
Abstract
Overwhelming clinical and experimental data demonstrate that tumour hypoxia is associated with aggressive disease and poor treatment outcome as hypoxic cells are refractive to radiotherapy and some forms of chemotherapy. However, hypoxia is rare in physiologically normal tissues representing a tumour-specific condition. To selectively target this therapeutically refractive cell population, we have combined bioreductive chemotherapy with hypoxia-directed gene therapy. We have transfected the human fibrosarcoma cell line, HT1080, with a hypoxia-regulated expression vector encoding the human flavoprotein cytochrome c P450 reductase (HRE-P450R). This conferred hypoxia-dependent sensitivity to the alkylating nitroimidazole prodrug RSU1069 in vitro, with a greater than 30-fold increase in oxic/hypoxic cytotoxicity ratio compared with controls. Xenografts of both the HRE-P450R and empty vector transfectants had comparable hypoxic fractions and were refractive to single dose radiotherapy of up to 15 Gy. However, combining a prodrug of RSU1069 with a reduced radiotherapy dose of 10 Gy represents a curative regimen (50% tumour-free survival; day 100) in the HRE-P450R xenografts. In complete contrast, 100% mortality was apparent by day 44 in the empty vector control xenografts treated in the same way. Thus, an oxygen-sensitive gene-directed enzyme prodrug therapy approach may have utility when incorporated into conventional radiotherapy and/or chemotherapy protocols for loco-regional disease in any tissue where hypoxia is a contra-indication to treatment success.
Similar content being viewed by others

Introduction
Poor tumour oxygenation, due to an inadequate blood supply, is a well-documented feature of most solid tumours leading to regions of acute and/or chronic hypoxia. Conclusive experimental and clinical research demonstrates the impact of hypoxic tumour cells upon the therapeutic outcome of cancer radiotherapy and some chemotherapy.12345 Hypoxic cells are approximately three times more radio-resistant than normoxic cells which is thought to result primarily from the ability of oxygen to react with, and chemically modify the initial radiation-induced DNA radicals. Although less clinical evidence is available regarding the impact of hypoxia on the failure of chemotherapy there is direct evidence that hypoxic cells within tumour xenografts are refractive to clinically relevant chemotherapeutic agents and it is reasonable to infer that the quiescent nature of hypoxic cells may render them insensitive to agents that target rapidly dividing cells.6 Experimental and clinical evidence also points to hypoxia as a potent physiological stimulus that promotes tumour progression, driving genetic change and selecting for aggressive neoplastic phenotypes.347
Consequently, an important goal for cancer research has been to develop treatment strategies that selectively target this resistant tumour cell population. One approach has been the use of chemotherapeutic agents that are specifically cytotoxic to hypoxic cells.8 These bioreductive ‘prodrugs’ can be reduced by cellular reductases to species that cause DNA damage under hypoxic conditions, but in the presence of molecular oxygen they are efficiently back-oxidized to the non-toxic parent compound, such that little or no DNA damage occurs. Of importance is the relative toxicity of bioreductive drugs under hypoxic versus aerobic conditions. Preferably, the drug should possess a high hypoxic cell cytotoxicity ratio (HCR; the ratio of the dose required to kill a proportion of aerobic cells to that required to kill an equal proportion of the same cells exposed to the drug under hypoxic conditions), leading to maximum hypoxic tumour cell killing with minimal toxicity to aerobic tissues.
Alternative strategies to exploit hypoxia have been developed in the field of gene therapy. Hypoxia not only provides a powerful physiological stimulus, but also results in a complex transcriptional cascade in order to facilitate cell survival under hypoxic stress. This is achieved by the up-regulation of genes supporting anaerobic metabolism and new blood vessel development, such as phosphoglycerate kinase-1 (PGK-1), lactate dehydrogenase A (LDH-A) glucose transporter-1 (Glut-1) and vascular endothelial growth factor (VEGF), amongst many others. This coordinated hypoxic gene activation is mediated through the transcription factor HIF-1 (hypoxia-inducible factor-1) that binds to a common hypoxia-responsive element (HRE) found within the enhancer region of all HIF-1 responsive genes.910 Furthermore, by introducing an HRE sequence into an appropriate expression cassette, hypoxic regulation of an exogenous therapeutic gene can be achieved. The oxygen ‘sensing’ pathway has been demonstrated to be universally functional in mammalian cells11 and unlike the limited occurrence of other tumour-specific transcription factors, HIF-1 is reported to be expressed in 70% of solid tumours,12 making HRE-regulated gene therapy an attractive strategy that can be applied to a broad range of cancers.
Hypoxia-selective gene therapy was first exemplified using HRE-regulated expression of the suicide gene E. coli cytosine deaminase, demonstrating hypoxia-specific sensitisation of tumour cells to the prodrug 5-fluorocytosine.13 Subsequent studies have similarly demonstrated the efficacy of HRE-regulated Herpes simplex virus 1-thymidine kinase activity to mediate ganciclovir sensitisation.14 Although HRE-mediated prodrug activating enzyme expression offers transcriptional selectivity over non-targeted enzyme prodrug therapies, we report a novel strategy that further enhances this approach by making hypoxia a prerequisite for both gene expression and drug activation. This is achieved by using an oxygen-sensitive bioreductive agent as a prodrug in an O2-sensitive gene directed enzyme prodrug therapy (GDEPT) approach. For this we have used the nitroimidazole RSU1069 (1-[3-aziridinyl-2-hydroypropyl]-2-nitroimidazole) that is reduced by a series of single electron transfers to generate highly reactive toxic species.1516This results in a large oxic/hypoxic selectivity being recorded for RSU1069 being 30–200-fold more potent in the absence of oxygen. This compares with a value of two reported for mitomycin C.17 Several endogenous enzymes may catalyse the reduction of RSU1069, however we have harnessed the reductive capacity of a dominant nitroreductase, NADPH cytochrome P450 (c) reductase (P450R). This flavoenzyme will readily donate single electrons to any prodrug with an appropriate redox potential and we have previously established that constitutive over-expression of P450R confers sensitivity to most classes of bioreductive agents, including nitro-compounds such as RSU1069.18
In this paper we restrict over-expression of P450R to hypoxic tumour cells by including the HRE from PGK-1 within our therapeutic gene cassette. We have previously demonstrated that PGK-1 HRE-mediated gene expression is activated in tumour cells shown to be radio-resistant in vivo.13 By combining HRE-driven P450R expression and the bioreductive drug RSU1069 with radiation, we aim to kill the remaining radiation-resistant hypoxic tumour subpopulation. In addition, by employing this refined GDEPT approach in which both enzyme expression and drug toxicity are dependent upon hypoxia, we can simultaneously increase the therapeutic index of the therapy without increasing systemic toxicity.
Results
Hypoxia-induced human cytochrome c P450 reductase expression in a human tumour cell line
A bicistronic vector, pCIneo PGK SV40mp P450R IRES EGFP (pHRE-P450R/EGFP), encoding for the co-expression of human P450 reductase (P450R) and enhanced green fluorescent protein (EGFP) was constructed (Figure 1). A trimer of the hypoxia-responsive element (HRE) of the mouse phosphoglycerate kinase gene-1 (PGK-1) was used as the hypoxic enhancer and cloned in the positive orientation upstream of the SV40 minimal promoter.13 A control vector, lacking only the P450R cDNA, was also constructed (pHRE-EGFP) and each vector was independently transfected into the HT1080 cell line to allow the selection of EGFP matched P450 reductase-expressing and empty vector control clones. Clones were isolated from each transfected population and a detailed comparison of growth kinetics (both in vitro and when grown as xenografts in vivo) and hypoxic gene induction was made in order to avoid cloning artifacts. This led to the selection of transcriptionally equivalent GFP/R9 (P450R expressing) and the GFP5 (empty vector) clones.

Structure of the hypoxia-regulated therapeutic cassette showing the mechanism of prodrug activation only under hypoxic conditions. The expression cassette was stably inserted into the genome of HT1080 fibrosarcoma cells to generate the GFP/R9 clone. Hypoxic regulation of human cytochrome P450 reductase (P450R) expression is afforded by a trimer of the HRE sequence from the PGK-1 gene. P450R expression is mediated via the transcription factor HIF-1 and yields the cytotoxic moiety from the bioreductive prodrug only under hypoxic conditions. The construct used to generate the control GFP-5 clone lacked the P450R cDNA.
In vitro parameters of the human cytochrome c P450 reductase-expressing and empty vector clones
The growth characteristics of both of these cell lines were identical in vitro (population doubling time 15.5 h (±1 h)) and expression of EGFP in air and following 18 h exposure to hypoxia was also matched. Analysis of EGFP presentation by flow cytometry (refer to Materials and methods for details) revealed a median peak fluorescence of 48 ± 3 and 42 ± 1 for the GFP/R9 and GFP-5 clones, respectively, in air. This was induced approximately two-fold in both cell lines in response to hypoxia and 3 h re-oxygenation (median peak fluorescence of 79.2 ± 8.7 for GFP/R9, and 72.7 ± 10 for GFP-5 cells), rising to 3.5-fold following 24 h re-oxygenation. The ability of re-oxygenation to enhance output from HRE-reporter constructs has been documented previously.13 P450R activity levels were also assessed in air and hypoxia. Aerobic P450R enzyme activity in the GFP/R9 cells was 137 ± 39 nmol cytochrome c reduced min−1 mg−1 protein. Eighteen hours of hypoxic exposure induced the activity by 2.2-fold and re-oxygenation led to further increases in activity to a maximum at 6 h re-oxygenation of 540 ± 63 nmol cytochrome c reduced min-1 mg-1 protein (Figure 2). P450R activity remained above basal aerobic levels for approximately 4 days after hypoxic exposure Figure 2. Basal P450R expression in the GFP-5 cells was 12 nmol cytochrome c reduced min−1 mg−1 protein which was further suppressed upon hypoxic exposure to 7 nmol cytochrome c reduced min−1 mg−1 protein.

A time course profiling P450R activity in the GFP/R9 during and post-hypoxic exposure. The level of P450R activity (represented as cytochrome c reduced min-1 mg-1 protein) in the GFP/R9 cells was determined during hypoxic exposure (open symbols) and upon subsequent re-oxygenation (closed symbols). 18 h hypoxic exposure induced P450R activity by 2.2-fold and re-oxygenation led to further increases in activity to a maximum at 6 h re-oxygenation of 540 ± 63 nmol cytochrome c reduced min-1 mg-1 protein. P450R activity remained above basal aerobic levels for approximately 4 days after hypoxic exposure. Data represent the mean of three independent experiments ± standard deviation.
The relatively low EGFP expression in response to hypoxic exposure, compared with this strong induction in P450R expression, probably reflects the fact that the IRES used to generate the constructs expressed by the two cell lines was attenuated to give preferentially higher out-put from the primary coding sequence, and that EGFP protein folding and consequently functionality is influenced by oxygen availability.19
In vivo parameters of the human cytochrome c P450 reductase-expressing and empty vector clones
As the purpose of these studies was to validate the potential of combining bioreductive chemotherapy and hypoxia-directed gene therapy both in vitro and in vivo, it was important that we did not select cell lines for in vitro analysis that then differed markedly when grown as xenografts in nude mice. Again, the inherent tumourigenecity and subsequent growth rate of both the GFP/R9 and GFP-5 cells was almost identical (Figure 3), and no significant difference was seen in the time taken to achieve two volume doublings between the two tumour types (8 ± 1.7 days for GFP/R9 (n = 12) versus 9.3 ± 1.4 days for GFP-5 (n=6), P = 0.213). In addition, the growth pattern mirrored that of un-transfected HT1080 cells Figure 3.

Comparative growth rate of GFP/R9, GFP-5 and HT1080 wild-type (wt) xenografts. Tumours were initiated from the intra-dermal injection of 5 × 106 cells and measured daily once a palpable tumour formed. Data represent the mean ± standard error of the mean plotted until the first tumour within each group achieved the maximum designated volume of 1000 mm3. All data sets were accrued from two or more independent experiments.
Further studies were undertaken to address the vascularity and extent of hypoxia within tumours derived from each cell type. The hypoxic marker, pimonidazole, was administered to mice bearing tumours of approximately 250 mm3. This represented the designated treatment size for use in subsequent studies, allowing a relative tumour volume four times that at the time of treatment (RTV4) to be used as the experimental endpoint, whilst staying within the maximum tumour burden allowed for ethical reasons. Quantitative analysis of the hypoxic fraction (HF) and vessel density (VD) following immunohistochemical staining of pimonidazole adducts and mouse endothelial structures revealed no significant differences between the two tumour types (Figure 4). However, there was greater variability in both parameters in the GFP/R9 tumours compared with GFP-5 tumours (GFP/R9 (n = 8), HF 6.2% tumour area ± 5.0%, VD 89 ± 43 mm−2; GFP-5 (n = 5), HF 3.3% ± 1.4%, VD 108 ± 18 mm−2; P = 0.196 and P = 0.245 for HF and VD, respectively). Further immunohistochemical analyses to simultaneously detect P450R and pimonidazole binding revealed that P450R was restricted to the hypoxic regions of the GFP/R9 tumours (Figure 5a). In addition, expression of P450R in the GFP/R9 tumours co-localised with the endogenous HIF-1 regulated protein Glut-12021 (data not shown). No P450R expression was detected in the hypoxic regions of GFP-5 tumours Figure 5c. This demonstrates that the transcriptional regulation afforded by the HRE that we have used specifically restricts P450R expression to the target tumour population. Reflecting the variability in hypoxic fraction (pimonidazole binding) in the GFP/R9 tumours, P450R activity measurements obtained for tumours excised at the designated treatment size ranged from 50–225 nmol cytochrome c reduced min−1 mg−1 protein.

Representative composite binary images of vascularity and hypoxic fraction in the GFP/R9 and GFP-5 tumour xenografts. Immunohistochemical studies determined the vascularity and extent of hypoxia within tumours derived from the GFP/R9 and GFP-5 cell types. Vascularity was determined by staining of mouse endothelial cells (red staining). The hypoxic marker, pimonidazole was administered before tumour extraction allowing for pimonidazole adduct labelling (green staining). Quantitative analysis of the hypoxic fraction (HF) and vessel density (VD) following immunohistochemical staining of pimonidazole adducts and mouse endothelial structures revealed no significant differences between the two tumour types.

Relative spatial distribution of P450R and pimonidazole binding in GFP/R9 and GFP5 tumour xenografts. The distribution of P450R (panels a and c) and pimonidazole binding (panels b and d) was assessed by immunohistochemical staining on 3–4 μM sections taken from formalin-fixed, paraffin-embedded GFP/R9 (panels a and b) and GFP-5 tumour xenografts (panels c and d). P450R expression in the GFP/R9 tumours (a) co-localises with hypoxic regions revealed by pimonidazole staining (b). No P450R expression was detected in the GFP-5 tumours (c).
Hypoxia-regulated human cytochrome c P450 reductase expression sensitises cells to bioreductive drug treatment in vitro
The relative sensitivity of the GFP/R9 and GFP-5 cells to bioreductive drug treatment was then analysed. Cells were exposed to the bioreductive drug RSU1069,1516 under aerobic or hypoxic conditions for a period of 18 h and proliferation relative to control cells ascertained 72 h later by MTT assay (Figure 6). Despite the difference in aerobic P450R activity in the two cell types, there was no difference in the mean IC50 value obtained for RSU1069 in air (47 μM for the GFP/R9, versus 43 μM for the GFP-5 cells; Figure 6). This is in part due to the stringency in the level of hypoxia required for RSU1069 activation, with maximal cytotoxicity being achieved below oxygen concentrations of 0.1%.17 Exposure to RSU1069 under hypoxic conditions increased toxicity by 30-fold in the GFP-5 cells, and a mean IC50 concentration of 1.4 μM was achieved. However, in the GFP/R9 cells the enhanced cytotoxicity achieved under hypoxic conditions was markedly increased to 1000-fold (mean IC50 hypoxia, 0.046 μM; Figure 6).

Hypoxia-regulated P450R expression hypersensitises GFP/R9 to treatment with the bioreductive drug RSU1069 in vitro. GFP-5 (□, ▪) and GFP/R9 (○, •) cells were exposed to RSU1069 for 18 h in air (closed symbols) or under anoxic conditions (open symbols). Proliferation relative to untreated controls was ascertained 3 days after drug exposure by MTT assay. Data represent mean ± standard deviation values obtained from three independent experiments.
Hypoxia-driven human cytochrome c P450 reductase expression sensitises tumours to bioreductive drug treatment in combination with radiotherapy in vivo
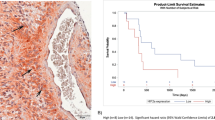
We next sought to evaluate the efficacy of bioreductive chemotherapy and hypoxia-directed gene therapy in tumour xenografts derived from the GFP/R9 and GFP-5 cells. For these studies, the prodrug RB6145 (1-[3-(2-bromoethylamino)-2-hydroxypropyl]-2-nitroimidazole) was used, which when hydrolysed in vivo releases RSU1069.22 As highlighted above, RSU1069 shows preferential toxicity towards hypoxic cells. Radiotherapy was chosen to control the oxic cell population within the tumours. From the pimonidazole studies, approximately 5% of the tumour cells in both xenograft types were radiobiologically hypoxic. In spite of the well-oxygenated nature of both the GFP/R9 and GFP-5 tumour types, a single dose of 10 or 15 Gy X-irradiation was not curative. RTV4 was achieved within a mean time of 28 days after treatment with 10 Gy and 40 days after treatment with 15 Gy for both tumour types. Administering RB6145 immediately after 10Gy radiotherapy in the GFP-5 tumours increased tumour response compared with radiotherapy alone, but this did not achieve statistical significance (P = 0.06). In striking contrast, 50% of the GFP/R9 tumour-bearing mice treated with this regimen were tumour-free 100 days after therapy. Data are the mean of two independent experiments, ≥5 xenografts per treatment group (Figure 7). In addition, the combination of RB6145 and 10 Gy radiotherapy was well tolerated. Although one mouse was excluded, 14 out of the 15 GFP-5 and GFP/R9 tumour-bearing mice treated exhibited no loss of condition or weight. Further, those mice whose GFP/R9 tumours were cured by the combination of radiotherapy and RB6145 showed no acute or late-normal tissue damage in the field of radiotherapy.

Hypoxia-regulated P450R expression enhances the efficacy of combined bioreductive chemotherapy and radiotherapy in vivo. The graphs in panel (a) represent the time taken for GFP/R9 and GFP-5 tumours to reach RTV4. Three different treatment groups are shown: untreated (control), radiotherapy alone (10 Gy) and combined with bioreductive chemotherapy (10 Gy + RB6145). Total tumour eradication was achieved in 50% of the GFP/R9 tumours treated with 10 Gy + RB6145. This is illustrated in the growth profiles for each individual GFP/R9 and GFP-5 tumour given this treatment (panel b). RB6145 was administered at a dose of 250 mg/kg immediately after radiotherapy.
Discussion
One of the major goals of cancer therapy is to achieve tumour-selective toxicity. To this end hypoxia provides a powerful tumour-specific condition towards which the therapy can be targeted. In addition, hypoxic cells within a tumour represent the most resistant cell population being refractive to radiotherapy and probably many forms of chemotherapy. Here we have sought to specifically hypersensitise this resistant population to bioreductive drug treatment using an oxygen-sensitive gene therapy approach. This significantly increased the efficacy of radiotherapy in vivo, achieving a 50% cure rate using radiation doses that consistently failed to control the primary tumour when administered alone.
Throughout these experiments, we have stringently selected clones that are transcriptionally equivalent using a bicistronic vector encoding for HRE driven P450R and EGFP reporter gene expression to generate the GFP/R9 clone and an almost identical vector, lacking only P450R cDNA, to generate the control GFP-5 cells. We have carefully evaluated these clones to match EGFP expression in hypoxic and oxic conditions and cell growth rates in vitro and in vivo. This provided us with confidence that no observable artifacts, inherent to the process of cloning, have been introduced during the selection procedures.
To test the validity of our approach in vitro, we selected the bioreductive drug RSU1069 for its exquisite oxygen sensitivity. The oxygen cytotoxicity profile of this drug is a mirror image of that for radiotherapy in that cells at oxygen concentrations rendering them resistant to radiotherapy are sensitive to RSU1069. This is also reflected in the very high hypoxic cytotoxicity ratio of RSU1069 being 30–200-fold more toxic to hypoxic cells dependent upon the cell type used.17 In addition, we have preliminary data to demonstrate a limited bystander effect with RSU1069 in vitro,23 which is consistent with earlier reports, although this may not have been expected from an activated nitroimidazole. The results obtained demonstrated that our gene therapy approach hypersensitised the GFP/R9 cells to RSU1069 in vitro specifically under anoxic conditions.
GFP/R9 and GFP5 cells grown as xenografts were allowed to reach exponential growth (Figure 3, tumour doubling time of approximately 4 days) before combined treatment with radiotherapy and RB6145 – a chemical precursor of RSU1069 that exhibits reduced systemic toxicity in vivo.22 This allowed us to evaluate the ability of our therapeutic approach to potentiate the radioresponse of an established tumour with a clinically relevant hypoxic fraction.24 Consistent with our in vitro data, hypoxia-regulated P450R expression proved a successful approach to enhance bioreductive hypoxic cell cytotoxicity and hence improve the efficacy of radiotherapy. Our combined therapy yielded a 50% cure rate. The inability to cure all tumours may reflect the number of cells within the mass exhibiting intermediate O2 tensions insufficient to invoke killing with radiotherapy, but too high to achieve adequate P450R expression and RSU1069 toxicity. This may also reflect the five-fold range in P450R activity levels measured in the tumours. In addition, this may suggest that the bystander effect of RSU1069 in vivo may be insufficient to kill these cells. Nevertheless, we have provided a proof of principle for an oxygen-sensitive GDEPT approach and envisage that with the rationale design of prodrugs specifically activated by P450R and exhibiting greater in vivo bystander properties than RSU1069 (which we are actively developing) a total tumour response would be achievable.
Several enzyme prodrug systems are available for enhancing the selective toxicity of cancer chemotherapy. Of these, the most evaluated pairings are cytosine deaminase/5-fluorocytosine (5-FC) and thymidine kinase/ganciclovir (GCV), the latter having demonstrated no activity when included with standard therapy (both surgical resection and radiotherapy) for treatment of patients with glioblastoma multiforme.25 Indeed, in this randomised multicentre phase III trial, standard therapy plus adjuvant gene therapy neither increased time to tumour progression nor improved overall survival when compared with radiotherapy and surgery alone. A possible contributing factor to the ineffectiveness of this gene therapy approach is the quiescent nature of hypoxic cells rendering them resistant to the cytotoxic metabolites of GCV. In addition, hypoxic cells have also been shown to be refractive to 5-fluorouracil, the active metabolite of 5-FC,6 suggesting that hypoxia may restrict the efficacy of these approaches.
Taken together this paper provides a persuasive argument for exploiting hypoxia using an oxygen-sensitive GDEPT approach. In addition, the inadequate response of poorly oxygenated tumours to radiotherapy and chemotherapy has set a precedent for evaluating the extent of tumour hypoxia before treatment. The growing availability of technologies for measuring the level of hypoxia in human tumours using bioreductive agents, eg pimonidazole;26 intrinsic markers, eg Glut-1;27 and the development of probes for use with non-invasive imaging techniques, such as magnetic resonance spectroscopy and positron emission tomography, is beginning to revolutionise the individualisation of patient treatment.28 This would identify a cohort of patients for which an oxygen-sensitive gene therapy approach would have great benefit.
Materials and methods
Vector construction
The full-length cDNA for human P450 reductase (2.1 kb) has previously been constitutively expressed within the vector pBabe/puro.18 To achieve co-expression of both the human P450R cDNA and enhanced green fluorescent protein (EGFP), under the transcriptional control of the HRE from the PGK-1 gene, we constructed a bicistronic expression cassette employing pCIneo as the vector backbone (Promega, Southampton, UK). A synthetic oligonucleotide containing three copies of the minimal 18 bp PGK-1 HRE sequence TGTCACGTCCTGCACGAC and BglII compatible 5’ and 3’ ends was cloned in the forward orientation directly upstream of the SV40 minimal promoter (SV40mp) in the pGL3 vector (Promega). The PGK-HRE-SV40 promoter region was then isolated from pGL3 by Nhe1/HindIII digestion and cloned between SpeI and HindIII restriction sites within pCIneo, to replace its CMV enhancer/promoter sequence, creating pCIneo PGK SV40mp. The P450R cDNA was excised from pBabe/puro by EcoRI/SalI restriction digestion and inserted into pCIneo creating pCIneo PGK SV40mp P450R (pHRE-P450R). To allow co-expression of the EGFP reporter an internal ribosome entry site (IRES) and EGFP fragment was created by combining pIRES and pEGFP-NI (Clontech, Palo Alto, CA, USA). Specifically EGFP was restricted from pEGFP-NI and directionally cloned into pIRES using SmaI/NotI. To facilitate cloning of IRES EGFP at the 3’ end of P450R the IRES EGFP fragment was excised by NheI/NotI digestion and inserted into pCIneo PGK SV40mp P450R (previously linearised with XbaI/NotI) to create the final vector pCIneo PGK SV40mp P450R IRES EGFP (pHRE-P450R/EGFP). This vector was used to generate the stable HT1080 cell line GFP/R9. A control vector lacking the therapeutic gene P450R was also created, pCIneo PGK SV40mp IRES EGFP, by cloning the NheI/NotI IRES EGFP fragment into the multiple cloning site of pCIneo PGK SV40mp (pHRE-EGFP). This vector allowed the generation of the transcriptionally equivalent control HT1080 cell line, GFP-5.
Cell culture
HT1080 (human fibrosarcoma) cells were maintained in exponential growth phase in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% (v/v) foetal calf serum (FCS), 2 mM glutamine and 0.2 mM non-essential amino acids in a 95% air:5% CO2 environment. HT1080 clones GFP-5 and GFP/R9 were selected and maintained in DMEM supplemented with 10% FCS, 2 mM glutamine, 0.2 mM non-essential amino acids and 0.2 mg/ml geneticin 418 sulphate (G418). All cell lines and clones were free from mycoplasma contamination (assayed using Mycotect; Gibco BRL).
Transfections and clonal selection of HT1080 cells
5 × 106 cells in exponential growth were transfected with 10 μg of linearised pCIneo PGK min SV40 P450R IRES EGFP or pCIneo PGK min SV40 IRES EGFP control vector DNA using standard electroporation techniques.29 Cells were plated at low density and 48 h later were exposed to 0.2 mg/ml G418. Individual colonies were then isolated and EGFP expression was employed to monitor the stability of vector expression within clonal populations for at least 8 weeks by carrying out multiple passages either in the presence or absence of G418. The clones were then analysed for PGK-1 HRE-mediated EGFP expression levels allowing the selection of trancriptionally equivalent control EGFP and P450R EGFP-expressing HT1080 cell lines. Briefly, cells were incubated in air or hypoxia (catalyst-induced anoxia, <1 ppm oxygen; Bactron anaerobic chamber, Sheldon Manufacturing, Cornelius, OR, USA) for 18 h followed by 3 h of re-oxygenation. Cells were then detached using 2 mM EDTA and resuspended in FACS flow (Becton Dickinson, Mountain View, CA, USA). EGFP expression was determined by flow cytometry using a Becton Dickinson FACSort with a single laser excitation wavelength of 488 nm. EGFP fluorescence (emission wavelength 508 nm) was measured in the FL-1 channel that detects emitted light of wavelength 500–600 nm. EGFP fluorescence was plotted versus number of cells and median peak values obtained for the air and hypoxic cell samples.
Reagents
RSU1069 (1-[3-aziridinyl-2-hydroypropyl]-2-nitroimidazole) and its brominated precursor RB6145 (1-[3-(2-bromoethylamino)-2-hydroxypropyl]-2-nitroimidazole) were prepared according to described methods.22
NADPH:cytochrome P450 reductase activity determination
NADPH:cytochrome P450 reductase activity was determined spectrophotometrically as the NADPH dependent reduction of cytochrome c. The reaction comprised 400 μl of cytochrome c (final concentration 50 μM), 100 μl of 10 mM potassium cyanide (final concentration 1 mM), 10–300 μg protein lysate (10–100 μl volume) and 100 mM phosphate buffer pH 7.6, to a total volume of 0.98 ml. The reaction was equilibrated to 37°C and was initiated by addition of 20 μl of 10 mM NADPH to the test cuvette (final concentration 200 μM) and the rate of reduction of cytochrome c was monitored at 550 nm for 3 min against a blank without NADPH. Initial rates of reaction were based on the extinction co-efficient of 21 mM/cm and expressed as nmol cytochrome c reduced min/mg/protein. Protein concentrations were determined by Bradford analysis using bovine serum albumen as a protein standard.
In vitro drug sensitivity
Dose response curves were determined using the MTT proliferation assay, which is based on the ability of viable cells to convert a soluble tetrazolium salt, MTT, into purple formazan crystals. For the hypoxic experiments, all plastics and media were pre-incubated in catalyst-induced anoxia for 72 h before use to remove any residual oxygen. RSU1069 (10 mM stock in dimethyl sulphoxide (DMSO)) was serially diluted using culture medium and aliquoted into 96-well plates (100 μl per well) at twice the required final concentration. GFP-5 and GFP/R9 cells (4 × 104 cells/ml) were seeded directly into the drug (100 μl per well) and cultured for 18 h under either hypoxic or aerobic conditions. To prevent growth arrest of the cells under hypoxia, the medium was also supplemented with 300 μM 2-deoxycytidine. The cells were then washed free of drug and allowed to grow in fresh medium in air for 72 h. MTT was added (to a final concentration of 0.5 mg/ml medium) and the cells incubated for a further 3 h after which the culture medium and unconverted MTT was removed. The formazan crystals were dissolved in 0.2 ml of DMSO and the optical density at 540 nm was measured on a multiwell spectrophotometer. The optical density of the dissolved crystals is proportional to the number of viable cells allowing IC50 values (the concentration of drug required to reduce optical density by 50% compared with untreated controls) to be calculated and used as a measure of cellular sensitivity to the given treatment.
Xenograft studies
Tumour xenografts of the GFP/R9 and GFP-5 clones were initiated from the interdermal injection, 1 cm from the tail base on the midline of the back of female nude mice (cba nu/nu, aged 9–12 weeks), of 5 × 106 cells prepared in a 0.1 ml volume of serum-free DMEM. Once palpable tumours were established, daily measurements of tumour dimensions were taken using calipers. Tumour bearing mice were randomly allocated into treatment groups (n = 5–10 per group) and all treatments were performed on tumours of approximately 250 mm3 in volume and experiments were terminated when tumours reached a volume four times that at the time of treatment (RTV4). Mice were restrained whilst localised radiotherapy (X-rays) was delivered at a dose rate of 2 Gy/min. RB6145 was prepared in 0.9% w/v saline at pH 4 and administered immediately after radiotherapy at a dose of 250 mg/kg by intra-peritoneal injection. The bioreductive hypoxic marker, pimonidazole (Hydroxyprobe-1; Natural Pharmacia International, USA)26 was administered at a dose of 80 mg/kg in 0.9% w/v saline to mice carrying 250 mm3 tumours by intra-peritoneal injection, 2 h before death. All procedures were carried out in accordance with the Scientific Procedures Act 1986 and in line with the UKCCCR guidelines 1999, by approved protocols (Home Office Project Licence number 40-1770).
Determination of tumour vascularity and hypoxic fraction (pimonidazole binding)
Staining for endothelial structures, pimonidazole binding and subsequent quantitative analysis of vessel density and hypoxic fraction have been extensively described elsewhere.30 Briefly, 5-μm sections were cut from snap frozen tumour specimens, fixed with acetone and incubated for 45 min with 9F1 antibody (rat monoclonal antibody raised against mouse endothelium). Sections were then sequentially treated for 30 min with tetramethyl rhodamine isothiocyanate (TRITC)-conjugated goat anti-rat and TRITC-conjugated donkey anti-goat antisera. Pimonidazole adducts in the same sections were disclosed by overnight incubation with rabbit anti-pimonidazole antiserum followed by 2 h incubation with FITC-conjugated donkey anti-rabbit antiserum. TRITC- and FITC-conjugated antibodies were supplied by Jackson Immuno Research Laboratories, West Grove, PA, USA. All quantitative assessment was made relative to the viable tumour area, ascertained from haemotoxylin and eosin-stained adjacent sections.
Immunohistochemical staining
Formalin-fixed, paraffin-embedded tumour specimens were sectioned at 3–4 μm thickness. P450R and pimonidazole adduct presentation was analysed in adjacent sections. The rabbit polyclonal anti-human P450 reductase antibody used18 was applied at a 1/1000 dilution. Controls were treated with an identical dilution of rabbit serum. The secondary antibody treatment (horseradish peroxidase-conjugate) and visualisation of immunoreactivity (3,3-diaminobenzidine hydrochloride, DAB) was achieved using reagents supplied in the Envision anti-rabbit kit (DAKO, Glostrup, Denmark), according to the manufacturer's instructions. To visualize pimonidazole adduct formation, sections were sequentially treated with 3% H2O2 in water (5 min, room temperature; RT), 0.05% pronase in tris-buffered saline (TBS; pH 7.2; 5 min, RT) and 10% casein (10 min, RT), with TBS washes in between. Hydroxyprobe-1MAb1 (mouse monoclonal; Natural Pharmacia) was applied at a 1/100 dilution for 30 min at RT and staining visualised using the appropriate Envision kit (DAKO).
References
Gatenby RA et al. Oxygen distribution in squamous cell carcinoma metastases and its relationship to outcome of radiation therapy Int J Radiat Oncol Biol Phys 1988 14: 831–838
Okunieff P et al. Oxygen tension distributions are sufficient to explain the local response of human breast tumors treated with radiation alone Int J Radiat Oncol Biol Phys 1993 26: 631–636
Teicher BA . Hypoxia and drug resistance Cancer Metastasis Rev 1994 13: 39–68
Hockel M et al. Association between tumor hypoxia and malignant progression in advanced cancer of the uterine cervix Cancer Res 1996 56: 4509–4515
Brizel DM et al. Tumor hypoxia adversely affects the prognosis of carcinoma of the head and neck Int J Radiat Oncol Biol Phys 1997 38: 285–289
Grau C, Overgaard JE . Effect of etoposide, carmustine, vincristine, 5-fluorouracil, or methotrexate on radiobiologically oxic and hypoxic cells in a C3H mouse mammary carcinoma in situ Cancer Chemother Pharmacol 1992 300: 277–280
Graeber TG et al. Hypoxia-mediated selection of cells with diminished apoptotic potential in solid tumours Nature 1996 379: 88–91
Stratford IJ, Workman PB . Bioreductive drugs into the next millennium Anticancer Drug Des 1998 13: 519–528
Ratcliffe PJ, O'Rourke JF, Maxwell PH, Pugh CW . Oxygen sensing, hypoxia-inducible factor-1 and the regulation of mammalian gene expression J Exp Biol 1998 201: 1153–1162
Semenza GL . Hypoxia-inducible factor 1: oxygen homeostasis and disease pathophysiology Trends Mol Med 2001 7: 345–350
Maxwell PH, Pugh CW, Ratcliffe PJ . Inducible operation of the erythropoietin 3’ enhancer in multiple cell lines: evidence for a widespread oxygen-sensing mechanism Proc Natl Acad Sci USA 1993 90: 2423–2427
Zhong H et al. Overexpression of hypoxia-inducible factor 1alpha in common human cancers and their metastases Cancer Res 1999 59: 5830–5835
Dachs GU et al. Targeting gene expression to hypoxic tumour cells Nature Med 1997 3: 515–520
Koshikawa N, Takenaga K, Tagawa M, Sakiyama S . Therapeutic efficacy of the suicide gene driven by the promoter of vascular endothelial growth factor gene against hypoxic tumour cells Cancer Res 2000 60: 2936–2941
Adams GE, Ahmed I, Sheldon PW, Stratford IJ . RSU 1069, a 2-nitroimidazole containing an alkylating group: high efficiency as a radio- and chemosensitizer in vitro and in vivo Int J Radiat Oncol Biol Phys 1984 10: 1653–1656
Stratford IJ et al. RSU 1069, a nitroimidazole containing an aziridine group. Bioreduction greatly increases cytotoxicity under hypoxic conditions Biochem Pharmacol 1986 35: 105–109
Koch CJ . Unusual oxygen concentration dependence of toxicity of SR-4233, a hypoxic cell toxin Cancer Res 1993 53: 3992–3997
Patterson AV et al. Overexpression of human NADPH:cytochrome c (P450) reductase confers enhanced sensitivity to both tirapazamine (SR4233) and RSU1069 Br J Cancer 1997 76: 1338–1347
Coralli C et al. Limitations of the reporter green fluorescent protein under simulated tumour conditions Cancer Res 2001 61: 4784–4790
Ebert BL, Firth JD, Ratcliffe PJ . Hypoxia and mitochondrial inhibitors regulate expression of glucose transporter-1 via distinct Cis-acting sequences J Biol Chem 1995 270: 29083–29089
Wiesener MS et al. Constitutive activation of hypoxia-inducible genes related to overexpression of hypoxia-inducible factor-1alpha in clear cell renal carcinomas Cancer Res 2001 61: 5215–5222
Jenkins TC et al. Synthesis and evaluation of alpha-[[(2-haloethyl)amino]methyl]-2- nitro-1H-imidazole-1-ethanols as prodrugs of alpha-[(1-aziridinyl)methyl]-2- nitro-1H-imidazole-1-ethanol (RSU-1069) and its analogues which are radiosensitizers and bioreductively activated cytotoxins J Med Chem 1990 33: 2603–2610
Cowen RL et al. Viral mediated gene delivery of hypoxia regulated reductive enzymes to activate bioreductive drugs and thus improve the therapeutic index of these anti-tumour agents P295 Mol Ther 2001 3: S103
Aquino-Parsons C, Luo C, Vikse CM, Olive PL . Comparison between the comet assay and the oxygen microelectrode for measurement of tumor hypoxia Radiother Oncol 1999 51: 179–185
Rainov NG . A phase III clinical evaluation of herpes simplex virus type 1 thymidine kinase and ganciclovir gene therapy as an adjuvant to surgical resection and radiation in adults with previously untreated glioblastoma multiforme Hum Gene Ther 2000 11: 2389–2401
Raleigh JA, Chou SC, Arteel GE, Horsman MR . Comparisons among pimonidazole binding, oxygen electrode measurements, and radiation response in C3H mouse tumors Radiat Res 1999 151: 580–589
Airley R et al. Glucose transporter glut-1 expression correlates with tumor hypoxia and predicts metastasis-free survival in advanced carcinoma of the cervix Clin Cancer Res 2001 7: 928–934
Aboagye EO, Kelson AB, Tracy M, Workman P . Pre-clinical development and current status of the fluorinated 2-nitroimidazole hypoxia probe N-(2-hydroxy-3trifluoropropyl)-2-(2-nitro-1-imidazolyl) acetamide (SR 4554, CRC 94/17): a non-invasive diagnostic probe for the measurement of tumor hypoxia by magnetic resonance spectroscopy and imaging, and by positron emission tomography Anticancer Drug Des 1998 13: 703–730
van den Hoff MJ, Moorman AF, Lamers WH . Electroporation in ‘intracellular’ buffer increases cell survival Nucleic Acids Res 1992 20: 2902
Bussink J et al. Optical sensor-based oxygen tension measurements correspond with hypoxia marker binding in three human tumor xenograft lines Radiat Res 2001 154: 547–555
Acknowledgements
The collaboration with Albert van der Kogel was initiated via the Biomed II Concerted Action Programme (Development of Methods for the Rapid Analysis of Tumour Oxygenation to Allow Treatment Stratification; contract BMH4983006). Many thanks are due to Hans Peters. The Medical Research Council funded this work.
Author information
Authors and Affiliations
Rights and permissions
About this article
Cite this article
Patterson, A., Williams, K., Cowen, R. et al. Oxygen-sensitive enzyme-prodrug gene therapy for the eradication of radiation-resistant solid tumours. Gene Ther 9, 946–954 (2002). https://doi.org/10.1038/sj.gt.3301702
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.gt.3301702
Keywords
This article is cited by
-
Disturbance in the regulation of miR 17-92 cluster on HIF-1-α expression contributes to clinically relevant radioresistant cells: an in vitro study
Cytotechnology (2020)
-
Targeting acute hypoxic cancer cells by doxorubicin-immunoliposomes directed by monoclonal antibodies specific to RON receptor tyrosine kinase
Cancer Chemotherapy and Pharmacology (2011)
-
Adenovirus-mediated hypoxia-targeting cytosine deaminase gene therapy enhances radiotherapy in tumour xenografts
British Journal of Cancer (2007)
-
Transcriptional targeting of acute hypoxia in the tumour stroma is a novel and viable strategy for cancer gene therapy
Gene Therapy (2005)
-
Exploiting tumour hypoxia in cancer treatment
Nature Reviews Cancer (2004)
