidoreductase subunit was increased in subjects with ongoing lithium treatment (Sun et al, 2006), which may play a role in lithium's neuroprotective action (discussed below).
In subjects with a history of alcohol abuse or dependence, changes in gene expression from temporal cortex encoding mitochondrial proteins, the ubiquitin system, and signal transduction have also been observed (Sokolov et al, 2003). Thus, caution is needed when interpreting the results of postmortem brain studies due to the numerous potentially confounding factors, including substance abuse/dependence (a comorbidity often seen in BPD subjects), antemortem medication history, postmortem interval, and cause of death. In this respect, it is important to note that the phosphorylation state of proteins is extremely sensitive to postmortem interval (Li et al, 2003), as are decreased tissue pH and increased RNA degradation (Catts et al, 2005). Observed changes in pH could be related to the disease, or to agonal factors (Li et al, 2004; Iwamoto et al, 2005), or be due to postmortem changes.
Whether reduced pH in the postmortem tissue itself may be due to mitochondrial dysfunction is currently an area of ongoing research. Recent work by Vawter et al (2006) speaks to this issue. The work carefully contrasted control brains with low versus high pH, showing that 28% of genes in mitochondrially related pathways meet criteria for differential expression. Among several interesting results, they confirmed the relevance of the agonal-pH state effect in postmortem brain studies involving mitochondrial gene expression. Notably, when these authors controlled for these factors, potential candidate genes emerged; these genes, confirmed by Q-PCR, included the NR4A1 (mitochondrial) and HSPA2 (apoptotic chaperone) genes (Vawter et al, 2006). Considering that in humans there is no control for several antemortem factors (including medication status), it is not surprising that post-mortem findings are not entirely consistent. Thus, it is possible that lower postmortem pH findings—due to agonal factors known to be related to lower RNA integrity—may explain the gene expression changes. This issue remains to be addressed in subsequent studies.
The postmortem brain findings described above receive additional, indirect support from a recent peripheral cell study that effectively demonstrated a deficient adaptation to an energy–stress paradigm on peripheral cells from BPD subjects (Naydenov et al, 2007). They showed that lymphocytes from BPD individuals, cultured in medium with glucose deprivation, downregulated nuclear transcripts for proteins of the electron transfer chain, whereas the opposite effect was observed in the control samples (Naydenov et al, 2007).
MITOCHONDRIAL DYSFUNCTION AND OTHER NEUROPSYCHIATRIC CONDITIONS
Alterations in mitochondrial function are likely to play a role in the pathophysiology of neurodegenerative conditions including Huntington's disease, Parkinson's disease, amyotrophic lateral sclerosis (ALS), and, possibly, schizophrenia. Of interest, results from postmortem studies in schizophrenia include changes in cytochrome c oxidase activity (complex IV) in several cortical areas and subcortical nucleus (Cavelier et al, 1995; Maurer et al, 2001; Prince et al, 1999), complexes I and III (Maurer et al, 2001), as well as mRNA and protein levels of complex I proteins (Karry et al, 2004). Other studies have confirmed changes in complex I (Ben-Shachar et al, 1999), but, conversely, not complex IV activity (Dror et al, 2002) in platelets from schizophrenic patients. However, this evidence should be interpreted with caution considering the reported effect of antipsychotic medications on mitochondrial function (Balijepalli et al, 1999; Modica-Napolitano et al, 2003).
In toto, the data for alterations in the oxidative phosphorylation in brains of patients with schizophrenia are not entirely consistent. However, it is possible that common mitochondrial dysfunction may underlie pathophysiological changes and phenotypic expression across a number of neuropsychiatric conditions, particularly schizophrenia and BPD; these observations are noteworthy because, although these are undoubtedly discrete illnesses, a growing body of evidence suggests genetic and pathophysiological overlap among them (DePaulo, 2004).
We now turn to a discussion of the converging evidence that mood stabilizers regulate mitochondrial function.
MOOD STABILIZERS EXERT MAJOR EFFECTS ON PROTEINS KNOWN TO REGULATE MITOCHONDRIAL FUNCTION
The possible involvement of bcl-2 in the pathophysiology and treatment of BPD initially arose from mRNA differential display studies that suggested that bcl-2 might represent a common target for the actions of both chronic lithium and VPA (Chen et al, 1999). Chronic treatment of rats with therapeutic doses of lithium and VPA doubled bcl-2 levels in the frontal cortex, an effect due primarily to a marked increase in the number of bcl-2 immunoreactive cells in layers II and III of the anterior cingulated cortex (Chen et al, 1999; Manji et al, 1999, 2000a). Interestingly, the importance of neurons in the anterior cingulate has recently been emphasized in neuroimaging studies of BPD, particularly because these areas provide connections with other cortical regions and are targets for subcortical input (Rajkowska, 2000). Chronic lithium was also found to markedly increase the number of bcl-2 immunoreactive cells in the dentate gyrus and striatum (Manji et al, 1999). Subsequent to these findings, lithium was shown to increase bcl-2 levels in C57BL/6 mice (Chen and Chuang, 1999), in human neuroblastoma SH-SY5Y cells in vitro (Manji et al, 2000b) and in rat cerebellar granule cells in vitro (Chen and Chuang, 1999).
Overall, the data clearly show that chronic lithium robustly increases levels of the neuroprotective protein bcl-2 in areas of rodent frontal cortex, hippocampus, and striatum in vivo, and in cultured cells of both rodent and human neuronal origin in vitro. Furthermore, at least in cultured cell systems, lithium reduces levels of the proapoptotic protein p53. As demonstrated recently, repeated electroconvulsive shock also significantly increases precursor cell proliferation in the dentate gyrus of the adult monkey, an effect that appears to be due to increased expression of bcl-2 (Perera et al, 2007).
Another target of mood stabilizers that may be relevant to mitochondrial function is glycogen synthase kinase 3 (GSK-3), a constitutively active kinase that is known to inhibit the mitochondrial multiprotein complex pyruvate dehydrogenase (PDH) (Hoshi et al, 1996)—notably, lithium and possibly VPA are known to inhibit this kinase (Gould and Manji, 2002). PDH acts as a key linkage (and point of regulation) between glycolysis and the tricarboxylic acid cycle. Inhibition of GSK-3 by mood stabilizers may lead to disinhibition of PDH, thus to an increase in maximal metabolic rate, and a more adequate supply of ATP in tissues with high energy requirements, such as brain.
Recent data also suggest that the mitochondrion is likely one of the main sites of action of GSK-3. Although GSK-3 is present throughout the cell, analysis of subcellular fractions has shown that GSK-3 in nuclei and mitochondria has a higher level of basal activity (Bijur and Jope, 2003; Jope, 2003); these pools are selectively activated by proapoptotic stimuli. GSK-3 exerts proapoptotic neuronal effects by regulating mitochondrial localization of bax (Linseman et al, 2004), whereas GSK-3 inhibition has been shown to inhibit or delay activation of PTP (Juhaszova et al, 2004; Murphy, 2004). Thus, the observation that lithium exerts its inhibitory effects on GSK-3 within mitochondria is noteworthy (Bijur and Jope, 2003). In a complementary fashion, lithium and VPA may also modulate intracellular Ca2+ signaling (an effect probably associated with the antiapoptotic properties of these mood stabilizers) by suppressing the activational phosphorylation of the NR2B subunit of the NMDA receptor (Hashimoto et al, 2002). Consistent with these effects on major antiapoptotic proteins, several studies have now demonstrated that lithium and VPA attenuate the activation of pro-apoptotic cascades (Pan et al, 2005; Lai et al, 2006; Yeste et al, 2007; Chen et al, 2006; Shao et al, 2005; Cui et al, 2007).
Lithium exerts robust neuroprotective effects in preclinical paradigms
In view of its major effects on BDNF, bcl-2 and GSK-3, it is not surprising that recent studies have investigated lithium's potential neuroprotective effects in a variety of preclinical paradigms, demonstrating robust neuroprotective properties against a variety of insults (reviewed by Bachmann et al, 2005; Chuang and Priller, 2006; Manji et al, 2000a; Schloesser et al, 2007).
Notably, lithium pretreatment has been demonstrated to protect cultured brain neurons from glutamate-induced, NMDA receptor-mediated apoptosis (reviewed by (Chuang and Priller, 2006). Excessive NMDA throughput is likely involved in stress-induced hippocampal atrophy, and has been implicated in the pathogenesis of a variety of neurodegenerative diseases such as stroke, Huntington's disease, ALS, spinal cord injury, brain trauma, and cerebellar degeneration. In cultured neurons, lithium-induced neuroprotection against glutamate excitotoxicity occurs within the therapeutic concentration range of this drug and requires 5–6 days of pretreatment for maximal effects. The lithium neuroprotection requires BDNF induction and activation of its receptor TrkB, and is associated with upregulation of bcl-2, downregulation of the proapoptotic proteins p53 and Bax, and inhibition of caspase-3. Treatment of cultured neurons with other GSK-3 inhibitors or transfection with GSK-3 siRNA mimics the neuroprotective effects of lithium (Liang and Chuang, 2007), again suggesting a critical role of GSK-3 in mediating neuroprotection.
Lithium also shows beneficial effects in a number of animal models of neurodegenerative diseases. For example, pre- or post-insult treatment with lithium suppresses cerebral ischemia-induced brain infarction, caspase-3 activation, and neurological deficits in rats, and these neuroprotective effects are associated with induction of heat shock protein 70 and decreased expression of Bax (Ren et al, 2003; Xu et al, 2003). Several independent studies demonstrated that lithium has neuroprotective effects in animal and cellular models of Alzheimer's disease, Huntington's disease, Parkinson's disease, retinal degeneration, spinal cord injury, and HIV infection (reviewed by (Chuang and Priller, 2006). Notably, Phiel et al (2003) demonstrated that therapeutic concentrations of lithium, by acting on GSK-3, blocked the production of Aβ peptides by interfering with amyloid peptide precursor protein (APP) cleavage at the γ-secretase step. Importantly, lithium also blocked the accumulation of Aβ peptides in the brains of mice that overproduce APP.
Similarly, lithium administration has been shown to significantly lower levels of phosphorylation at several epitopes of tau known to be hyperphosphorylated in Alzheimer's disease and to significantly reduce levels of aggregated, insoluble tau (Noble et al, 2005). Furthermore, levels of aggregated tau correlated strongly with degree of axonal degeneration, and lithium-treated mice showed less degeneration if administration was started during early stages of tangle development. Most recently, it has been demonstrated that lithium is neuroprotective in APP transgenic mice (Rockenstein et al, 2007). Thus, mice treated with lithium exhibited improved performance in the water maze, preservation of the dendritic structure in the frontal cortex and hippocampus, and decreased tau phosphorylation (Rockenstein et al, 2007). Chronic lithium treatment also protects against neurodegeneration and improves spatial learning deficits in rats perfused with Aβ fibrils (de Ferrari et al, 2003).
HUMAN EVIDENCE SUPPORTING THE NEUROTROPHIC EFFECTS OF MOOD STABILIZERS
Almost a decade ago, volumetric MRI studies demonstrated that familial BPD patients had approximately 40% lower gray matter volumes in the left subgenual prefrontal cortex; a subsequent re-analysis of these data showed that patients treated with chronic lithium or VPA did not exhibit the subgenual prefrontal cortex volumetric reduction (discussed by Gould et al, 2004; Quiroz et al, 2004).
To examine potential neurotrophic effects of lithium more directly, several longitudinal studies have recently been undertaken. Chronic lithium administration at therapeutic doses was found to increase NAA concentrations in the human brain in vivo (Moore et al, 2000); furthermore, a correlation of approximately 0.97 between lithium-induced NAA increases and regional voxel gray matter content was observed. Interestingly, the region-specific bcl-2 increases induced by lithium in rodent brain cortices (eg, gray versus white matter) colocalize with the aforementioned areas. In follow-up studies, it was hypothesized that lithium-induced mitochondrially-mediated trophic effects would lead to neuropil increases and consequently to increased brain gray matter volume in BPD patients. Thus, gray matter volumes were quantified at baseline and then repeated after 4 weeks of lithium at therapeutic doses (Moore et al, 2000). Chronic lithium was found to significantly increase total gray matter content; no significant changes were observed in brain white matter volume or in quantitative measures of the regional cerebral water content, thereby providing evidence that the observed increases in gray matter content were due to neurotrophic effects as opposed to any possible cell swelling and/or osmotic effects associated with lithium treatment (Moore et al, 2000). In addition, several independent cross-sectional studies have now demonstrated that lithium-treated patients with BPD show increased gray matter volumes compared to untreated BPD patients (Sassi et al, 2002, 2004; Chang et al, 2005; Bearden et al, 2007).
Most recently, another study was undertaken in well-characterized BPD-depressed subjects (n=28) at baseline (medication free), and following chronic lithium administration (4 weeks) (Moore et al, 2005). Significant increases in total gray matter volume were observed in BPD subjects following chronic lithium administration, confirming the previous preliminary study. Moreover, only responders showed increases in gray matter in the left subgenual prefrontal cortex (Moore et al, 2005). These data suggest that lithium—likely through its effect on mitochondrial functioning—produces a reversal of illness-related atrophy. These findings may have implications not only for long-term functional outcome, but also for ‘here and now’ symptom resolution.
CONCLUDING REMARKS
We have outlined here the evidence supporting the contention that mitochondrial function is integral to many facets of BPD. Mitochondria are intracellular organelles best known for their critical roles in regulating energy production through oxidative phosphorylation, regulation of Ca2+, and as critical mediators of cellular apoptosis. However, increasing evidence suggests that mitochondria may be integrally involved in the general processes of synaptic plasticity. Indeed, increased synaptic activity has been shown to induce the expression of mitochondrially encoded genes, suggesting that the regulation of metabolism is an important component in the long-term regulation of synaptic strength. These findings suggest that mitochondrial Ca2+ sequestration plays a key role in modulating the tone of synaptic plasticity in a variety of neuroanatomical circuitry and regions mediating complex behaviors, including those implicated in the pathophysiology of mood disorders.
It needs to be reiterated that although some studies suggest a parent-of-origin effect, we are not implying that BPD is a classic mitochondrial disorder, but rather that many upstream abnormalities (likely nuclear genome coded) converge to regulate mitochondrial function implicated both in abnormalities of neurotransmitter synaptic plasticity and long-term cellular resilience.
Hovatta et al (2005) recently used a combination of behavioral analysis and quantitative gene expression profiling of several brain regions in six inbred mouse strains. They found that genes involved in oxidative stress metabolism were related to complex affective behaviors. Together, these results suggest that the mitochondria-mediated impairments of plasticity observed in BPD may have ramifications not only for long-term disease progression/course of illness/functional impairments, but also for BPD symptomatology. Indeed, as previously mentioned, it has recently been demonstrated that short-term lithium-induced increases in subgenual prefrontal cortex gray matter were related to treatment response (Moore et al, 2005).
There has, unfortunately, been little progress in developing truly novel drugs specifically for the treatment of BPD, and most recent additions to the pharmacopeia are brain-penetrant medications developed for the general treatment of epilepsy or schizophrenia. Here, we have outlined evidence—derived from neuroimaging, postmortem brain, biochemical, and pharmacological studies—to support the contention that mitochondrial function is integral to many facets of BPD. These observations raise the intriguing possibility that enhancing mitochondrial vigor may represent an important adjunctive strategy for the optimal long-term treatment of BPD. Novel molecular targets to improve mitochondrial function include pharmacological attempts to bypass defects in the respiratory chain, scavenging excessive oxygen radicals (reviewed by Dimauro et al, 2004), and enhancers of mitochondrial membrane stabilization, including, theoretically, inhibitors of PTP. These developments hold much promise for the discovery of new therapeutics for this devastating illness.
References
Bachmann RF, Schloesser RJ, Gould TD, Manji HK (2005). Mood stabilizers target cellular plasticity and resilience cascades: implications for the development of novel therapeutics. Mol Neurobiol 32: 173–202.
Balijepalli S, Boyd MR, Ravindranath V (1999). Inhibition of mitochondrial complex I by haloperidol: the role of thiol oxidation. Neuropharmacology 38: 567–577.
Bates TE, Strangward M, Keelan J, Davey GP, Munro PM, Clark JB (1996). Inhibition of N-acetylaspartate production: implications for 1H MRS studies in vivo. Neuroreport 7: 1397–1400.
Bearden CE, Thompson PM, Dalwani M, Hayashi KM, Lee AD, Nicoletti M et al (2007). Greater cortical gray matter density in lithium-treated patients with bipolar disorder. Biol Psychiatry 62: 7–16.
Ben-Shachar D, Zuk R, Gazawi H, Reshef A, Sheinkman A, Klein E (1999). Increased mitochondrial complex I activity in platelets of schizophrenic patients. Int J Neuropsychopharmcol 2: 245–253.
Benes FM, Matzilevich D, Burke RE, Walsh J (2006). The expression of proapoptosis genes is increased in bipolar disorder, but not in schizophrenia. Mol Psychiatry 11: 241–251.
Berk M, Bodemer W, van Oudenhove T, Butkow N (1994). Dopamine increases platelet intracellular calcium in bipolar affective disorder and controls. Int Clin Psychopharmacol 9: 291–293.
Bernardi P, Colonna R, Costantini P, Eriksson O, Fontaine E, Ichas F et al (1998). The mitochondrial permeability transition. Biofactors 8: 273–281.
Bertolino A, Frye M, Callicott JH, Mattay VS, Rakow R, Shelton-Repella J et al (2003). Neuronal pathology in the hippocampal area of patients with bipolar disorder: a study with proton magnetic resonance spectroscopic imaging. Biol Psychiatry 53: 906–913.
Bijur GN, Jope RS (2003). Glycogen synthase kinase-3beta is highly activated in nuclei and mitochondria. Neuroreport 14: 2415–2419.
Billups B, Forsythe ID (2002). Presynaptic mitochondrial calcium sequestration influences transmission at mammalian central synapses. J Neurosci 22: 5840–5847.
Bothwell RA, Eccleston D, Marshall E (1994). Platelet intracellular calcium in patients with recurrent affective disorders. Psychopharmacology (Berl) 114: 375–381.
Bowden CL, Huang LG, Javors MA, Johnson JM, Seleshi E, McIntyre K et al (1988). Calcium function in affective disorders and healthy controls. Biol Psychiatry 23: 367–376.
Catts VS, Catts SV, Fernandez HR, Taylor JM, Coulson EJ, Lutze-Mann LH et al (2005). A microarray study of post-mortem mRNA degradation in mouse brain tissue. Post-mortem interval effects on the phosphorylation of signaling proteins. Brain Res Mol Brain Res 138: 164–177.
Cavelier L, Jazin EE, Eriksson I, Prince J, Bave U, Oreland L et al (1995). Decreased cytochrome-c oxidase activity and lack of age-related accumulation of mitochondrial DNA deletions in the brains of schizophrenics. Genomics 29: 217–224.
Cecil KM, DelBello MP, Morey R, Strakowski SM (2002). Frontal lobe differences in bipolar disorder as determined by proton MR spectroscopy. Bipolar Disord 4: 357–365.
Chalmers S, Nicholls DG (2003). The relationship between free and total calcium concentrations in the matrix of liver and brain mitochondria. J Biol Chem 278: 19062–19070.
Chang K, Adleman N, Dienes K, Barnea-Goraly N, Reiss A, Ketter T (2003). Decreased N-acetylaspartate in children with familial bipolar disorder. Biol Psychiatry 53: 1059–1065.
Chang K, Barnea-Goraly N, Karchemskiy A, Simeonova DI, Barnes P, Ketter T et al (2005). Cortical magnetic resonance imaging findings in familial pediatric bipolar disorder. Biol Psychiatry 58: 197–203.
Chen CL, Lin CF, Chiang CW, Jan MS, Lin YS (2006). Lithium inhibits ceramide- and etoposide-induced protein phosphatase 2A methylation, Bcl-2 dephosphorylation, caspase-2 activation, and apoptosis. Mol Pharmacol 70: 510–517.
Chen G, Zeng WZ, Yuan P, Huang LD, Jiang YM, Zhao ZH et al (1999). The mood-stabilizing agents lithium and valproate robustly increase the levels of the neuroprotective protein bcl-2 in the CNS. J Neurochem 72: 879–882.
Chen RW, Chuang DM (1999). Long term lithium treatment suppresses p53 and Bax expression but increases Bcl-2 expression: a prominent role in neuroprotection against excitotoxicity. J Biol Chem 274: 6039–6042.
Chuang DM, Priller J (2006). Potential use of lithium in neurodegenerative disorders. In: Bauer M, Grof P, Muller-Oerlinghausen B (eds). Lithium in Neuropsychiatry: the Comprehensive Guide. Informa UK Ltd: Abingdon: Oxon. pp 381–398.
Cui J, Shao L, Young LT, Wang JF (2007). Role of glutathione in neuroprotective effects of mood stabilizing drugs lithium and valproate. Neuroscience 144: 1447–1553.
Culmsee C, Mattson MP (2005). p53 in neuronal apoptosis. Biochem Biophys Res Commun 331: 761–777.
de Ferrari GV, Chacon MA, Barria MI, Garrido JL, Godoy JA, Olivares G et al (2003). Activation of Wnt signaling rescues neurodegeneration and behavioral impairments induced by beta-amyloid fibrils. Mol Psychiatry 8: 195–208.
Deicken RF, Fein G, Weiner MW (1995). Abnormal frontal lobe phosphorous metabolism in bipolar disorder. Am J Psychiatry 152: 915–918.
Deicken RF, Pegues MP, Anzalone S, Feiwell R, Soher B (2003). Lower concentration of hippocampal N-acetylaspartate in familial bipolar I disorder. Am J Psychiatry 160: 873–882.
DePaulo Jr JR (2004). Genetics of bipolar disorder: where do we stand? Am J Psychiatry 161: 595–597.
Dimauro S, Mancuso M, Naini A (2004). Mitochondrial encephalomyopathies: therapeutic approach. Ann NY Acad Sci 1011: 232–245.
Drevets WC (2000). Neuroimaging studies of mood disorders. Biol Psychiatry 48: 813–829.
Drevets WC, Ryan CM, Bogers WD, Birmaher B, Axelson D, Dahl RE (2004). Subgenual prefrontal cortex volume decreased in healthy humans at high familial risk for mood disorders. Society for Neuroscience. Washington, DC: Program No. 799.19.
Dror N, Klein E, Karry R, Sheinkman A, Kirsh Z, Mazor M et al (2002). State-dependent alterations in mitochondrial complex I activity in platelets: a potential peripheral marker for schizophrenia. Mol Psychiatry 7: 995–1001.
Dubovsky SL, Christiano J, Daniell LC, Franks RD, Murphy J, Adler L et al (1989). Increased platelet intracellular calcium concentration in patients with bipolar affective disorders. Arch Gen Psychiatry 46: 632–638.
Dubovsky SL, Lee C, Christiano J, Murphy J (1991). Elevated platelet intracellular calcium concentration in bipolar depression. Biol Psychiatry 29: 441–450.
Dubovsky SL, Murphy J, Thomas M, Rademacher J (1992). Abnormal intracellular calcium ion concentration in platelets and lymphocytes of bipolar patients. Am J Psychiatry 149: 118–120.
Dubovsky SL, Thomas M, Hijazi A, Murphy J (1994). Intracellular calcium signalling in peripheral cells of patients with bipolar affective disorder. Eur Arch Psychiatry Clin Neurosci 243: 229–234.
Emamghoreishi M, Schlichter L, Li PP, Parikh S, Sen J, Kamble A et al (1997). High intracellular calcium concentrations in transformed lymphoblasts from subjects with bipolar I disorder. Am J Psychiatry 154: 976–982.
Fadic R, Johns DR (1996). Clinical spectrum of mitochondrial diseases. Semin Neurol 16: 11–20.
Forstner U, Bohus M, Gebicke-Harter PJ, Baumer B, Berger M, van Calker D (1994). Decreased agonist-stimulated Ca2+ response in neutrophils from patients under chronic lithium therapy. Eur Arch Psychiatry Clin Neurosci 243: 240–243.
Glazner GW, Chan SL, Lu C, Mattson MP (2000). Caspase-mediated degradation of AMPA receptor subunits: a mechanism for preventing excitotoxic necrosis and ensuring apoptosis. J Neurosci 20: 3641–3649.
Goodwin FK, Jamison KR (2007). Manic-Depressive Illness: Bipolar Disorder and Recurrent Depression Vol 2nd edn. Oxford University Press: New York.
Gould TD, Manji HK (2002). The Wnt signaling pathway in bipolar disorder. Neuroscientist 8: 497–511.
Gould TD, Quiroz JA, Singh JB, Zarate Jr CA, Manji HK (2004). Emerging experimental therapeutics for bipolar disorder: insights from the molecular and cellular actions of current mood stabilizers. Mol Psychiatry 9: 734–755.
Hamakawa H, Kato T, Murashita J, Kato N (1998). Quantitative proton magnetic resonance spectroscopy of the basal ganglia in patients with affective disorders. Eur Arch Psychiatry Clin Neurosci 248: 53–58.
Hamakawa H, Murashita J, Yamada N, Inubushi T, Kato N, Kato T (2004). Reduced intracellular pH in the basal ganglia and whole brain measured by 31P-MRS in bipolar disorder. Psychiatry Clin Neurosci 58: 82–88.
Hashimoto R, Hough C, Nakazawa T, Yamamoto T, Chuang DM (2002). Lithium protection against glutamate excitotoxicity in rat cerebral cortical neurons: involvement of NMDA receptor inhibition possibly by decreasing NR2B tyrosine phosphorylation. J Neurochem 80: 589–597.
Heckers S, Stone D, Walsh J, Shick J, Koul P, Benes FM (2002). Differential hippocampal expression of glutamic Acid decarboxylase 65 and 67 messenger RNA in bipolar disorder and schizophrenia. Arch Gen Psychiatry 59: 521–529.
Hoshi M, Takashima A, Noguchi K, Murayama M, Sato M, Kondo S et al (1996). Regulation of mitochondrial pyruvate dehydrogenase activity by tau protein kinase I/glycogen synthase kinase 3beta in brain. Proc Natl Acad Sci USA 93: 2719–2723.
Hough C, Lu SJ, Davis CL, Chuang DM, Post RM (1999). Elevated basal and thapsigargin-stimulated intracellular calcium of platelets and lymphocytes from bipolar affective disorder patients measured by a fluorometric microassay. Biol Psychiatry 46: 247–255.
Hovatta I, Tennant RS, Helton R, Marr RA, Singer O, Redwine JM et al (2005). Glyoxalase 1 and glutathione reductase 1 regulate anxiety in mice. Nature 438: 662–666.
Iosifescu DV, Renshaw PE (2003). 31P-magnetic resonance spectroscopy and thyroid hormones in major depressive disorder: toward a bioenergetic mechanism in depression? Harv Rev Psychiatry 11: 51–63.
Iwamoto K, Bundo M, Kato T (2005). Altered expression of mitochondria-related genes in postmortem brains of patients with bipolar disorder or schizophrenia, as revealed by large-scale DNA microarray analysis. Hum Mol Gemt 14: 241–253.
Jope RS (2003). Lithium and GSK-3: one inhibitor, two inhibitory actions, multiple outcomes. Trends Pharmacol Sci 24: 441–443.
Juhaszova M, Zorov DB, Kim SH, Pepe S, Fu Q, Fishbein KW et al (2004). Glycogen synthase kinase-3beta mediates convergence of protection signaling to inhibit the mitochondrial permeability transition pore. J Clin Invest 113: 1535–1549.
Kakiuchi C, Iwamoto K, Ishiwata M, Bundo M, Kasahara T, Kusumi I et al (2003). Impaired feedback regulation of XBP1 as a genetic risk factor for bipolar disorder. Nat Genet 35: 171–175.
Kandel ER, Schwartz JH, Jessell TM (2000). Principles of Neural Sciences 4th edn. McGraw-Hill: New York, NY.
Karry R, Klein E, Ben Shachar D (2004). Mitochondrial complex I subunits expression is altered in schizophrenia: a postmortem study. Biol Psychiatry 55: 676–684.
Kato T, Ishiwata M, Mori K, Washizuka S, Tajima O, Akiyama T et al (2003). Mechanisms of altered Ca2+ signalling in transformed lymphoblastoid cells from patients with bipolar disorder. Int J Neuropsychopharmacol 6: 379–389.
Kato T, Kato N (2000). Mitochondrial dysfunction in bipolar disorder. Bipolar Disord 2: 180–190.
Kato T, Kunugi H, Nanko S, Kato N (2001). Mitochondrial DNA polymorphisms in bipolar disorder. J Affect Disord 62: 151–164.
Kato T, Murashita J, Kamiya A, Shioiri T, Kato N, Inubushi T (1998). Decreased brain intracellular pH measured by 31P-MRS in bipolar disorder: a confirmation in drug-free patients and correlation with white matter hyperintensity. Eur Arch Psychiatry Clin Neurosci 248: 301–306.
Kato T, Shioiri T, Murashita J, Hamakawa H, Takahashi Y, Inubushi T et al (1995). Lateralized abnormality of high energy phosphate metabolism in the frontal lobes of patients with bipolar disorder detected by phase-encoded 31P-MRS. Psychol Med 25: 557–566.
Kato T, Takahashi S, Shioiri T, Inubushi T (1992). Brain phosphorous metabolism in depressive disorders detected by phosphorus-31 magnetic resonance spectroscopy. J Affect Disord 26: 223–230.
Kim H, McGrath BM, Silverstone PH (2005). A review of the possible relevance of inositol and the phosphatidylinositol second messenger system (PI-cycle) to psychiatric disorders—focus on magnetic resonance spectroscopy (MRS) studies. Hum Psychopharmacol Clin Exp 20: 309–326.
Konradi C, Eaton M, MacDonald ML, Walsh J, Benes FM, Heckers S (2004). Molecular evidence for mitochondrial dysfunction in bipolar disorder. Arch Gen Psychiatry 61: 300–308.
Kusumi I, Koyama T, Yamashita I (1992). Thrombin-induced platelet calcium mobilization is enhanced in bipolar disorders. Biol Psychiatry 32: 731–734.
Kusumi I, Koyama T, Yamashita I (1994). Serotonin-induced platelet intracellular calcium mobilization in depressed patients. Psychopharmacology (Berl) 113: 322–327.
Kusumi I, Suzuki K, Sasaki Y, Kameda K, Koyama T (2000). Treatment response in depressed patients with enhanced Ca mobilization stimulated by serotonin. Neuropsychopharmacology 23: 690–696.
Lai JS, Zhao C, Warsh JJ, Li PP (2006). Cytoprotection by lithium and valproate varies between cell types and cellular stresses. Eur J Pharmacol 539: 18–26.
Li J, Gould TD, Yuan P, Manji HK, Chen G (2003). Post-mortem interval effects on the phosphorylation of signaling proteins. Neuropsychopharmacology 28: 1017–1025.
Li J, Vawter MP, Walsh DM, Tomita H, Evans SJ, Choudary PV et al (2004). Systematic changes in gene expression in postmortem human brains associated with tissue pH and terminal medical conditions. Hum Mol Genet 13: 609–616.
Liang M-H, Chuang DM (2007). Regulation and function of glycogen synthase kinase-3 isoforms in neuronal survival. J Biol Chem 282: 3904–3917.
Linseman DA, Butts BD, Precht TA, Phelps RA, Le SS, Laessig TA et al (2004). Glycogen synthase kinase-3beta phosphorylates Bax and promotes its mitochondrial localization during neuronal apoptosis. J Neurosci 24: 9993–10002.
Manji HK, Moore GJ, Chen G (1999). Lithium at 50: have the neuroprotective effects of this unique cation been overlooked. Biol Psychiatry 46: 929–940.
Manji HK, Moore GJ, Chen G (2000a). Clinical and preclinical evidence for the neurotrophic effects of mood stabilizers: implications for the pathophysiology and treatment of manic-depressive illness. Biol Psychiatry 48: 740–754.
Manji HK, Moore GJ, Chen G (2000b). Lithium up-regulates the cytoprotective protein Bcl-2 in the CNS in vivo: a role for neurotrophic and neuroprotective effects in manic depressive illness. J Clin Psychiatry 61 (Suppl 9): 82–96.
Manji HK, Quiroz JA, Sporn J, Payne JL, Denicoff K, Gray NA et al (2003). Enhancing neuronal plasticity and cellular resilience to develop novel, improved therapeutics for difficult-to-treat depression. Biol Psychiatry 53: 707–742.
Mattson MP, Gary DS, Chan SL, Duan W (2001). Perturbed endoplasmic reticulum function, synaptic apoptosis and the pathogenesis of Alzheimer's disease. Biochem Soc Symp 67: 151–162.
Mattson MP, Liu D (2003). Mitochondrial potassium channels and uncoupling proteins in synaptic plasticity and neuronal cell death. Biochem Biophys Res Commun 304: 539–549.
Maurer I, Zierz S, Moller H (2001). Evidence for a mitochondrial oxidative phosphorylation defect in brains from patients with schizophrenia. Schizophr Res 48: 125–136.
McMahon FJ, Stine OC, Meyers DA, Simpson SG, DePaulo JR (1995). Patterns of maternal transmission in bipolar affective disorder. Am J Hum Genet 56: 1277–1286.
Modica-Napolitano JS, Lagace CJ, Brennan WA, Aprille JR (2003). Differential effects of typical and atypical neuroleptics on mitochondrial function in vitro. Arch Pharm Res 26: 951–959.
Moore GJ, Bebchuk JM, Hasanat K, Chen G, Seraji-Bozorgzad N, Wilds IB et al (2000). Lithium increases N-acetyl-aspartate in the human brain: in vivo evidence in support of bcl-2's neurotrophic effects? Biol Psychiatry 48: 1–8.
Moore GJ, Cortese BM, Glitz DA, Zajac-Benitez C, Quiroz JA, Uhede TW et al (2005). Lithium increases gray matter in the preformtal and subgenual preforntal cortices in treatment responsive bipolar disorder patients. ACNP 44th Annual Meeting. Waikoloa, Hawaii.
Murashita J, Kato T, Shioiri T, Inubushi T, Kato N (2000). Altered brain energy metabolism in lithium-resistant bipolar disorder detected by photic stimulated 31P-MR spectroscopy. Psychol Med 30: 107–115.
Murphy AN, Bredesen DE, Cortopassi G, Wang E, Fiskum G (1996). Bcl-2 potentiates the maximal calcium uptake capacity of neural cell mitochondria. Proc Natl Acad Sci USA 93: 9893–9898.
Murphy E (2004). Inhibit GSK-3beta or there's heartbreak dead ahead. J Clin Invest 113: 1526–1528.
Naydenov AV, MacDonald ML, Ongur D, Konradi C (2007). Differences in lymphocyte electron transport gene expression levels between subjects with bipolar disorder and normal controls in response to glucose deprivation stress. Arch Gen Psychiatry 64: 555–564.
Nicholls DG, Ward MW (2000). Mitochondrial membrane potential and neuronal glutamate excitotoxicity: mortality and millivolts. Trends Neurosci 23: 166–174.
Noble W, Planel E, Zehr C, Olm V, Meyerson J, Suleman F et al (2005). Inhibition of glycogen synthase kinase-3 by lithium correlates with reduced tauopathy and degeneration in vivo. Proc Natl Acad Sci USA 102: 6990–6995.
Okamoto Y, Kagaya A, Shinno H, Motohashi N, Yamawaki S (1995). Serotonin-induced platelet calcium mobilization is enhanced in mania. Life Sci 56: 327–332.
Pae CU, Choe BY, Joo RH, Lim HK, Kim TS, Yoo SS et al (2004). Neuronal dysfunction of the frontal lobe in schizophrenia. Neuropsychobiology 50: 211–215.
Pan T, Li X, Xie W, Jankovic J, Le W (2005). Valproic acid-mediated Hsp70 induction and anti-apoptotic neuroprotection in SH-SY5Y cells. FEBS Lett 579: 6716–6720.
Perera TD, Coplan JD, Lisanby SH, Lipira CM, Arif M, Carpio C et al (2007). Antidepressant-induced neurogenesis in the hippocampus of adult nonhuman primates. J Neurosci 27: 4894–4901.
Phiel CJ, Wilson CA, Lee VM, Klein PS (2003). GSK-3alpha regulates production of Alzheimer's disease amyloid-beta peptides. Nature 423: 435–439.
Prince J, Blennow K, Gottfries CG, Karlsson I, Oreland L (1999). Mitochondrial function is differentially altered in the basal ganglia of chronic schizophrenics. Neuropsychopharmacology 21: 372–379.
Quiroz JA, Singh JB, Gould TD, Denicoff K, Zarate, Jr CA. et al (2004). Emerging experimental therapeutics for bipolar disorder: clues from the molecular pathophysiology. Mol Psychiatry 9: 756–776.
Rajkowska G (2000). Postmortem studies in mood disorders indicate altered numbers of neurons and glial cells. Biol Psychiatry 48: 766–777.
Ren M, Senatorov VV, Chen RW, Chuang DM (2003). Post-insult treatment with lithium reduces brain damage and facilitates neurological recovery in a rat ischemia/reperfusion model. Proc Natl Acad Sci USA 100: 6210–6215.
Richter C, Park JW, Ames BN (1988). Normal oxidative damage to mitochondrial and nuclear DNA is extensive. Proc Natl Acad Sci USA 85: 6465–6467.
Rockenstein E, Torrance M, Adame A, Mante M, Bar-on P, Rose JB et al (2007). Neuroprotective effects of regulators of the glycogen synthase kinase-3beta signaling pathway in a transgenic model of Alzheimer's disease are associated with reduced amyloid precursor protein phosphorylation. J Neurosci 27: 1981–1991.
Rowland L, Bustillo JR, Lauriello J (2001). Proton magnetic resonance spectroscopy (H-MRS) studies of schizophrenia. Semin Clin Neuropsychiatry 6: 121–130.
Sapolsky RM (2000). The possibility of neurotoxicity in the hippocampus in major depression: a primer on neuron death. Biol Psychiatry 48: 755–765.
Sassi RB, Brambilla P, Hatch JP, Nicoletti M, Mallinger AG, Frank E et al (2004). Reduced left anterior cingulate volumes in untreated bipolar patients. Biol Psychiatry 56: 467–475.
Sassi RB, Nicoletti M, Brambilla P, Mallinger AG, Frank E, Kupfer DJ et al (2002). Increased gray matter volume in lithium-treated bipolar disorder patients. Neurosci Lett 329: 243–245.
Schloesser RJ, Song J, Klein PS, Manji HK (2007). Cellular plasticity cascades in the pathophysiology and treatment of bipolar disorder. Neuropsychopharmacology Rev e-pub ahead of print, October 3, 2007; doi:10.1038/sj.npp.1301575.
Shao L, Young LT, Wang JF (2005). Chronic treatment with mood stabilizers lithium and valproate prevents excitotoxicity by inhibiting oxidative stress in rat cerebral cortical cells. Biol Psychiatry 58: 879–884.
Simpson PB, Russell JT (1998). Mitochondrial Ca2+ uptake and release influence metabotropic and ionotropic cytosolic Ca2+ responses in rat oligodendrocyte progenitors. J Physiol 508: 413–426.
Sokolov BP, Jiang L, Trivedi NS, Aston C (2003). Transcription profiling reveals mitochondrial, ubiquitin and signaling systems abnormalities in postmortem brains from subjects with a history of alcohol abuse or dependence. J Neurosci Res 72: 756–767.
Stine OC, Xu J, Koskela R, McMahon FJ, Gschwend M, Friddle C et al (1995). Evidence for linkage of bipolar disorder to chromosome 18 with a parent-of-origin effect. Am J Hum Genet 57: 1384–1394.
Stork C, Renshaw PE (2005). Mitochondrial dysfunction in bipolar disorder: evidence from magnetic resonance spectroscopy research. Mol Psychiatry 10: 900–919.
Stout AK, Raphael HM, Kanterewicz BI, Klann E, Reynolds JJ (1998). Glutamate-induced neuron death requires mitochondrial calcium uptake. Nat Neurosci 1: 366–373.
Sun X, Wang JF, Tseng M, Young LT (2006). Downregulation in components of the mitochondrial electron transport chain in the postmortem frontal cortex of subjects with bipolar disorder. J Psychiatry Neurosci 31: 189–196.
Suzuki K, Kusumi I, Sasaki Y, Koyama T (2001). Serotonin-induced platelet intracellular calcium mobilization in various psychiatric disorders: is it specific to bipolar disorder. J Affect Disord 64: 291–296.
Tan CH, Javors MA, Seleshi E, Lowrimore PA, Bowden CL (1990). Effects of lithium on platelet ionic intracellular calcium concentration in patients with bipolar (manic-depressive) disorder and healthy controls. Life Sci 46: 1175–1180.
Tan CH, Lee HS, Kua EH, Peh LH (1995). Resting and thrombin-stimulated cytosolic calcium in platelets of patients with alcoholic withdrawal, bipolar manic disorder and chronic schizophrenia. Life Sci 56: 1817–1823.
Theberge J, Al-Semaan Y, Drost DJ, Malla AK, Neufeld RW, Bartha R et al (2004). Duration of untreated psychosis vs N-acetylaspartate and choline in first episode schizophrenia: a 1H magnetic resonance spectroscopy study at 4.0 Tesla. Psychiatry Res 131: 107–114.
Torok TL (1989). Neurochemical transmission and the sodium-pump. Prog Neurobiol 32: 11–76.
van Calker D, Forstner U, Bohus M, Gebicke-Harter P, Hecht H, Wark HJ et al (1993). Increased sensitivity to agonist stimulation of the Ca2+ response in neutrophils of manic-depressive patients: effect of lithium therapy. Neuropsychobiology 27: 180–183.
Vawter MP, Tomita H, Meng F, Bolstad B, Li J, Choudary PV et al (2006). Mitochondrial-related gene expression changes are sensitive to agonal-pH state: implications for brain disorders. Mol Psychiatry 11: 663–679.
Volz HP, Rzanny R, Riehemann S, May S, Hegewald H, Preussler B et al (1998). 31P magnetic resonance spectroscopy in the frontal lobe of major depressed patients. Eur Arch Psychiatry Clin Neurosci 248: 289–295.
Washizuka S, Kakiuchi C, Mori K, Kunugi H, Tajima O, Akiyama T et al (2003). Association of mitochondrial complex I subunit gene NDUFV2 at 18p11 with bipolar disorder. Am J Med Genet 120B: 72–78.
Weeber EJ, Levy M, Sampson MJ, Anflous K, Armstrong DL, Brown SE et al (2002). The role of mitochondrial porins and the permeability transition pore in learning and synaptic plasticity. J Biol Chem 277: 18891–18897.
Williams JM, Thompson VL, Mason-Parker SE, Abraham WC, Tate WP (1998). Synaptic activity-dependent modulation of mitochondrial gene expression in the rat hippocampus. Brain Res Mol Brain Res 60: 50–56.
Winsberg ME, Sachs N, Tate DL, Adalsteinsson E, Spielman D, Ketter TA (2000). Decreased dorsolateral prefrontal N-acetyl aspartate in bipolar disorder. Biol Psychiatry 47: 475–481.
Wolff DJ, Poirier PG, Brostrom CO, Brostrom MA (1977). Divalent cation binding properties of bovine brain Ca2+-dependent regulator protein. J Biol Chem 252: 4108–4117.
Xu J, Culman J, Blume A, Brecht S, Gohlke P (2003). Chronic treatment with a low dose of lithium protects the brain against ischemic injury by reducing apoptotic death. Stroke 34: 1287–1292.
Yamawaki S, Kagaya A, Okamoto Y, Shimizu M, Nishida A, Uchitomi Y (1996). Enhanced calcium response to serotonin in platelets from patients with affective disorders. J Psychiatry Neurosci 21: 321–324.
Yang F, He XP, Russell J, Lu B (2003). Ca2+ influx-independent synaptic potentiation mediated by mitochondrial Na(+)-Ca2+ exchanger and protein kinase C. J Cell Biol 163: 511–523.
Yeste M, Alvira D, Verdaguer E, Tajes M, Folch J, Rimbau V et al (2007). Evaluation of acute antiapoptotic effects of Li+ in neuronal cell cultures. J Neural Transm 114: 405–416.
Yildiz-Yesiloglu A, Ankerst DP (2006). Review of 1H magnetic resonance spectroscopy findings in major depressive disorder: a meta-analysis. Psychiatry Res 147: 1–25.
Yoon IS, Li PP, Siu KP, Kennedy JL, Macciardi F, Cooke RG et al (2001). Altered TRPC7 gene expression in bipolar-I disorder. Biol Psychiatry 50: 620–626.
Zamzami N, Brenner C, Marzo I, Susin SA, Kroemer G (1998). Subcellular and submitochondrial mode of action of Bcl-2-like oncoproteins. Oncogene 16: 2265–2282.
Acknowledgements
We acknowledge the support of the Intramural Research Program of the National Institute of Mental Heath, NARSAD, and the Stanley Medical Research Institute. We thank Alex Noury, Lisa Catapano, and Ioline Henter for outstanding editorial assistance and critical review. Due to space limitations, we often cited review papers and apologize to those authors whose original data could not be included.
Author information
Authors and Affiliations
Corresponding author
Additional information
DISCLOSURE/CONFLICT OF INTEREST
We acknowledge the support of the Intramural Research Program of the National Institute of Mental Health. The author(s) declare that, except for income received from our primary employer, no financial support or compensation has been received from any individual or corporate entity over the past three years for research or professional service and there are no personal financial holdings that could be perceived as constituting a potential conflict of interest. JAQ is currently an employee of Johnson & Johnson PRD. This work was performed during his tenure at the Laboratory of Molecular Pathophysiology, National Institute of Mental Health, NIH, HHS.
Rights and permissions
About this article
Cite this article
Quiroz, J., Gray, N., Kato, T. et al. Mitochondrially Mediated Plasticity in the Pathophysiology and Treatment of Bipolar Disorder. Neuropsychopharmacol 33, 2551–2565 (2008). https://doi.org/10.1038/sj.npp.1301671
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.npp.1301671
Keywords
This article is cited by
-
Comparative evaluation of natural neuroprotectives and their combinations on chronic immobilization stress-induced depression in experimental mice
3 Biotech (2023)
-
Cellular calcium in bipolar disorder: systematic review and meta-analysis
Molecular Psychiatry (2021)
-
Mitochondrial DNA copy number is associated with psychosis severity and anti-psychotic treatment
Scientific Reports (2018)
-
The Neurobiology of Depression: an Integrated Overview from Biological Theories to Clinical Evidence
Molecular Neurobiology (2017)
-
Psychiatric disorders biochemical pathways unraveled by human brain proteomics
European Archives of Psychiatry and Clinical Neuroscience (2017)
