
Abstract
Hurler syndrome (mucopolysaccharidosis type I, MPS IH) is characterized by a deficiency of α-L-iduronidase resulting in progressive multiorgan dysfunction. We sought to determine whether enzyme replacement therapy (ERT) with iduronidase in the peritransplant period affects outcome of hematopoietic stem cell transplantation (HSCT) for MPS IH. Seven children with MPS IH at a median age of 1.5 years at the time of myeloablative HSCT were eligible. All patients had null mutations in IDUA gene. Iduronidase (0.58 mg/kg per dose) was administered intravenously in 11–14 weekly doses before HSCT and 8 weekly doses after HSCT. The infusions were well tolerated. All patients developed antibodies to iduronidase but all engrafted with >90% donor hematopoiesis. A majority of patients had significant pulmonary complications before ERT and HSCT but all are alive and well with a median follow-up of more than 1 year after HSCT. This suggests that ERT prior to HSCT is unlikely to alter engraftment. In addition, morbidity was acceptable, despite a previous history of pulmonary difficulties that suggested that these patients were high risk for these complications. Therefore, we recommend treatment of MPS IH patients with combination of ERT and HSCT therapy to further investigate its potential to enhance outcomes with HSCT.
Similar content being viewed by others


Introduction
Hurler syndrome (mucopolysaccharidosis type I, MPS IH) is an autosomal recessive inborn error of metabolism due to mutations in the IDUA gene. Deficiency of α-L-iduronidase enzyme activity results in accumulation of complex sugars glycosaminoglycans (GAG) in the lysosomes throughout the cells of the body.1
While clinical findings may be apparent at birth, the onset of initial symptoms usually occurs by 6–8 months of age. Corneal clouding, chronic rhinitis and otitis, obstructive airway disease, umbilical or inguinal hernia, progressive hepatosplenomegaly, cardiac disease, severe skeletal abnormalities, hydrocephalus and neurocognitive deterioration result in substantial morbidity and early death, often by 5 years of age.
The potential treatment of lysosomal storage diseases through the replacement of missing enzymes was proposed initially by De Duve.2 Delivery of α-L-iduronidase may be achieved by either the exogenous administration of α-L-iduronidase or through the endogenous production of α-L-iduronidase following stable engraftment of cells producing enzyme within the affected individual.3 The therapeutic basis for both treatment options is that α-L-iduronidase in the environment can be taken up by cells via the mannose 6-phosphate receptor and routed to the lysosome where it can reduce storage material.4, 5
The development of recombinant α-L-iduronidase has led to its testing in patients with MPS I. Documented benefits of enzyme replacement therapy (ERT) include decreased organomegaly, clearance of urinary GAG, increased joint range of motion and lessening of sleep apnea.6, 7 However, i.v. delivery of enzyme has been unsuccessful in achieving delivery to the central nervous system.
The first experience with hematopoietic stem cell transplantation (HSCT) as therapy for Hurler was reported by Hobbs,8 and since that time HSCT has been accepted as the standard of care for patients with MPS IH, as this results in engraftment of cells within the brain, providing enzyme to the central nervous system, halting neurocognitive decline. We have considered matched unaffected siblings as the optimal graft source, but results with matched unrelated donors, especially from cord blood, are encouraging. While outcomes after HSCT have improved with the availability of improved methods for stem cell graft typing and supportive care, concerns remain regarding the toxicity of transplantation.9, 10, 11, 12
Data from our institution as well as others indicate that there is an increased risk of pulmonary and cardiac complications after HSCT in patients with MPS IH.13, 14 Thus, we and others15, 16 had postulated that pre-HSCT and post-HSCT administration of recombinant α-L-iduronidase may provide benefit to patients being considered for transplantation by decreasing the burden of GAG in the visceral organs.
To date, we have treated seven MPS IH patients with ERT and HSCT combination therapy who have been followed for at least 6 months after transplantation. Here we report that they are engrafted and are well. Moreover, our study suggests that this combination treatment strategy may result in fewer pulmonary complications than would be expected after HSCT alone.
Patients
Transplant and ERT protocols were approved by the Institutional Review Board at the University of Minnesota, and informed consent was obtained in all cases before the procedures were performed. The diagnosis of MPS IH was based on clinical phenotype, mutation analysis of the IDUA gene and absence of α-L-iduronidase activity in peripheral blood leukocytes (data not shown).
Between 2004 and 2007, seven patients received allogeneic HSCT with ERT (Table 1). Outcomes were assessed at least 6 months after transplantation. Follow-up is reported through August 2007 (median 1.1 years).
For ERT, α-L-iduronidase (Laronidase; Genzyme Corporation, Cambridge, MA, USA) was administered weekly at a dose of 0.58 mg/kg per dose i.v. for 11–14 weeks prior to HSCT and for additional 8 weeks after HSCT. The goal of this dosing, which was not interrupted for the period of the conditioning regimen, was to provide a source of exogenous α-L-iduronidase from the time of diagnosis until stable donor chimerism was achieved.
The HSCT myeloablative preparative regimen consisted of busulfan 0.8 mg/kg per dose (1.1 mg/kg per dose for patients <12 kg of body weight; based on the increased metabolism of busulfan in younger children18) every 6 h i.v. for 4 consecutive days (days −9 to −6; total dose 12.8 mg/kg), cyclophosphamide 50 mg/kg per dose once daily i.v. for four consecutive days (days −4 to −1; total dose 200 mg/kg), and antithymocyte globulin 15 mg/kg per dose twice daily for 4 consecutive days (days −4 to −1; total dose 120 mg/kg). No chemotherapy was administered on day −5. Busulfan pharmacokinetics were monitored and dosing was adjusted when serum levels obtained on day −8 were outside the desired target range (area under curve, AUC) of 900–1350 μM per min. The adjusted dose was calculated as follows: adjusted dose (mg)=actual dose (mg) × target AUC (μM min)/actual AUC (μM min).
GvHD prophylaxis consisted of cyclosporine A and mycophenylate mofetil for unrelated cord blood recipients, and cyclosporine A and methotrexate for related bone marrow donors. The Glucksberg scoring system and International Bone Marrow Transplant Registry severity index were used to grade acute GvHD.19, 20 All patients received unmanipulated (non–T cell depleted) stem cells grafts, either bone marrow from related sibling donors or cord blood from unrelated donors (Table 1).
Hematopoietic chimerism was assessed on peripheral blood leukocyte DNA by competitive PCR analysis of variable tandem repeat regions.21 Anti-α-L-iduronidase antibody titers were determined and positivity was confirmed by radioimmune precipitation (Genzyme Corporation).
Results
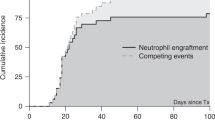
Patients' age, enzyme replacement, transplant and graft characteristics are summarized in the Table 1. The median age at HSCT was 1.5 years (range 237–686 days). Complete donor engraftment (defined as donor chimerism of more ⩾90% 2 months after HSCT) was observed in all seven evaluable patients.
Two patients developed skin GvHD. Patient 1 had stage 3 grade C GvHD and patient 4 had stage 2 grade B GvHD. Both were treated successfully with systemic and topical steroids. No evidence of acute intestinal or liver GvHD, and no chronic GvHD was observed.
ERT prior to HSCT does not compromise donor engraftment
Mutational analysis showed that all seven patients had severe IDUA mutations that resulted in no α-L-iduronidase production. This is relevant because of the theoretical risk of immune-mediated elimination of donor IDUA-producing cells by IDUA-naive host immune cells. Although all patients generated serum antibodies against the α-L-iduronidase protein (average positive titer 1:1800, range 1:100–1:102 400), no impact on early engraftment has been observed. In addition, the post transplant α-L-iduronidase activity in all patients, including the ones with high α-L-iduronidase antibody levels, was within normal range, suggesting that α-L-iduronidase antibodies even when present were not neutralizing.22, 23
ERT limits pulmonary complications after HSCT
In a recent analysis we have observed that MPS IH patients with a history of pulmonary disease including a history of pneumonia or reactive airway disease had worse outcomes than those who were free from pulmonary issues before HSCT (manuscript in preparation). We reasoned that ERT before transplant could have a potential to decrease the GAG burden in the lung and other visceral tissues. As a result, regimen-related morbidity and mortality of HSCT could potentially be decreased, as regimen-related toxicities after HSCT in MPS IH patients may be a result of GAG-associated pathology.14
Thus, we have correlated pretransplant pulmonary compromise with post transplant lung-related complications in all MPS IH patients in this study. Four pretransplant risk factors (pneumonia, abnormal sleep study with apnea, reactive airway disease and need for supplemental oxygen) were assessed (Table 2). Five out of seven MPS IH patients had two or more risk factors, a finding that we have observed to be associated with significant morbidity and mortality in MPS IH patients treated with HSCT alone.
In addition, all patients were noisy breathers and had a history of rhinorrhea (as is typical of MPS IH). One patient (patient 4) had a ventricular shunt placed, one (patient 5) underwent tonsillectomy and adenoidectomy and four (patients 4, 5, 6 and 7) had myringotomy tubes placed prior to HSCT. The noisy breathing and rhinorrhea improved in all patients after ERT/HSCT. Myringotomy tubes had to be replaced in patients 4, 5 and 7.
Combination therapy results in low overall morbidity and normal α-L-iduronidase levels
Significant transplant-related morbidity (in addition to the lung-related complications summarized in Table 2) consisted of bacteremia in three patients (patients 1, 2 and 5); and hemorrhagic cystitis and pericardial effusion requiring drainage in one patient (patient 3). In the patients for which the size measurements by computed tomography of visceral organs was available (liver volumetrics for patients 4, 5 and 7; and spleen volumetrics for patients 1, 5 and 7) before ERT and after 11–14 weeks ERT (before HCT), less prominent organomegaly has been observed (average±s.d. before ERT versus after ERT; liver: 642±85 cm3 versus 441±22 cm3, P=0.02; spleen: 97±14 cm3 versus 72±9 cm3, P=0.06) suggesting an overall decrease in GAG visceral burden. α-L-iduronidase leukocyte levels were measured following HSCT. As expected on the basis of robust donor engraftment in all patients, enzyme levels were within the normal range after HSCT (Table 1).
Discussion
This prospective, single-institution trial evaluating the effect of ERT and HSCT provides evidence that this combination therapy is safe, and does not negatively impact on donor stem cell engraftment and GvHD. Moreover, we have observed low morbidity after ERT+HSCT combination therapy despite significant pulmonary risk factors present before HSCT.
Several studies confirmed the safety of ERT in MPS I.6, 7, 15, 24, 25 Though beneficial in the mild form of MPS IS (Scheie syndrome), the role of i.v. ERT in addition to HSCT for the severe form of MPS IH was far from clear.
In the largest study to date, Cox-Brinkman et al.15 concluded that the use of ERT could not be demonstrated to affect the outcome of HSCT. These patients had a follow-up of at least 3 months (range 3–17.5 months). The authors studied patients from multiple institutions. The patient population was heterogeneous in the choice of stem cell grafts (cord blood, bone marrow, peripheral blood stem cells; matched and mismatched family donors and unrelated donors), conditioning regimens (four different myeloablative and one reduced-intensity regimen) and the number of cell infusions (37% received second transplants and 9% third transplants). In addition, donor lymphocyte infusions were given to one patient for progressively decreasing donor chimerism, and duration of ERT before and after HSCT varied between 7–24 weeks and 0–12 weeks, respectively. Of the 22 patients that received ERT+HSCT, 13 were alive and engrafted after the HSCT; 6 additional patients engrafted after the second or third HSCT (engraftment was defined as a donor chimerism of >10%, with a concomitant α-L-iduronidase level above the lower level of normal for heterozygous individuals).
We chose to investigate the combination of ERT and HSCT in patients treated using a unified conditioning regimen, receiving a similar number of enzyme infusions with consistent supportive care at a single institution. This limits the number of possible study confounders. A majority of patients had significant pulmonary complications before receiving ERT and HSCT, confirming that this group was not a low-risk population (Table 2). Our unpublished data show that in the group of patients with Hurler syndrome undergoing HSCT alone, patients who had a history of pneumonia or reactive airway disease prior to HSCT had a significantly poorer 1-year and 5-year survival expectancy when compared to patients with no history of pulmonary disease. Nevertheless, all patients in the current study are alive and well with a median follow-up of more than 1 year after HSCT. We observed that all children in our study engrafted with >90% donor hematopoiesis as assessed at 2 months after their initial HSCT. All but one (patient 6; Table 1) remain >90% engrafted at the time of this writing. Between 3 and 6 months after HSCT donor engraftment of patient 6 decreased from 85 to 60%.
Furthermore, our data indicate that despite the expected formation of anti-α-L-iduronidase antibodies,6, 7, 24 no evidence of immune-mediated graft rejection was observed in MPS IH patients receiving pre-HSCT ERT. Historically, the engraftment rates after HSCT (without ERT) for MPS IH at the University of Minnesota have been 77% after the first and 50% after the second HCT, which are similar to the engraftment rates reported form other institutions (33–85%).9, 10, 15, 26, 27, 28, 29 Of note, patient 6 with 60% donor engraftment at 6 months after HSCT had normal levels of leukocyte α-L-iduronidase and decreasing titers of serum anti-α-L-iduronidase antibody (1:12 800–1:200) over 6 months after HSCT. This argues against immune-mediated mechanism of his/her decreased donor chimerism, as antibody-mediated rejection of allogeneic donor bone marrow in primed animals has been noted to occur within several hours.30 In fact, it is possible that ERT-mediated GAG clearance in the bone marrow niche could create a more permissive environment for donor engraftment31 when compared to patients who are recipients of HSCT alone. This is potentially of significant clinical value as the requirement for a second or third HSCT is associated with high rates of morbidity and mortality.29
In addition, achieving expedient donor chimerism and concomitant higher α-L-iduronidase levels are likely to result in more rapid and more complete clearance of GAG from visceral organs, which would be expected to translate into clinical benefit to the MPS IH patients.
A previous report15 recommended treating only patients in a poor clinical condition with the combination therapy. In contrast, our recommendation is to treat all severe MPS IH patients with ERT and HSCT to improve morbidity and mortality.
References
Bach G, Friedman R, Weissmann B, Neufeld EF . The defect in the Hurler and Scheie syndromes: deficiency of -L-iduronidase. Proc Natl Acad Sci USA 1972; 69: 2048–2051.
De Duve C . From cytases to lysosomes. Fed Proc 1964; 23: 1045–1049.
Boelens JJ . Trends in haematopoietic cell transplantation for inborn errors of metabolism. J Inherit Metab Dis 2006; 29: 413–420.
Fratantoni JC, Hall CW, Neufeld EF . Hurler and Hunter syndromes: mutual correction of the defect in cultured fibroblasts. Science 1968; 162: 570–572.
Fratantoni JC, Hall CW, Neufeld EF . The defect in Hurler's and Hunter's syndromes: faulty degradation of mucopolysaccharide. Proc Natl Acad Sci USA 1968; 60: 699–706.
Kakkis ED, Muenzer J, Tiller GE, Waber L, Belmont J, Passage M et al. Enzyme-replacement therapy in mucopolysaccharidosis I. N Engl J Med 2001; 344: 182–188.
Wraith JE, Clarke LA, Beck M, Kolodny EH, Pastores GM, Muenzer J et al. Enzyme replacement therapy for mucopolysaccharidosis I: a randomized, double-blinded, placebo-controlled, multinational study of recombinant human alpha-L-iduronidase (laronidase). J Pediatr 2004; 144: 581–588.
Hobbs JR, Hugh-Jones K, Barrett AJ, Byrom N, Chambers D, Henry K et al. Reversal of clinical features of Hurler's disease and biochemical improvement after treatment by bone-marrow transplantation. Lancet 1981; 2: 709–712.
Peters C, Balthazor M, Shapiro EG, King RJ, Kollman C, Hegland JD et al. Outcome of unrelated donor bone marrow transplantation in 40 children with Hurler syndrome. Blood 1996; 87: 4894–4902.
Staba SL, Escolar ML, Poe M, Kim Y, Martin PL, Szabolcs P et al. Cord-blood transplants from unrelated donors in patients with Hurler's syndrome. N Engl J Med 2004; 350: 1960–1969.
Whitley CB, Belani KG, Chang PN, Summers CG, Blazar BR, Tsai MY et al. Long-term outcome of Hurler syndrome following bone marrow transplantation. Am J Med Genet 1993; 46: 209–218.
Boelens JJ, Wynn RF, O'Meara A, Veys P, Bertrand Y, Souillet G et al. Outcomes of hematopoietic stem cell transplantation for Hurler's syndrome in Europe: a risk factor analysis for graft failure. Bone Marrow Transplant 2007; 40: 225–233.
Gassas A, Sung L, Doyle JJ, Clarke JT, Saunders EF . Life-threatening pulmonary hemorrhages post bone marrow transplantation in Hurler syndrome. Report of three cases and review of the literature. Bone Marrow Transplant 2003; 32: 213–215.
Kharbanda S, Panoskaltsis-Mortari A, Haddad IY, Blazar BR, Orchard PJ, Cornfield DN et al. Inflammatory cytokines and the development of pulmonary complications after allogeneic hematopoietic cell transplantation in patients with inherited metabolic storage disorders. Biol Blood Marrow Transplant 2006; 12: 430–437.
Cox-Brinkman J, Boelens JJ, Wraith JE, O'Meara A, Veys P, Wijburg FA et al. Haematopoietic cell transplantation (HCT) in combination with enzyme replacement therapy (ERT) in patients with Hurler syndrome. Bone Marrow Transplant 2006; 38: 17–21.
Wraith JE . Enzyme replacement therapy in mucopolysaccharidosis type I: progress and emerging difficulties. J Inherit Metab Dis 2001; 24: 245–250.
deGasperi R, Raghavan SS, Sosa MG, Kolodny EH, Carrier C, Rubenstein P et al. Measurements from normal umbilical cord blood of four lysosomal enzymatic activities: alpha-L-iduronidase (Hurler), galactocerebrosidase (globoid cell leukodystrophy), arylsulfatase A (metachromatic leukodystrophy), arylsulfatase B (Maroteaux-Lamy). Bone Marrow Transplant 2000; 25: 541–544.
Jacobson P, Park JJ, DeFor TE, Thrall M, Abel S, Krivit W et al. Oral busulfan pharmacokinetics and engraftment in children with Hurler syndrome and other inherited metabolic storage diseases undergoing hematopoietic cell transplantation. Bone Marrow Transplant 2001; 27: 855–861.
Glucksberg H, Storb R, Fefer A, Buckner CD, Neiman PE, Clift RA et al. Clinical manifestations of graft-versus-host disease in human recipients of marrow from HL-A-matched sibling donors. Transplantation 1974; 18: 295–304.
Rowlings PA, Przepiorka D, Klein JP, Gale RP, Passweg JR, Henslee-Downey PJ et al. IBMTR Severity Index for grading acute graft-versus-host disease: retrospective comparison with Glucksberg grade. Br J Haematol 1997; 97: 855–864.
Scharf SJ, Smith AG, Hansen JA, McFarland C, Erlich HA . Quantitative determination of bone marrow transplant engraftment using fluorescent polymerase chain reaction primers for human identity markers. Blood 1995; 85: 1954–1963.
Kakavanos R, Turner CT, Hopwood JJ, Kakkis ED, Brooks DA . Immune tolerance after long-term enzyme-replacement therapy among patients who have mucopolysaccharidosis I. Lancet 2003; 361: 1608–1613.
Soni S, Hente M, Breslin N, Hersh J, Whitley C, Cheerva A et al. Pre-stem cell transplantation enzyme replacement therapy in Hurler syndrome does not lead to significant antibody formation or delayed recovery of the endogenous enzyme post-transplant: a case report. Pediatr Transplant 2007; 11: 563–567.
Grewal SS, Wynn R, Abdenur JE, Burton BK, Gharib M, Haase C et al. Safety and efficacy of enzyme replacement therapy in combination with hematopoietic stem cell transplantation in Hurler syndrome. Genet Med 2005; 7: 143–146.
Sifuentes M, Doroshow R, Hoft R, Mason G, Walot I, Diament M et al. A follow-up study of MPS I patients treated with laronidase enzyme replacement therapy for 6 years. Mol Genet Metab 2007; 90: 171–180.
Peters C, Shapiro EG, Anderson J, Henslee-Downey PJ, Klemperer MR, Cowan MJ et al. Hurler syndrome: II. Outcome of HLA-genotypically identical sibling and HLA-haploidentical related donor bone marrow transplantation in fifty-four children. The storage disease collaborative study group. Blood 1998; 91: 2601–2608.
Vellodi A, Young EP, Cooper A, Wraith JE, Winchester B, Meaney C et al. Bone marrow transplantation for mucopolysaccharidosis type I: experience of two British centres. Arch Dis Child 1997; 76: 92–99.
Souillet G, Guffon N, Maire I, Pujol M, Taylor P, Sevin F et al. Outcome of 27 patients with Hurler's syndrome transplanted from either related or unrelated haematopoietic stem cell sources. Bone Marrow Transplant 2003; 31: 1105–1117.
Grewal SS, Krivit W, Defor TE, Shapiro EG, Orchard PJ, Abel SL et al. Outcome of second hematopoietic cell transplantation in Hurler syndrome. Bone Marrow Transplant 2002; 29: 491–496.
Taylor PA, Ehrhardt MJ, Roforth MM, Swedin JM, Panoskaltsis-Mortari A, Serody JS et al. Preformed antibody, not primed T cells, is the initial and major barrier to bone marrow engraftment in allosensitized recipients. Blood 2007; 109: 1307–1315.
Baxter MA, Wynn RF, Schyma L, Holmes DK, Wraith JE, Fairbairn LJ et al. Marrow stromal cells from patients affected by MPS I differentially support haematopoietic progenitor cell development. J Inherit Metab Dis 2005; 28: 1045–1053.
Acknowledgements
We thank Eileen Hanson and Teresa Kivisto for their dedication and persistence in the care of the families and in obtaining the necessary data. This study was supported by Children's Cancer Research Fund, Minneapolis, MN, USA.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Tolar, J., Grewal, S., Bjoraker, K. et al. Combination of enzyme replacement and hematopoietic stem cell transplantation as therapy for Hurler syndrome. Bone Marrow Transplant 41, 531–535 (2008). https://doi.org/10.1038/sj.bmt.1705934
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.bmt.1705934
Keywords
This article is cited by
-
In vivo adenine base editing corrects newborn murine model of Hurler syndrome
Molecular Biomedicine (2023)
-
Enzyme replacement therapy for mucopolysaccharidoses; past, present, and future
Journal of Human Genetics (2019)
-
Open issues in Mucopolysaccharidosis type I-Hurler
Orphanet Journal of Rare Diseases (2017)
-
Long term survival and cardiopulmonary outcome in children with Hurler syndrome after haematopoietic stem cell transplantation
Journal of Inherited Metabolic Disease (2017)
-
Increased longevity and metabolic correction following syngeneic BMT in a murine model of mucopolysaccharidosis type I
Bone Marrow Transplantation (2012)
