
Abstract
Deficiency of the apoptosome component Apaf1 leads to accumulation of supernumerary brain cells in mouse embryos. We observed that neural precursor cells (NPCs) in Apaf1−/− embryos escape programmed cell death, proliferate and retain their potential to differentiate. To evaluate the circumstances of Apaf1−/− NPC survival and investigate their fate under neurodegenerative conditions, we established cell lines of embryonic origin (ETNA). We found that Apaf1−/− NPCs resist common apoptotic stimuli and neurodegenerative inducers such as amyloid-β peptide (typical of Alzheimer's disease) and mutant G93A superoxide dismutase 1 (typical of familial amyotrophic lateral sclerosis). Similar results were obtained in Apaf1−/− primary cells. When death is prevented by Apaf1 deficiency, cytochrome c is released from mitochondria and rapidly degraded by the proteasome, but mitochondria remain intact. Under these conditions, neither activation by cleavage of initiator caspases nor release of alternative apoptotic inducers from mitochondria takes place. In addition, NPCs can still differentiate, as revealed by neurite outgrowth and expression of differentiation markers. Our findings imply that the mitochondrion/apoptosome pathway is the main route of proneural and neural cells to death and that its inhibition prevents them from dismantling in neurodegenerative conditions. Indeed, the ETNA cell model is ideally suited for exploring the potential of novel cell therapies for the treatment of human neurodegenerations.
Similar content being viewed by others


Introduction
The apoptosome is a large molecular complex operating as an engine of caspase activation that, in turn, initiates and/or executes apoptosis.1 Apaf1 is a scaffold protein participating in apoptosome formation in mammals.2 Apaf1 and the apoptosome are involved in many aspects of mouse development, regulated by morphogenetic, histogenetic and phylogenetic apoptosis.3, 4, 5 In particular, Apaf1 deficiency leads to brain hyperplasia, possibly due to accumulation of neural precursor cells (NPCs) that escape death and retain their potential to proliferate.6 This phenotype is highly similar to Caspase-3 (Casp3) and Casp9 knockout phenotypes. It is widely believed that activation of the apoptosome, followed by Casp9 and Casp3 activation, plays a central role in cell death in the nervous system.7 This pathway of death is distinct from those occurring in other embryonic tissues, such as the immune system8 and the haematopoietic system, where Casp2 activation seems to be more relevant and the apoptosome-mediated pathway represents a mere amplification loop.9 Moreover, in the brain, cell death is not simply a mechanism to match the neuron population to its target fields. Rather, cell death plays an additional and important role in adjusting the initial progenitor pool needed for proper morphogenesis of the nervous system, the mechanism of which is still to be investigated. Based on the preliminary finding that Apaf1−/− neuroepithelium proliferates in vivo for a few days of embryonic life and then starts to differentiate, we established cell lines of embryonic origin, which express or lack Apaf1 and show several features of bona fide NPCs. To shed more light on Apaf1−/− NPC circumstances of survival and fate, we applied several death inducers to them and checked if cells which escape death are still able to differentiate properly. Additionally, we extended the analysis to other developing CNS cell types, obtaining comparable results. Amid the various death stimuli we used, amyloid-β1–42 peptide (Aβ) and a mutant form of superoxide dismutase 1 (G93A-SOD1) are particularly significant. In fact, inappropriate activation of apoptotic pathways mediated by these stimuli is a contributing event in two neurodegenerative diseases, Alzheimer's disease (AD) and familial amyotrophic lateral sclerosis (fALS).7, 10 The two stimuli are very different: Aβ forms aggregate and lead mainly to an extracellular death induction, while mutant SOD1 acts at the intracellular level. Furthermore, the stimuli's neuronal targets in adult life are different, since hippocampal, striatal, cortical and cerebellar neurons are typically affected in AD, whereas motor neurons are selectively affected in fALS.
Results
The Embryonic Telencephalic Naïve Apaf1 (ETNA) cells represent a model to study the behaviour of NPCs
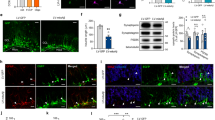
We analysed cell proliferation in the neural tube of Apaf1−/− and wild-type (wt) littermate embryos at embryonic day 12. 5 (e12.5) and found that mutant embryos showed a proliferation index twice as high as wt littermates in the rostral part of the neural tube [74.9±3.9 and 37.2±3.6% (forebrain), 28.3±4.1% and 26.1±3.0% (hindbrain), respectively; see Figure 1a]. We then checked mutant embryos at a later stage (e13.5–e14) for expression of proneural markers such as nestin, delta1, notch1, or a later marker of nerve cells (class III β-tubulin). We found that they were all expressed in conserved domains even when the cyto-architecture of the brain structure was profoundly disturbed (Figure 1b). We observed a high level of nestin mRNA expression in the mutant neuroepithelium at e14, indicating the presence of progenitor cells.11 For this reason, we decided to establish cell lines from the striatum primordia isolated from e14 wt and mutant embryos. This makes it possible to overcome problems occurring when using primary cultures, ranging from the heterogeneity of primary progenitor populations to the small amount of cells obtained with that method. Cells were infected with a retrovirus transducing the tsA58/U19 Large T Antigen and selected.12 Cell lines derived in this manner are grown at 33°C (permissive temperature) and contain a normal complement of chromosomes without contamination from a neuroblastoma line, this being a consequence of the somatic fusion strategy.13 After appropriate selection, several surviving cell clones were picked and named Embryonic Telencephalic Naïve Apaf1 (ETNA)+/+ and ETNA−/− cells, respectively (Figure 1c). Analysis of all clones gave consistent results: data from clones NDR1 (ETNA+/+) and ADR5 (ETNA−/−) are shown. When induced to differentiate, both ETNA+/+ and ETNA−/− cells displayed neurite outgrowth and the upregulation of some neural markers, such as choline acetyltransferase (ChAT) and class III β-tubulin (βIII-tubulin) (Figure 1d, e).

Apaf1-deficiency leads to accumulation of NPCs expressing neural markers in vivo and in vitro. (a) BrdU staining on coronal sections from e12.5 wt and Apaf1−/− (−/−) littermates embryos. nt: neural tube (hindbrain), fb: forebrain. (b) Analysis of differentiation markers from e14 wt and Apaf1−/− littermate embryos. nestin, delta1, notch1 mRNA levels were revealed by in situ mRNA hybridization as previously described (Stoykova and Gruss, 1994) on embryonic coronal sections. Class III β-tubulin was revealed by immunohistochemistry on embryonic e14.5 coronal sections with a mouse monoclonal anti-class III β-tubulin antibody. The corresponding histological structures are indicated in the sketch and in the right panel as follows: I: intermediate zone, M: marginal zone, V: ventricular germinal zone, ve: brain ventricle. Scale bar: 0.4 mm. (c) ETNA+/+ (+/+) and ETNA−/− (−/−) cell lines were isolated from e14 wt and mutant embryos, respectively, and immortalized with a retrovirus transducing the tsA58/U19 large T antigen. Total extracts from both cell lines were immunoprecipitated with an anti-Apaf1 rabbit polyclonal antibody and analysed with a rat monoclonal antibody anti-Apaf1. Ctrl: human neuroblastoma cells (SH-SY5Y) stably overexpressing Apaf1. (d) Total cell lysates from ETNA+/+ and ETNA−/− cells, either naïve (N) or incubated for 48 h with a differentiation medium (D), were analysed by Western blotting for the expression of NeuN, class III β-tubulin, ChAT, PSD95, tau; β-actin was also assayed as a control of equal protein loading. (e) ETNA+/+ and ETNA−/− cells, either naïve or differentiated, were immunostained with an antibody recognizing the 160 kDa form of neurofilament (NF-M). Scale bar: 40 μm
ETNA−/− cells resist several death stimuli
Cell death was induced in ETNA+/+ and ETNA−/− cells by means of several common proapoptotic stimuli, such as staurosporine (STS), serum starvation (not shown) and transcriptional inhibition (actinomycin D, Act D). As a result of all these stimuli, ETNA+/+ cells died within a few hours, as revealed by nuclear condensation, DNA fragmentation, cleavage of poly ADP ribose polymerase (PARP) and phosphatidylserine exposure, while ETNA−/− cells were clearly resistant to all assayed stimuli (Figure 2a, c, e). Few, if any, apoptotic cells were observed in the several optical fields analysed. This behaviour of ETNA−/− cells could be recapitulated in ETNA+/+ cells by treating them with zVAD-fmk, a wide-range caspase inhibitor (Figure 2b). In order to verify the activity of Casp3 in ETNA+/+ cells, its capacity to bind a fluoresecent substrate was tested; we observed that Casp3 is indeed involved in ETNA cell death (Figure 2d).

ETNA−/− cells are resistant to apoptotic stimuli. (a) Hoechst 33342 nuclear staining of ETNA+/+ and ETNA−/− cells either treated for 24 h with 5 μM STS, 40 h with 20 μM Act D, 48 h after transient transfection with plasmids coding for wt SOD1 (wtSOD1), or the fALS mutant G93A-SOD1 (G93A) or 72 h with 50 μM Aβ1–42 peptide (Aβ). Scale bar: 20 μm. (b) Quantification of apoptotic nuclei of ETNA cells treated as in (a) in the absence or in the presence of the pan-caspase inhibitor zVAD-fmk (100 μM). (c) Quantification of apoptotic cells by Annexin V-FITC binding of exposed phosphatidylserine in ETNA+/+ and ETNA−/− cells treated as in (a). (d) Lack of Casp3 activity in ETNA−/− cells exposed to different apoptotic stimuli. ETNA+/+ and −/− cells were either treated for 24 h with 5 μM STS, 40 h with 20 μM Act D, 72 h with 50 μM Aβ, or 48 h after transient transfection with plasmids coding for wtSOD1, or the fALS mutant G93A-SOD1 (G93A). Equal amounts (30 μg) of clear cell lysates were incubated with 20 μM Ac-DEVD-AMC in lysis buffer for 30′ at 37°C. The amount of AMC release was quantified using a fluorescence plate reader. Casp3 activity is expressed as arbitrary fluorescence units (AFU). (e) Total cell lysates from ETNA+/+ and ETNA−/− treated for 24 h with 5 μM STS, 40 h with 20 μM Act D, or 72 h with 50 μM Aβ were subjected to SDS-PAGE and analysed for PARP processing
ETNA+/+ and ETNA−/− cells were then exposed to neurodegenerative stimuli typical of AD and fALS. First, we checked for apoptotic signals upstream of Apaf1. Upon induction of cell death, cytochrome c is released from the mitochondria, binds to Apaf1 and activates the apoptosome. At 72 h after Aβ treatment, cytochrome c was released from the mitochondria of both ETNA+/+ and ETNA−/− cells (Figure 3). As a consequence of apoptosome formation, Casp3 was activated and death occurred in ETNA+/+ cells (Figures 2 and 3). In contrast, Casp3 activation and apoptotic death did not follow cytochrome c release in the absence of Apaf1 (ETNA−/− cells) (Figures 2 and 3). Cells transfected with the G93A mutant form of SOD1, SOD1 mutant forms being the cause of 20% of fALS cases,14 followed a similar pathway. Cytochrome c was released in both ETNA+/+ and ETNA−/− cells, while Casp3 activation and apoptotic death took place only in ETNA+/+ cells (Figures 2 and 3). As a control, wtSOD1 transfected in parallel was unable to cause cell death in both cell lines (Figures 2 and 3). In some experiments, the green fluorescent protein (GFP) was used as a reporter of both wild-type and G93A-SOD1 expression (see Materials and Methods).

Cytochrome c is released but Casp3 is not activated in ETNA−/− cells upon neurodegenerative stimuli. Left panel: double-labeling confocal immunofluorescence microscopy of ETNA+/+ and ETNA−/− cells treated for 72 h with 50 μM Aβ. Pattern of active Casp3 (red), cytochrome c (green) and merged patterns of the same representative fields. Right panel: double-labeling confocal immunofluorescence microscopy of ETNA+/+ and ETNA−/− cells 24 h after transient transfection with either wtSOD1-GFP or G93A-SOD1-GFP. Pattern of cytochrome c (red), GFP (green) and merged patterns of the same representative fields. White arrowheads point to cells with released cytochrome c. Scale bar: 20 μm. A quantitative analysis of the numbers of cells releasing cytochrome c is represented by histograms, each indicating the mean ±SD of three independent experiments. In all, 100 cells were analysed in each examined field and five randomly chosen fields for each experimental condition were counted
The protection of ETNA−/− cells from death is solely due to the Apaf1 deficiency
In order to demonstrate that the protection from death observed in ETNA−/− cells under several stimuli was a monogenic phenotype, we transformed them with a construct carrying the Apaf1 cDNA under the control of the strong constitutive promoter CAGGs. ETNA−/− cells transfected with Apaf1 cDNA were sensitive to death induced by STS, Act D, Aβ and transfected G93A-SOD1 (Figure 4a, b), as revealed by Casp3 activation and nuclear condensation. In addition to the genetic complementation experiment, we also analysed the cells after inactivation of the tsA58/U19 Large T Antigen gene expression by shifting the growth temperature from 33 to 39°C (nonpermissive) (Figure 4c, d). This was done to verify that the viral gene expression was not responsible for the observed phenotype. In fact, ETNA−/− cells at 39°C resist apoptotic induction with all the applied stimuli. In conclusion, we were able to link the observed phenotype to Apaf1 deficiency only in the cell clones analysed.

The protection of ETNA−/− cells from death is uniquely due to the Apaf1 deficiency. (a) Double-labeling confocal immunofluorescence microscopy of ETNA−/− cells transiently transfected with a plasmid coding for Apaf1 and treated for 24 h with 5 μM STS, for 40 h with 20 μM Act D or for 72 h with 50 μM Aβ. Pattern of Apaf1 (red), SYTOX nuclear staining (green) and merged patterns of the same representative fields are shown. White arrowheads point to cells overexpressing Apaf1 and showing apoptotic nuclei. Histogram represents the mean ±SD of three independent experiments. Scale bar: 20 μm. (b) ETNA−/− cells were treated as in (a) or co-transfected with a decreasing amount of a plasmid coding for Apaf1 and with either a plasmid coding for wtSOD1 or G93A mutant. Total cell lysates were subjected to SDS-PAGE and analysed for Apaf1, active Casp3 and SOD1 expression. (c) Immunofluorescence staining of LTAg expression and Hoechst nuclear staining of ETNA+/+ and ETNA−/− cells cultured at 33°C (permissive temperature) or shifted to 39°C (nonpermissive temperature) for 24 h and then treated for an additional 24 h with 5 μM STS, for 40 h with 20 μM Act D or for 72 h with 50 μM Aβ Scale bar: 40 μm. (d) Western blot analysis of active Casp3 and SOD1 expression in ETNA+/+ and ETNA−/− cells incubated at 39°C and treated as in (c) or transfected with wtSOD1 or G93A mutant
The main route of ETNA cells to death is along the mitochondrion/apoptosome pathway
In order to better understand which caspases participate in the apoptosome-mediated death pathway in ETNA cells, we analysed the involvement of Casp9 by means of a specific inhibitor. The activation of Casp8 and Casp2 was also assessed by determining the cleaved forms of the respective pro-enzymes in Western blots. Casp9 was activated, consistently with Casp3 cleavage described above, upon G93A-SOD1 (Figure 5a) and Aβ (not shown) stimuli. In contrast, Casp2 was not cleaved in this system, differing from what observed in the haematopoietic system and in transformed fibroblasts, where Casp2 plays a role upstream of the apoptosome9, 15 (Figure 5b). Similarly, Casp8 also remained uncleaved upon G93A-SOD1 or Aβ treatment, although its expression levels were substantially increased with the latter stimulus (Figure 5b). Modulation of pro-Casp8 expression without significant cleavage was previously shown as a hallmark of severe traumatic head injury in humans.16 Together, these results demonstrate a pivotal role for the apoptosome-mediated pathway in cell death induced by neurodegenerative stimuli.

The apoptosome pathway is the main route of ETNA cells to death. (a) ETNA+/+ and ETNA−/− cells were transiently transfected with G93A-SOD1 in the absence or in the presence of 100 μM zLEHD-fmk, a specific inhibitor of Casp9. TUNEL assay and Hoechst staining were performed 48 h after transfection. The same representative field is shown here. Scale bar: 20 μm. Histogram represents the mean ±SD of three independent experiments. In all, 100 cells were analysed in three randomly chosen fields. (b) Total cell lysates from ETNA+/+ and ETNA−/− cells untreated (ctrl), treated for 72 h with 50 μM Aβ or 24 h after transfection with an unrelated plasmid (mock) or G93A-SOD1, were subjected to SDS-PAGE and analysed for Casp3, Casp2 and Casp8 processing. WtSOD1 or G93A-SOD1 transfected cells were also analysed for SOD1 expression. mSOD1: endogenous mouse SOD1; hSOD1: transfected human SOD1. Casp8 antibody sensitivity was assessed in CEM cells treated with 250 ng/ml of anti-Fas antibody (Upstate biotechnology) for the indicated times
ETNA cells apoptosis induced by neurodegenerative stimuli is caspase-dependent and AIF- and EndoG-independent
Next, we followed the fate of Aβ-treated and G93A-SOD1-overexpressing ETNA−/− cells that do not undergo apoptosis due to the absence of Apaf1. It has been suggested that, when caspases are inhibited, other factors, released from mitochondria in a caspase-independent manner, can trigger a secondary nonapoptotic form of death.17 Apoptosis-inducing factor (AIF18) has been related to a nonapoptotic morphotype in several cell types and conditions, for example, Apaf1−/− interdigital web cells19 and primary hippocampal neurons.20 More recently, the independence of AIF release from caspase activation has been questioned.21, 22 We found that AIF continued to remain in the mitochondrial compartment upon neurodegenerative stimuli in ETNA cells 72 h after G93A-SOD1 transfection or 96 h after Aβ application (Figure 6a, b), both in the presence and absence of apoptosome. Similarly, when cytochrome c was released in ETNA cells, the Endonuclease G (EndoG), a factor also known to trigger a caspase-independent type of death,23 was retained in the mitochondria and did not contribute to any delayed forms of cell death (Figure 6c). Release of both AIF and EndoG from the mitochondria was induced in HeLa cells upon several stimuli, confirming that such a release is cell type-dependent (Figure 6d and data not shown). In addition, ETNA−/− cells showed no necrotic morphology after 72 and 96 h of G93A-SOD1 transfection and Aβ treatment, respectively (data not shown). Therefore, under these conditions, ETNA cells do not undergo alternative death pathways when neurodegenerative stimuli are applied.

ETNA−/− cells retain mitochondrial AIF and EndoG upon neurodegenerative stimuli. (a,b) Double-labeling confocal immunofluorescence microscopy staining of cytochrome c (green) and AIF (red) in ETNA+/+ cells (a) or ETNA−/− cells (b) either treated for 72 h with 50 μM Aβ or 48 h after transfection with G93A-SOD1. (c) Double-labelling confocal immunofluorescence microscopy staining of cytochrome c (green) and EndoG (red) in ETNA−/− cells after transfection with G93A-SOD1. White arrowheads point to cells with released cytochrome c. Scale bar: 20 μm. (d) Double-labelling confocal immunofluorescence microscopy staining of cytochrome c (green) and AIF (red, upper panel) or EndoG (red, lower panel) in HeLa cells untreated (ctrl) and treated for 8 h with 2 μM STS. White arrowheads point to cells with released cytochrome c and AIF (upper panel) or cytochrome c and EndoG (lower panel)
ETNA−/− cells mitochondria remain intact after cytochrome c release induced by neurodegenerative stimuli
At this point, we analysed whether mitochondria remain intact in ETNA+/+ and ETNA−/− cells induced to die by G93A-SOD1 transfection and Aβ treatment. We evaluated the effect of apoptosome inactivation on mitochondrial transmembrane potential (Δψm) by means of two reagents: CM-H2XROS and tetramethylrhodamine ethyl ester (TMRE). Both reagents, when added to the culture medium, can enter exclusively intact mitochondria in a voltage-dependent manner, and can be revealed by fluorescence. Apaf1 deficiency protected ETNA cells from the loss of mitochondrial potential (Figure 7a, b), as revealed by fluorescence localization and measurement, using the ETNA+/+ cells as positive control and valinomycin-treated cells as a standard of strong Δψm dissipation. To analyse the mitochondrial capability for energy production, ATP concentration in ETNA cells exposed to death stimuli was also measured. An [ATP] decrease was evident in ETNA+/+ cells; in contrast, Apaf1-deficient ETNA cells showed a physiological ATP concentration (Figure 7c). Notably, HeLa cells can recover from Δψm dissipation when caspases are inhibited, but in this cell line eventual loss of Δψm and drop of [ATP] precede cell death.24, 25 We then performed a morphological analysis of ETNA cells upon G93A-SOD1 death induction by EM (Figure 7d). Most ETNA+/+ cells transfected with G93A-SOD1 showed extensive mitochondrial damage with disarray of the cristae, as well as aspects of severe cytoplasmic degeneration and chromatin damage. Similar degenerative aspects were also seen in a few ETNA−/− cells. However, these cells mostly resembled nontransfected controls in their cytoplasmic and nuclear ultrastructure, suggesting again that apoptosome inhibition has the potential to protect cells from mitochondrial degeneration (Figure 7d).

ETNA−/− cells mitochondria are functional after cytochrome c release induced by neurodegenerative stimuli. (a) Aβ-treated or G93A-SOD1-transfected ETNA−/− cells were loaded for 15 min with 50 nM MitoTracker Red CM-H2XROS and then processed for immunofluorescence staining of cytochrome c. White arrowheads point to cells with released cytochrome c. Scale bar: 20 μm. (b) FACS analysis of Δψm in ETNA+/+ and ETNA−/− cells transfected with wtSOD1 or G93A-SOD1 for 24 h and then stained with 100 nM TMRE for 15 min in the absence or presence of 100 nM valinomycin as a control for mitochondrial depolarization. (c) ETNA−/− cells show a physiological ATP concentration when exposed to neurodegenerative stimuli. Total cellular ATP levels in ETNA+/+ and ETNA−/− cells 48 h after transfection with wtSOD1 or G93A-SOD1. (d) EM of ETNA+/+ and ETNA−/− cells 24 h after transfection with G93A-SOD1. Clear mitochondrial damage (arrows) and pathological chromatin distribution were observed in ETNA+/+ cells transfected with G93A-SOD1. ETNA−/− cells after transfection with G93A-SOD1 mostly resembled nontransfected ETNA cells in their nuclear and cytoplasmic morphology. Middle panels show higher magnification details. Mitochondria (M) in cultured ETNA−/− cells, as well as in nontransfected cells, mostly appeared as homogeneous structures, although a fine profile of the cristae was not clearly visible. N: cell nucleus. Scale bars: 0.2 μm
After release from mitochondria, cytochrome c is degraded by the proteasome in ETNA−/− cells escaping death
In order to follow the fate of cytochrome c, once it has been released from the mitochondria in the surviving ETNA−/− cells, we also analysed its levels in cell compartments after the application of death stimuli. We chose STS and Act D, since they are capable of rapidly inducing apoptosis in the vast majority of treated cells. Upon these stimuli, cytochrome c is released from the mitochondria and then degraded (Figure 8a,b). The degradation is carried out by the proteasome, as confirmed by the lack of cytochrome c degradation in the presence of the proteasome inhibitor Lactacystin (Figure 8b).

Released cytochrome c is degraded by the proteasome in ETNA−/− cells escaping death. (a) ETNA−/− cells were treated with 5 μM STS or 20 μM Act D for the indicated times. Cytosolic and membrane fractions were analysed by Western blotting for the presence of cytochrome c. β-Actin and Cox IV were used as control for equal protein loading in the cytosolic and membrane fractions, respectively. (b) Time-course analysis of cytochrome c expression in total cell lysates of ETNA−/− cells treated with 5 μM STS or 20 μM Act D, in the absence or in the presence of 10 μM lactacystin. β-Actin was also assayed as a control of equal protein loading
ETNA−/− cells induced to die by neurodegenerative stimuli can still differentiate
Finally, we analysed whether ETNA−/− cells induced to die by neurodegenerative stimuli retain their potential to differentiate. Aβ-treated (120 h) or G93A-SOD1-GFP-transfected (48 h) cells were exposed to differentiating conditions (48 h in differentiating medium). Aβ treatment could not be followed at a single-cell level, while transfected cells were detectable because of GFP fluorescence. After 120 h of Aβ treatment, many surviving ETNA−/− cells differentiated (as shown by neurite outgrowth from cells showing cytochrome c release; Figure 9a). Similarly, 96 h after G93A-SOD1-GFP transfection, about 70% of GFP-positive ETNA+/+ cells died, whereas about 70% of GFP-positive ETNA−/− cells survived and differentiated (Figure 9b). Due to the latter stimulus, when left in culture for an additional 48 h [total timeframe: 144 h (6 days)], transfected ETNA−/− cells eventually died with a phenotype characterized by massive overproduction and intracellular accumulation of G93A-SOD1-GFP (not shown). In conclusion, cells that escape death induced by neurodegenerative stimuli are able to differentiate and maintain a sustainable lifespan until aggregates or inclusions induce a dramatic cellular collapse.

ETNA−/− cells retain the potential to differentiate upon neurodegenerative stimuli. (a) Aβ-treated (120 h) ETNA−/− cells were exposed to a differentiating medium for an additional 48 h, still in the presence of Aβ. Cells were immunostained with anti-cytochrome c antibody (red) and the differentiated morphology was assessed by NF-M immunostaining (green). Three different fields are shown. (b) G93A-SOD1-GFP-transfected (48 h) ETNA−/− cells were exposed to differentiating medium for additional 48 h. Differentiated morphology was assessed by NF-M immunostaining (red) of SOD1-GFP-positive cells (green). Three different fields are shown. Scale bar: 20 μm
Apaf1−/− primary cortical cells behave similarly to ETNA−/− cells when induced to die by several death stimuli
In order to further exclude a clonal specificity of the observed cellular phenotypes and to extend the analysis to other developing CNS cell types, we applied several death stimuli to primary cortical cells from Apaf1−/− and wt littermate dissected brains (e14). Wt primary cells, upon induction by Act D, Aβ and STS, underwent death by cytochrome c release, Casp3 activation and nuclear condensation, as expected (Figure 10a, b). In contrast, Apaf1−/− primary cells from cortical primordia do not show Casp3 activation induced by the same stimuli after several hours from the treatment and after release of cytochrome c. We could not test the G93A-SOD1 death induction conditions, due to the low levels of transfection efficiency in this culture system (not shown). Since we could observe some morphological alterations in the surviving cell nuclei of Apaf1−/− cells, we tested their ability to express differentiation markers as a proof of their viability. Indeed, class III β-tubulin was expressed in the soma and neurites of most treated cells, independently of their morphological appearance (Figure 10c).

Apaf1−/− primary cortical cells resist to several death stimuli similarly to ETNA−/− cells. Double-labelling confocal immunofluorescence microscopy of primary cortical cells from Apaf1−/− and wt embryonic brains (e14) treated for 48 h with 20 μM Act D (a), for 72 h with 50 μM Aβ (a, c) or for 24 h with 5 μM STS (b). Patterns of cytochrome c and SYTOX nuclear staining (a), active Casp3 and cytochrome c (b) and βIII-tubulin and SYTOX nuclear staining (c) are shown. Scale bars: 20 μm. Histograms represent the mean ±SD of three independent experiments. In all, 100 cells were analysed in each examined field and three randomly chosen fields for each experimental condition were counted
Discussion
In this work, we have followed the fate of embryonic or embryonic-derived cells in vivo and in vitro, where cell death has been induced by developmental, stress-related and neurodegenerative stimuli in the presence or absence of the apoptosome. To date, Apaf1 and the apoptosome have been related to developmental cell death in the nervous system, eye, limbs, inner ear, craniofacial structures3, 4, 5 and in several pathological conditions, such as malignant melanoma, prostate cancer and leukaemia.26 The ability of Apaf1-, Casp9- or Casp3-knockout brains to overgrow was previously reported.3, 4 Apaf1 involvement in brain-mass control in a dosage-dependent manner was also confirmed by the analysis of the hypomorphic mutant fog (forebrain overgrowth), in which Apaf1 levels are strongly reduced.27 We wondered whether Apaf1 is involved in the main route of NPCs to death, as suggested by the knockout mouse phenotype, and whether the path to death could be somehow prevented or reverted in Apaf1−/− cells. Additionally, we wanted to know what was the quality of surviving Apaf1−/− cells committed to die, and whether they retained their potential to differentiate.
Our results here suggest that (i) NPCs are able to die predominantly, if not exclusively, by means of the mitochondrion–apoptosome pathway during development; (ii) cell lines derived from NPCs behave similarly when induced to die by several stimuli, including neurodegenerative ones; (iii) cytochrome c release from mitochondria is not a ‘point of no return’ in the NPC death pathway, since mitochondria on the verge of disruption do not lose their Δψm unless downstream steps of the apoptotic program are executed; however, cytochrome c becomes degraded by the proteasome after release; (iv) ETNA−/− cells escaping death, similarly to NPCs in vivo, are able to differentiate; (v) all the observed phenotypes are exclusively due to Apaf1 deficiency in the cell lines and can be recapitulated in primary cortical cells derived from the mutant embryos.
From e12 onwards, the brain of Apaf1−/− embryos shows severe morphological distortions. A huge brain overgrowth, due to an enlargement of proliferative zones, is observed as a bilateral protrusion of the forebrain or as an exencephalic brain mass. Basically, three cell populations die during neural development: neuronal precursors, glial cells and postmitotic neurons. Our findings confirm that an apoptosome-dependent death regulates the size of NPC pools. This is certainly not the case in other regions of the developing organism. For instance, Apaf1−/− thymocytes have been shown to be normally susceptible to Fas-mediated cell death, demonstrating that Apaf1 is dispensable for the Fas-mediated apoptotic pathways in thymocytes, involving Casp8 as an initiator caspase.4 More recently, we reported that caspase activity is still discernible in embryonic haematopoietic cells lacking Apaf1 or Casp9, and that a Casp2 antagonist both inhibits apoptosis and retards cytochrome c release.9 This Casp2-dependent caspase activation program acts independently of the cytochrome c/Apaf1/Casp9 apoptosome, which seems to amplify rather than initiate the caspase cascade. This seems to be the case also in human tumor cell lines, where cytotoxic stress was shown to cause activation of Casp2, required for mitochondria permeabilization.15 We conclude that the main death pathways may vary among cell types in development and that a Casp8-, Casp2-dependent or nonapoptotic pathway of death is unlikely to occur or cannot compensate for the apoptosome in NPCs. This is in line with the absolute preponderance of the nervous phenotype in Apaf1-, Casp9- and Casp3-knockout mice.
We observed that, in ETNA cells, Casp8 expression is upregulated upon neurodegenerative stimuli, as happens in human brain after acute injury, as reported by others.16 In this work, the authors showed that induction, upregulation and proteolysis of Casp8 occurs in human brain after trauma, and that relative Casp8 levels are associated with levels of Fas. Although cleavage of Casp8 was undetectable in both ETNA cell clones, our evidence suggests that Casp8 may be involved in the apoptotic response to neurodegenerative stimuli, but strictly in an apoptosome-dependent way.
We also observed that Casp2 expression levels in ETNA−/− cells were lower than in ETNA+/+ cells. We cannot exclude that Casp2 expression might be downregulated in a given cell clone, since a slightly different panel of markers might be expressed in independent clones at a given step of differentiation. However, activation of Casp2 was undetectable in both ETNA cell clones. It should be mentioned that Aβ-mediated death requires Casp2 in hippocampal neurons, sympathetic neurons and PC12 cells,28 and requires Casp12 in primary cortical neurons.29 This suggests that different pathways of death can be activated in different neuronal cell types. Since we showed the Apaf1 dependence of the Aβ death pathway also in this cell culture system, our results argue for an epistatic relationship between the apoptosome activation and Casp12 under these conditions.
In addition to HeLa cells, which have been shown to recover from cytochrome c release during apoptosis upon inhibition of caspases,24, 25 the definition of cytochrome c release as a ‘point of no return’ during cell death commitment in the nervous system has been previously confuted. Survival was observed in sympathetic neurons induced to die and deprived of apoptosome or caspase activity upon treatment with the trophic factor NGF. This factor seemed to induce de novo synthesis of protein in order to restore the mitochondrial pool of cytochrome c.30 In the absence of this treatment, vice versa, cells were secondarily committed to die (25 h later) by a nonapoptotic mechanism.31 ETNA−/− cells, however, seem to behave differently, surviving in normal growth condition (-NGF) for a period six times longer than 25 h and exhibiting properties of normal NPCs in culture. However, we show here that cytochrome c in these conditions is rapidly degraded by the proteasome. This could be due to the fact that, in contrast to NGF deprivation which could be reverted by simply adding NGF to the medium, other stimuli exert irreversible signals that cannot be removed and probably cause mitochondria to release cytochrome c persistently. This would happen even in the case that cytochrome c is newly synthesized and uptaken by the mitochondria: cytochrome c could be uptaken, released and degraded in a continuous loop. It would be interesting to investigate which bioenergetical pathways the maintenance of ATP concentration is based on, since it is conceivable that mitochondria deprived of cytochrome c cannot perform oxidative phosphorylation properly. As hypothesized elsewhere,32 the best candidate to play this role is, without doubt, the glycolytic pathway which is highly active in the nervous system.
To ETNA cells, along with common apoptotic inducers, we applied neurodegenerative stimuli linked to AD and fALS in humans. In principle, in chronic neurodegenerative diseases, the apoptotic molecules play the dominant role in regulating cell dysfunction and death (see Friedlander33 and reference therein). A spectrum of apoptosis mediators is seen in neurons vulnerable in AD and ALS post-mortem tissues.7, 10, 34 More recently, Casp9, a component of the apoptosome, has been found to be activated in spinal motor neurons of human ALS patients and to be involved in disease progression in transgenic mouse models.35 Cellular models mimic various aspects of the neurodegenerative diseases, none entirely recapitulating the human pathology, but most overcoming many of the limitations of human post-mortem tissues (including the nonfeasibility of manipulating mediators of apoptosis). In addition, new therapeutic nonpharmacological methodology in Parkinson's disease36, 37 and AD38 involves cell and synaptic renewal or replacement in order to restore the function of neuronal systems. ETNA cells exhibit properties of immature cells, including expression of proneural proteins, the ability of self-renewal and terminal differentiation. Therefore, they are an ideal model system for studying apoptosis modulation in vitro in neurodegenerative conditions, and for unravelling pathways of death in neural precursors, both of these being essential for devising novel cell therapy strategies for AD and fALS.
Materials and Methods
Plasmid constructs
pRc/CMV plasmids coding for wt or G93A mutant SOD1 (wtSOD1 or G93A-SOD1, respectively) were described elsewhere.39 Wt and mutant SOD1–GFP fusion protein (wtSOD1–GFP and G93A-SOD1–GFP, respectively) expression vectors were created by PCR cloning of the SOD1 cDNAs40 between the PmlI and PstI sites of pCMV/myc/mito/GFP (Invitrogen). This cloning step deleted the mitochondrial targeting sequence. Apaf1 expression plasmid was created by cloning of Apaf1 cDNA3 under control of the strong promoter CAGGs (a kind gift of Dr. Miyazaki from Osaka University).
Mouse stocks and manipulation
The generation of Apaf1−/− mice has been described previously.3 The embryos were taken from timed matings and the day on which a vaginal plug was detected was designated as e0.5. Pregnant Apaf1+/- females were killed by cervical dislocation and the embryos were dissected and fixed in 4% paraformaldehyde. The embryos or mice were genotyped from extraembryonic membranes or from tail biopsies.3
Immunohistochemistry and RNA in situ hybridization
Immunohistochemical detection of class III β-tubulin was performed according to the manufacturer's protocol. BrdU/PBS intraperitoneal injection and detection in proliferating cells were performed as previously described.41 The sections were counterstained with Hoechst 33258. The mean proliferation indexes (proportion of BrdU-positive nuclei to all nuclei in a section of neuroepithelium) with ±SD% have been reported (n=3). The differences between wt and Apaf1−/− mutant values were analysed using the two-tailed Student's t-test (P>0.0001). In situ hybridization on paraffin-embedded sections was also performed as previously described.42 After staining, the embryos were embedded in paraffin and sectioned. Photographs were taken using a Zeiss-Axioskop microscope.
Cell cultures
ETNA cells were obtained as follows: control mice or timed pregnant Apaf1+/− female crossed with an Apaf1+/− male3 were killed by decapitation at e14. The foetuses were removed and the striatum primordia from homozygous embryos (+/+ and −/−) were isolated. Cells were dissociated and plated in DMEM +10% foetal calf serum (FCS, Invitrogen). The following day, the primary culture was infected for 24 h with filtered conditioned medium from cells packaging a retrovirus transducing the tsA58/U19 Large T Antigen and the neomycin resistance gene.12 After infection, the virus-containing medium was replaced with fresh DMEM + 10% FCS, and the cells kept at 33°C. After 2 days, the cultures were passaged and fed with fresh complete medium in the presence of the selective agent G418 (Invitrogen) at 200 μg/ml. After selection, single colonies were picked and expanded. ETNA cells were routinely grown in DMEM +10% FCS, at 33°C (permissive temperature for large T antigen expression) in an atmosphere of 5% CO2 in air, and used between passages 2 and 7. Cell culture differentiation was obtained by shifting cells in growth medium containing 50 μM forskolin, 250 μM IBMX, 200 nM TPA, 10 μM dopamine and 10 ng/ml aFGF. The differentiation medium was replaced every 24 h. For the inactivation of the tsA58/U19 Large T Antigen gene expression, cells were cultured at 39°C.
Transient expression of each vector (1.5 μg DNA/5–7 × 105 cells) was obtained with LipofectAMINE Plus reagent (Invitrogen) according to the manufacturer's instructions. After a 3 h incubation with transfection reagents, the cells were cultured in normal growth medium for the indicated period of time. For pure cortical primary culture, e14 embryos from pregnant Apaf1+/− mice were killed, the cortical hemispheres were removed and neocortices were treated with 0.025% trypsin for 10 min at 37°C. After centrifugation, the pellets were resuspended in MEM with 1% FCS (Invitrogen) and gently triturated. Cell suspension was centrifugated and resuspended in MEM containing 5% FCS, 5% horse serum, 2 mM L-glutamine and 25 mM glucose and plated onto polylysine-coated dishes. After 24 h, the medium was replaced with Neurobasal-A (Invitrogen) supplemented with B-27 and 1 mM glutamine. After 3 days, 10 μM cytosine arabinoside (Ara-C, SIGMA-ALDRICH) was added to inhibit non-neuronal cell proliferation. Cultures were mantained at 37°C in an atmosphere of 5% CO2 in air.
Aβ peptide preparation
Aβ1–42 was purchased from PolyPeptide Laboratories GmbH. It was initially dissolved to 1 mM in hexafluoroisopropanol (HFIP, SIGMA). HFIP was then removed under vacuum and the dessicated Aβ1–42 was stored at −20°C. For peptide aggregation, Aβ1–42 was dissolved to 5 mM in dimethyl sulphoxide (DMSO, SIGMA) and then brought to a final concentration of 125 μM with phenol red-free Dulbecco's medium (Invitrogen). It was incubated at 33°C for 24 h and used at a final concentration of 50 μM.
Caspase activity assay and assessment of cell death
Casp3 activitiy was determined by measuring the rates of 7-amido-4-methylcoumarin (AMC) release from the synthetic caspase substrate Ac-DEVD-AMC (Calbiochem), as previously described.43 Quantification of apoptotic cells was obtained by direct visual counting either after nuclear staining of 4% paraformaldehyde-fixed cells with the fluorescent probe Hoechst 33342 (1 μg/ml) (SIGMA-ALDRICH), or after Annexin V-FITC binding according to the manufacturer's protocol (Santa Cruz Biotechnology, Inc). In all, 100 cells were analysed in each examined field at × 200 magnification, and eight randomly chosen fields for each experimental condition were counted. For apoptotic nuclei counting, only the cells containing clearly picnotic or fragmented nuclei were considered apoptotic. For terminal deoxynucleotidyltransferase-mediated UTP end labeling (TUNEL) assay, we used the in situ ApopTag detection kit (Intergen). TUNEL assay was carried out 48 h after transfection according to the manufacturer's protocol.
Immunoprecipitation and Western blotting
After rinsing the cultures with ice-cold PBS, cell lysis was performed in lysis buffer (20 mM Tris–HCl, pH 7.4, 1% Triton X-100, 150 mM NaCl, 1 mM EDTA, 5 mM MgCl2) containing 1 mM PMSF and a protease inhibitor cocktail (SIGMA-ALDRICH). A clear supernatant was obtained by centrifugation of lysates at 17 000 × g for 10 min. Protein content was determined using Bradford protein assay (Bio-Rad). For the detection of Apaf1 expression, 300 μg of protein was immunoprecipitated with anti-Apaf1 polyclonal antibody (AB16941, Chemicon Int.) coupled to protein-G–sepharose beads (SIGMA-ALDRICH). Western blots were performed on polyvinylidene difluoride membranes (Immobilon P, Millipore) as previously described (Casciati et al., 2002). Equal loading of samples was confirmed by Ponceau red staining (SIGMA-ALDRICH). Apaf1 was detected with a rat monoclonal anti-Apaf1 antibody (clone 18H2). Immunoreactive SOD1 was detected with a sheep polyclonal antibody (Upstate biotech). Cleaved Casp3 was detected with a polyclonal antibody against the large fragment of activated Casp3 (Cell Signaling). For the detection of both full-length and cleaved active form of Casp8, the monoclonal antibody anti-Casp8 1C12 was used (Cell Signaling). Both full-length and cleaved active fragments of Casp2 and PARP were detected with the anti-Casp2 monoclonal antibody 11B4 or with the anti-PARP rabbit polyclonal antibody (Cell Signaling), respectively. For the analysis of cytochrome c expression, the mouse monoclonal antibody anti-cytochrome c 7H8.2C12 (BD PharMingen) was used. To analyse the expression of cell differentiation markers, antibodies anti-NeuN (Chemicon Int.), -ChAt (Chemicon Int.), -tau (Biosource), class III β-tubulin (TUJ1, Covance Research Products) and -PSD95 (BD Biosciences) were used. Anti-β-actin antibody (clone AC15) was purchased from Sigma and anti-Cox IV antibody was purchased from Molecular Probes.
Subcellular fractionation
ETNA cells were harvested in hypotonic buffer (2 mM MgCl2, 10 mM KCl, 10 mM Tris–HCl, pH 7.6). supplemented with protease inhibitor cocktail (SIGMA) and incubated for 20 min on ice. In all, 1 V of 2 × Mitobuffer (450 mM sucrose, 10 mM Tris–HCl, pH 7.4, 400 μM EGTA, pH 7.4, 2 μM DTT) was added and the cell suspension was homogenized for 40 strokes with a Dounce homogenizer. Samples were centrifuged twice at 900 × g for 5 min at 4°C to eliminate cell nuclei and unbroken cells. The resulting supernatant was centrifuged at 10 000 × g for 30 min at 4°C to recover the heavy-membrane pellet enriched for mitochondria, and the resulting supernatant was stored as the cytosolic fraction.
Immunocytochemistry
Cells cultured in 35 mm Petri dishes were washed in PBS and fixed with 4% paraformaldehyde in PBS for 15 min. After permeabilization with 0.2% Triton X-100 in PBS for 5 min, cells were blocked in 2% horse serum in PBS and incubated for 1 h at 37°C with primary antibodies. We used an anti-cytochrome c mouse monoclonal antibody (clone 6H2.B4, BD PharMingen), an anti-cytochrome c rabbit polyclonal antibody (Santa Cruz), an anti-AIF rabbit polyclonal antibody (a generous gift of Guido Kroemer), an anti-EndoG rabbit polyclonal antibody (ProSci Incorporated), an anti-Neurofilament 160 mouse monoclonal antibody (clone NN18, SIGMA), anti-Apaf1 polyclonal antibody (AB16941, Chemicon Int.) and an anti- Large T Antigen monoclonal antibody (clone PAb419, Oncogene). Cells were then washed in blocking buffer and incubated for 1 h with labelled anti-mouse or anti-rabbit secondary antibodies (Alexa, Molecular Probes). After rinsing in blocking buffer, cell nuclei were stained with 1 μg/ml Hoechst 33342 (SIGMA-ALDRICH) or 25 nM SYTOX (Molecular Probes) and examined under a Zeiss LSM 510 Confocal Microscopy equipped with a × 40 objective. Fluorescence Images were adjusted for brightness, contrast and colour balance by using Adobe Photoshop 7.0.
Electron microscopy
ETNA−/− and ETNA+/+ cells were plated on Thermanox coverslips (Nunc), in a 24-well culture plate. After 24 h, cells were either left untransfected or transfected with G93A-SOD1 plasmid. Cells were fixed 24 h after transfection. The coverslips were treated with 1% osmium tetroxide for 90 min at 4°C. Cells were washed in buffer and dehydrated in a graded ethanol series at 4°C. The cell monolayers were treated with propylene oxide and then infiltrated in a mixture of epon and propylene oxide at 4°C and embedded in epon for 24 h at 60°C. Coverslips were removed and the adjacent surface of the epon block was cut into ultrathin sections of 85 nm with a Reichert-Jung Ultracut. Sections were collected, dried and stained with uranyl acetate and lead. The sections were observed with a LEO 906E electron microscope equipped with a HSC2 Digital Camera (Proscan). The microphotographs were acquired by Vario Vision, brought to the final size and resolution and mounted in a plate by Adobe Photoshop 7.0.
Quantitative assessment of mitochondrial impairment
Δψm was measured using TMRE (Molecular Probes) according to Waterhouse et al,24 with minor modifications. Transfected cells were detached and incubated at 33°C for 15 min in media containing TMRE (100 nM). As a control for Δψm dissipation, cells were treated with 100 nM valinomycin. TMRE fluorescence was detected by flow cytometry on a FACScan (Becton Dickinson) using FL-2. Aβ1–42-treated or wtSOD1 and G93A-SOD1-transfected ETNA cells were also incubated with 100 nM CM-H2XROS (Molecular Probes), for 15 min, 33°C, washed in fresh medium and then processed for immunofluorescence staining of cytochrome c as above. Measurement of cellular ATP was performed using the CytoLux kit (Perkin-Elmer) according to the manufacturer's instructions. A standard curve of known ATP concentration was used to calculate ATP concentration in the samples.
Abbreviations
- NPCs:
-
neural precursor cells
- Apaf:
-
apoptotic proteases-activating factor
- ETNA:
-
embryonic telencephalic naïve Apaf1
- Casp:
-
caspase
- Aβ:
-
amyloid-β1–42 peptide
- SOD1:
-
superoxide dismutase 1
- AD:
-
Alzheimer's disease
- fALS:
-
familial amyotrophic lateral sclerosis
- CNS:
-
central nervous system
- wt:
-
wild type
- e:
-
embryonic day
- ChAT:
-
choline acetyltransferase
- βIII-tubulin:
-
class III β-tubulin
- STS:
-
staurosporine
- Act D:
-
actinomycin D
- PARP:
-
poly ADP ribose polymerase
- fmk:
-
fluoromethylketone
- AIF:
-
apoptosis-inducing factor
- EndoG:
-
endonuclease G
- Δψm:
-
mitochondrial transmembrane potential
- TMRE:
-
tetramethylrhodamine ethyl ester
- EM:
-
electron microscopy
- GFP:
-
green fluorescent protein
- NGF:
-
nerve growth factor
- BrdU:
-
bromodeoxy-uridine
References
Adams JM and Cory S (2002) Apoptosomes: engines for caspase activation. Curr. Opin. Cell. Biol. 14: 715–720
Zou H, Henzel WJ, Liu X, Lutschg A and Wang X (1997) Apaf-1, a human protein homologous to C. elegans CED-4, participates in cytochrome c-dependent activation of caspase-3. Cell 90: 405–413
Cecconi F, Alvarez-Bolado G, Meyer BI, Roth KA and Gruss P (1998) Apaf1 (CED-4 homolog) regulates programmed cell death in mammalian development. Cell 94: 727–737
Yoshida H, Kong YY, Yoshida R, Elia AJ, Hakem A, Hakem R, Penninger JM and Mak TW (1998) Apaf1 is required for mitochondrial pathways of apoptosis and brain development. Cell 94: 739–750
Honarpour N, Du C, Richardson JA, Hammer RE, Wang X and Herz J (2000) Adult Apaf-1-deficient mice exhibit male infertility. Dev. Biol. 218: 248–258
Kuan CY, Roth KA, Flavell RA and Rakic P (2000) Mechanisms of programmed cell death in the developing brain. Trends Neurosci. 23: 291–297
Yuan J and Yankner BA (2000) Apoptosis in the nervous system. Nature 407: 802–809
Krammer PH (2000) CD95's deadly mission in the immune system. Nature 407: 789–795
Marsden VS, O'Connor L, O'Reilly LA, Silke J, Metcalf D, Ekert PG, Huang DC, Cecconi F, Kuida K, Tomaselli KJ, Roy S, Nicholson DW, Vaux DL, Bouillet P, Adams JM and Strasser A (2002) Apoptosis initiated by Bcl-2-regulated caspase activation independently of the cytochrome c/Apaf-1/caspase-9 apoptosome. Nature 419: 634–637
Guegan C and Przedborski S (2003) Programmed cell death in amyotrophic lateral sclerosis. J. Clin. Invest. 111: 153–161
Zimmerman L, Parr B, Lendahl U, Cunningham M, McKay R, Gavin B, Mann J, Vassileva G and McMahon A (1994) Independent regulatory elements in the nestin gene direct transgene expression to neural stem cells or muscle precursors. Neuron 12: 11–24
Cattaneo E and Conti L (1998) Generation and characterization of embryonic striatal conditionally immortalized ST14A cells. J. Neurosci. Res. 53: 223–234
Ehrlich ME, Conti L, Toselli M, Taglietti L, Fiorillo E, Taglietti V, Ivkovic S, Guinea B, Tranberg A, Sipione S, Rigamonti D and Cattaneo E (2001) ST14A cells have properties of a medium-size spiny neuron. Exp. Neurol. 167: 215–226
Cleveland DW and Rothstein JD (2001) From Charcot to Lou Gehrig: deciphering selective motor neuron death in ALS. Nat. Rev. Neurosci. 2: 806–819
Lassus P, Opitz-Araya X and Lazebnik Y (2002) Requirement for caspase-2 in stress-induced apoptosis before mitochondrial permeabilization. Science 297: 1352–1354
Zhang X, Graham SH, Kochanek PM, Marion DW, Nathaniel PD, Watkins SC and Clark RS (2003) Caspase-8 expression and proteolysis in human brain after severe head injury. FASEB J. 17: 1367–1369
Sperandio S, de Belle I and Bredesen DE (2000) An alternative, nonapoptotic form of programmed cell death. Proc. Natl. Acad. Sci. USA 97: 14376–14381
Susin SA, Lorenzo HK, Zamzami N, Marzo I, Snow BE, Brothers GM, Mangion J, Jacotot E, Costantini P, Loeffler M, Larochette N, Goodlett DR, Aebersold R, Siderovski DP, Penninger JM and Kroemer G (1999) Molecular characterization of mitochondrial apoptosis-inducing factor. Nature 397: 441–446
Cande C, Cecconi F, Dessen P and Kroemer G (2002) Apoptosis-inducing factor (AIF): key to the conserved caspase-independent pathways of cell death? J. Cell. Sci. 115: 4727–4734
Cregan SP, Fortin A, MacLaurin JG, Callaghan SM, Cecconi F, Yu SW, Dawson TM, Dawson VL, Park DS, Kroemer G and Slack RS (2002) Apoptosis-inducing factor is involved in the regulation of caspase-independent neuronal cell death. J. Cell. Biol. 158: 507–517
Arnoult D, Gaume B, Karbowski M, Sharpe JC, Cecconi F and Youle RJ (2003) Mitochondrial release of AIF and EndoG requires caspase activation downstream of Bax/Bak-mediated permeabilization. EMBO J. 22: 4385–4399
Arnoult D, Parone P, Martinou JC, Antonsson B, Estaquier J and Ameisen JC (2002) Mitochondrial release of apoptosis-inducing factor occurs downstream of cytochrome c release in response to several proapoptotic stimuli. J. Cell. Biol. 159: 923–929
van Loo G, Schotte P, van Gurp M, Demol H, Hoorelbeke B, Gevaert K, Rodriguez I, Ruiz-Carrillo A, Vandekerckhove J, Declercq W, Beyaert R and Vandenabeele P (2001) Endonuclease G: a mitochondrial protein released in apoptosis and involved in caspase-independent DNA degradation. Cell Death Differ. 8: 1136–1142
Waterhouse NJ, Goldstein JC, von Ahsen O, Schuler M, Newmeyer DD and Green DR (2001) Cytochrome c maintains mitochondrial transmembrane potential and ATP generation after outer mitochondrial membrane permeabilization during the apoptotic process. J. Cell. Biol. 153: 319–328
Ricci JE, Gottlieb RA and Green DR (2003) Caspase-mediated loss of mitochondrial function and generation of reactive oxygen species during apoptosis. J. Cell. Biol. 160: 65–75
Ferraro E, Corvaro M and Cecconi F (2003) Physiological and pathological roles of Apaf1 and the apoptosome. J. Cell. Mol. Med. 7: 21–34
Honarpour N, Gilbert SL, Lahn BT, Wang X and Herz J (2001) Apaf-1 deficiency and neural tube closure defects are found in fog mice. Proc. Natl. Acad. Sci. USA 98: 9683–9687
Troy CM, Rabacchi SA, Friedman WJ, Frappier TF, Brown K and Shelanski ML (2000) Caspase-2 mediates neuronal cell death induced by beta-amyloid. J. Neurosci. 20: 1386–1392
Nakagawa T, Zhu H, Morishima N, Li E, Xu J, Yankner BA and Yuan J (2000) Caspase-12 mediates endoplasmic-reticulum-specific apoptosis and cytotoxicity by amyloid-beta. Nature 403: 98–103
Martinou I, Desagher S, Eskes R, Antonsson B, Andre E, Fakan S and Martinou JC (1999) The release of cytochrome c from mitochondria during apoptosis of NGF-deprived sympathetic neurons is a reversible event. J. Cell. Biol. 144: 883–889
Deshmukh M, Kuida K and Johnson Jr EM (2000) Caspase inhibition extends the commitment to neuronal death beyond cytochrome c release to the point of mitochondrial depolarization. J. Cell. Biol. 150: 131–143
Chang LK, Schmidt RE and Johnson Jr EM (2003) Alternating metabolic pathways in NGF-deprived sympathetic neurons affect caspase-independent death. J. Cell. Biol. 162: 245–256
Friedlander RM (2003) Apoptosis and caspases in neurodegenerative diseases. N. Engl. J. Med. 348: 1365–1375
Troy CM and Salvesen GS (2002) Caspases on the brain. J. Neurosci. Res. 69: 145–150
Inoue H, Tsukita K, Iwasato T, Suzuki Y, Tomioka M, Tateno M, Nagao M, Kawata A, Saido TC, Miura M, Misawa H, Itohara S and Takahashi R (2003) The crucial role of caspase-9 in the disease progression of a transgenic ALS mouse model. EMBO J. 22: 6665–6674
Storch A and Schwarz J (2002) Neural stem cells and neurodegeneration. Curr. Opin. Investig. Drugs 3: 774–781
Isacson O, Bjorklund LM and Schumacher JM (2003) Toward full restoration of synaptic and terminal function of the dopaminergic system in Parkinson's disease by stem cells. Ann. Neurol. 53 (Suppl 3): S135–S146; discussion S146-S138
Mattson MP (2000) Emerging neuroprotective strategies for Alzheimer's disease: dietary restriction, telomerase activation, and stem cell therapy. Exp. Gerontol. 35: 489–502
Carri MT, Ferri A, Battistoni A, Famhy L, Gabbianelli R, Poccia F and Rotilio G (1997) Expression of a Cu,Zn superoxide dismutase typical of familial amyotrophic lateral sclerosis induces mitochondrial alteration and increase of cytosolic Ca2+ concentration in transfected neuroblastoma SH-SY5Y cells. FEBS Lett. 414: 365–368
Ding H, Schwarz DS, Keene A, Affar el B, Fenton L, Xia X, Shi Y, Zamore PD and Xu Z (2003) Selective silencing by RNAi of a dominant allele that causes amyotrophic lateral sclerosis. Aging Cell 2: 209–217
Salminen M, Meyer BI, Bober E and Gruss P (2000) Netrin 1 is required for semicircular canal formation in the mouse inner ear. Development 127: 13–22
Stoykova A and Gruss P (1994) Roles of Pax-genes in developing and adult brain as suggested by expression patterns. J. Neurosci. 14: 1395–1412
Casciati A, Ferri A, Cozzolino M, Celsi F, Nencini M, Rotilio G and Carri MT (2002) Oxidative modulation of nuclear factor-kappaB in human cells expressing mutant fALS-typical superoxide dismutases. J. Neurochem. 83: 1019–1029
Acknowledgements
We thank Tilmann Achsel, Claudia Bagni, Daniela Barilá, Stefano Bertuzzi, Fulvio Celsi, Carlo Rodolfo and Rainer Herken for helpful discussion. We are grateful to Silke Eckert, Natalie Obermayer, Fulvio Florenzano and Elke Heyder for technical assistance, Johan Lundqvist and Urban Lendhal for RNA probes, Lorraine O’Reilly and Andreas Strasser for anti-Casp2 and anti-Apaf1 antibodies. We are indebted to Peter Gruss, Barbara I Meyer, Kamal Chowdhury and Anastassia Stoykova for the generation of the Apaf1−/− mice. This work was supported by Telethon (Grant TCP99038 to FC and GGP030066 to MTC), MIUR (Grant RBAU01FZMZ), Compagnia di San Paolo, and Min. Salute (PF 2002-04).
Author information
Authors and Affiliations
Corresponding author
Additional information
Edited by JC Martinou
Rights and permissions
About this article
Cite this article
Cozzolino, M., Ferraro, E., Ferri, A. et al. Apoptosome inactivation rescues proneural and neural cells from neurodegeneration. Cell Death Differ 11, 1179–1191 (2004). https://doi.org/10.1038/sj.cdd.4401476
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.cdd.4401476
Keywords
This article is cited by
-
Transcriptomic analysis of glutamate-induced HT22 neurotoxicity as a model for screening anti-Alzheimer’s drugs
Scientific Reports (2023)
-
Reversible induction of mitophagy by an optogenetic bimodular system
Nature Communications (2019)
-
AMBRA1 is able to induce mitophagy via LC3 binding, regardless of PARKIN and p62/SQSTM1
Cell Death & Differentiation (2015)
-
Apaf1-deficient cortical neurons exhibit defects in axonal outgrowth
Cellular and Molecular Life Sciences (2015)
-
The DNA repair complex Ku70/86 modulates Apaf1 expression upon DNA damage
Cell Death & Differentiation (2011)
