
Abstract
In Caenorhabditis elegans, several distinct apoptosis pathways have been characterized in the germline. The physiological pathway is though to eliminate excess germ cells during oogenesis to maintain gonad homeostasis and it is activated by unknown mechanisms. The DNA damage-induced germ cell apoptosis occurs in response to genotoxic agents and involves the proteins EGL-1 and CED-13, and the DNA damage response protein p53. Germ cell apoptosis can also be induced in response to pathogen infection through an EGL-1 dependent pathway. To gain insight into the mechanism and functions of germ cell apoptosis, we investigated whether and how other forms of stress induce this cell death. We found that oxidative, osmotic, heat shock and starvation stresses induce germ cell apoptosis through a p53 and EGL-1 independent pathway. We also learned that the MAPK kinases MEK-1 and SEK-1, and the p53 antagonist protein ABL-1, are essential for stress-induced germ cell apoptosis. We conclude that in C. elegans responses to various stresses that do not involve genotoxicity include an increase in germ cell apoptosis through the physiological pathway.
Similar content being viewed by others


Introduction
In many organisms, gametogenesis is known to involve the death of a significant number of germ cells, but how this process is regulated is not known. The nematode Caenorhabditis elegans has proved to be an excellent model organism for studying many biological processes due to its easy manipulation and the high conservation rate of its genes during evolution. The C. elegans hermaphrodite gonad consists of two identical U-shaped tubes that are connected to the uterus (Figure 1a). In the most distal part of each tube, the gonad maintains a population of mitotically active stem cells. During the L4 larval stage, approximately the first 40 cells acquire a male fate and produce about 160 sperm, which are stored in the spermatheca. The next germ cells to undergo gametogenesis all adopt the female fate. Oocytes form in the loop region, and then grow significantly in size by accumulating large amounts of cytoplasm.1 During oogenesis, it has been estimated that close to 50% of germ cells die through the so-called physiological pathway.2 Germ cell apoptosis occurs in the loop area precisely where oogenesis takes place; its location suggests that some of these cells die in order to contribute their cytoplasm to forming oocytes, and to maintain gonad homeostasis.2 One of the most intriguing aspects of physiological germ cell apoptosis is that it is induced independently of the BH3-only domain protein EGL-1, which is involved in all other apoptosis in C. elegans.2 Physiological germ cell death also occurs independently of the transcription factor p53, known in C. elegans as CEP-1, which is required for DNA damage-induced germ cell apoptosis.3, 4 Mutations that specifically block physiological germ cell apoptosis have not been reported, but mutations in seven genes are known to result in elevated levels of physiological germ cell apoptosis: CGH-1, an RNA helicase that protects oocytes from apoptosis;5 CAR-1, a protein that interacts with CGH-1;6 and five other genes isolated in a genome-wide RNAi screening.7 Unfortunately, we still do not understand how any of these genes protect germ cells from apoptosis or if they have any direct role during this process.

The C. elegans hermaphrodite gonad and its cell death pathways. (a) The hermaphrodite gonad consists of two U-shaped tubes that are bound by the uterus (U). At the distal end of each gonad arm, the somatically derived DTC maintains a self-renewing mitotic stem cell population through direct contact. Although germline cells initially form a syncytium, we refer to them as cells for simplicity. The first 40 germ cells that enter meiosis form approximately 160 sperm (S); afterwards only oocytes (O) are produced. Cellularization occurs in the gonad loop, as germ cells progress from the pachytene to the diakinesis stages. It is estimated that 50% of germ cells die (corpse) through the so-called physiological pathway during oogenesis as they exit the pachytene region of the loop area. Fertilization occurs in the spermatheca (S) while eggs leave through the proximal gonad end. Embryos (E) are transiently stored in the uterus (U). (b) Several pathways can induce cell death in C. elegans and these converge in the core apoptosis machinery. This machinery consists of the antiapoptotic protein CED-9, which inhibits the caspase activator CED-4 and the caspase CED-3. Mammalian homologs for each protein are indicated in parentheses. Somatic cell death during early embryogenesis and throughout the larval stages is regulated by way of the proapoptotic protein EGL-1. The genotoxic pathway requires DNA damage checkpoint genes, the transcriptional activator CEP-1, and partially requires EGL-1 and CED-13 (not shown); the physiological pathway is induced through an unknown mechanism independent of EGL-1. Pathogen infection can also induce germ cell apoptosis through an EGL-1 dependent pathway (not shown)
A different type of germ cell apoptosis has been observed when nematodes are exposed to genotoxic agents. This type of apoptosis, known as DNA damage-induced germ cell apoptosis, requires checkpoint proteins, the transcriptional activator p53 and acts partially through EGL-1 and CED-13.3, 4, 8, 9A third mechanism of germ cell apoptosis is induced by pathogens through an EGL-1 dependent mechanism.10 The physiological,2 DNA damage8 and pathogen10 pathways converge on the classic apoptosis machinery: the antiapoptotic protein CED-9 (the human BCL-2 homolog); the caspase activator and APAF homolog CED-4; and CED-3, a C. elegans caspase essential for apoptosis (Figure 1b).
We used C. elegans to test how different stress conditions can affect germ cell apoptosis. We found that germ cell apoptosis can be induced in response to different stresses including oxidative, osmotic, heat shock and starvation. Unexpectedly, we also learned that this stress-induced apoptosis occurs independently of the proteins p53 and EGL-1, which are essential for DNA damage-induced germ cell apoptosis.
An additional finding was that the mitogen-activated kinase kinases (MAPKKs), MEK-1 and SEK-1, and the p53 antagonist tyrosine kinase, ABL-1, are all essential for germ cell apoptosis under oxidative, osmotic and heat shock stress conditions. In contrast, starvation-induced apoptosis is activated through a different pathway, which is independent of the MAPKKs and ABL-1.
Results
Multiple forms of stress induce germ cell apoptosis
To investigate whether stress affects the frequency of apoptosis in the C. elegans germline, we first exposed 1-day old adults to oxidative stress (10 mM paraquat for 1 h), and then assessed the number of dead cells in each gonad arm. We counted corpses, as revealed by Nomarski microscopy, and the number of dying cells that stained with the vital dye acridine orange (see Materials and Methods). Using each method, we observed that paraquat exposure significantly increased the number of dead germ cells present in the gonad (1.7- and 3.2-fold, respectively; Table 1, Figure 2f and data not shown). Paraquat treatment also significantly increased the number of germ cells that expressed transgenic green fluorescent protein (GFP) driven by the ced-1 promoter (Plim-7ced-1∷GFP), a sensitive marker of apoptotic cells that facilitates accurate scoring of corpses (Table 2 and Supplementary Figure S1).9

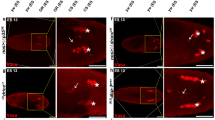
Different stress conditions induce germ cell apoptosis. Nomarski views of control and experimentally treated ced-1(e1735) hermaphrodite gonads. Distal part of the gonad is towards the top and to the left. One-day old animals were incubated for 1 h in untreated M9 (a); in M9 treated with 10 mM paraquat (b); or 70 mM sodium chloride (c). Following incubation, animals were transferred to seeded NGM plates, allowed to recover for 1 h, then anesthetized and mounted for corpse visualization under the microscope. For heat shock, 1-day old animals were incubated on seeded NGM plates in a water bath at 33°C for 1 h (d). To effect starvation conditions, animals were transferred to NGM plates without food and incubated at 20°C for 6 h (e) before they were mounted and observed under the microscope. White arrows point to corpses within insets that show loop gonad detail. Graph (f) shows fold increased germ cell apoptosis under the conditions and genetic backgrounds indicated
The steady-state number of dead cells present in the gonad is determined not only by the frequency of apoptosis but also by the rate of engulfment, a process that normally occurs within less than an hour in the gonad.2 To test whether paraquat treatment might have simply slowed the rate of corpse engulfment and not increased germ cell death, we counted the number of germline corpses present in ced-1(e1735), ced-1(RNAi) and ced-5(n1812) animals, in which engulfment is defective and corpses remain visible for several hours.11, 12 As observed in wild-type animals, in these backgrounds paraquat treatment also increased the number of corpses present in the gonad (Table 1, Figure 2b and f and Supplementary Figure S3b), indicating that oxidative stress increases the frequency of germ cell death. Further supporting this idea, time-lapse microscopy revealed that in wild-type animals corpses formed after paraquat treatment were removed within 1 h, indicating that oxidative stress did not slow engulfment (Supplementary Figure S2). To confirm that this cell death indeed derived from apoptosis, we tested two mutants: ced-3(n717), which is defective in the only known C. elegans caspase, and ced-4(n1162) a mutant in which CED-3 is present but cannot be activated because its activator, CED-4, is absent.13 Paraquat treatment did not increase the numbers of germline corpses in ced-3(n717); ced-1(RNAi) or ced-4(n1162); ced-1(RNAi) animals (Table 1 and Supplementary Figure S3g and l), demonstrating that it induces germ cell death through apoptosis. Together, these results demonstrate that oxidative stress significantly increases the frequency of germ cell apoptosis.
We next tested whether other types of stress increased germ cell apoptosis by subjecting C. elegans to osmotic shock (70 mM sodium chloride for 1 h), heat shock (33°C for 1 h) or starvation (6 h of food deprivation) (see Materials and Methods). In apparent contrast to paraquat treatment, these three conditions did not significantly increase the numbers of germ cell corpses detectable in wild-type animals (Figure 2f and Table 1). It is possible, however, that these stresses might increase germ cell death to a modest extent that is masked when corpse engulfment proceeds at a normal rate, or that they could simultaneously increase the rate of engulfment, a process that involves a complex network of signals.14 To circumvent these possibilities, we tested how these treatments affected engulfment-defective ced-1(RNAi), ced-1(e1735) and ced-5 (n1812) animals. In each case, these stresses significantly increased the numbers of germ cell corpses in these engulfment-defective backgrounds, suggesting that they increased germ cell death (Table 1 and Figure 2c–f). The highest increase in germ cell apoptosis (2.5-fold) was induced by starvation (Figure 2e and f), followed by two-fold increases linked to osmotic and heat shock stress (Figure 2c, d and f). For all three stresses, this cell death was abolished by ced-3 or ced-4 mutation, demonstrating that was apoptotic (Table 1 and Supplementary Figure S3f–o). We also noted that stress-induced apoptosis was not affected by an animals' age (Supplementary Table S1).
Importantly, these stresses also significantly increased the number of corpses that were apparent in the ced-1∷GFP strain (MD701), in which engulfment occurs normally but corpse detection is more sensitive (Table 2 and Supplementary Figure S1). This last result shows that osmotic, heat shock and starvation increase germ cell apoptosis under normal circumstances, thereby validating the findings obtained in engulfment-defective backgrounds. In the remainder of this study, we will investigate the extent to which various mechanisms are required for these stresses to induce germ cell apoptosis, as detected under engulfment-impaired conditions. We will refer to this type of apoptosis as stress-induced germ cell apoptosis.
We also examined stress-induced apoptosis in somatic cells and germ cells undergoing spermatogenesis. Such increases were not observed in somatic tissues (heads of ced-1(e1735) pretzel stage embryos) or in L4 hermaphrodites and male gonads (data not shown). We conclude that the increase in apoptosis observed following stress is specific to germ cells undergoing oogenesis.
The genetic requirements for stress-induced germ cell apoptosis differ from somatic apoptosis
The same basic machinery mediates somatic and physiological cell deaths, but is regulated differently in each case. In ced-9(n1950gf) gain of function and egl-1(n1084n3082) loss of function mutants, somatic cell apoptosis is prevented while the physiological cell death pathway occurs normally.2 To determine whether EGL-1 is involved in stress-induced apoptosis, we asked how ced-9(n1950gf) and egl-1(n1084n3082) mutants would behave when subjected to stress-inducing conditions. As shown in Supplementary Figures S3p–y and Table 1, stress-induced germline apoptosis occurs normally in ced-9 (n1950gf); ced-1(RNAi) and egl-1(n1084n3082); ced-1(RNAi) animals. These data suggest that genetic requirements for stress-induced germ cell apoptosis differ from the somatic apoptosis.
p53 and the DNA damage response machinery are not required to activate stress-induced apoptosis
Germ cell apoptosis is triggered when C. elegans is exposed to genotoxic agents like gamma radiation or N-ethyl-N-nitrosourea. This response, known as DNA damage-induced germ cell apoptosis, occurs through a genotoxic pathway that requires checkpoint genes and p53.3, 4, 8 The tumor suppressor protein p53 has been implicated in apoptosis induced by genotoxic agents in Drosophila, C. elegans and mammals.15, 3, 4 DNA damage-induced germ cell apoptosis is prevented when the C. elegans p53 homolog (cep-1) is eliminated.4, 3 To determine whether CEP-1 is also required for stress-induced apoptosis, we treated cep-1(w40); ced-1(RNAi) or cep-1(gk138); ced-1(RNAi) mutants under stress conditions (Figure 3, Supplementary Figure S4, Table 1 and data not shown). We found that an increase in germ cell death was still observed in these mutants indicating that p53 is not required for stress-induced germ cell apoptosis.

p53 and checkpoint genes are not required for stress-induced germ cell apoptosis. One-day old animals of the indicated genetic backgrounds were treated under stress-inducing conditions as indicated in the Materials and Methods section. Fold increased stress-induced apoptosis are shown in the graph. Pictures of cep-1(w40);ced-1(RNAi) gonads from animals treated under stress-inducing conditions are shown in Supplementary material (Supplementary Figure S4)
To further test the hypothesis, that stress-induced germ cell apoptosis occurs independently of the DNA damage pathway, we examined mutants mrt-2(e2663), him-7(e1480); hus-1(op236) and rad-5(mn159), which are all defective in the DNA damage response.8 The gene mrt-2, a homolog of S. pombe rad1 and S. cerevisae rad17, is required for telomere maintenance.16 Strain him-7(e1480); hus-1(op236) has a mutation in the hus-1 gene, which encodes a checkpoint protein required for the p53-dependent activation of EGL-1 in DNA damage-induced germ cell apoptosis.8, 17 rad-5/clk-2 encodes a protein that is required for telomere maintenance in yeast.18 On exposure to stress inducing conditions, we detected a 1.5- to three-fold induction in germ cell apoptosis in the gonads of mrt-2 (e2663); ced-1(RNAi), him-7 (e1480); hus-1(op236); ced-1(RNAi) and rad-5(mn159); ced-1(RNAi) as we did in ced-1(RNAi) animals (Figure 3 and Table 1). We concluded that stress-induced apoptosis does not depend on the regulators involved in DNA damage-induced apoptosis.
The MAPK kinases MEK-1 and SEK-1 and the p53 antagonist protein ABL-1 are essential for stress-induced germ cell apoptosis
In various organisms, conditions that include oxidative stress, heat, osmotic shock, antioxidants, and UV treatment will activate both the Jnk and p38 MAP kinase pathways.19 In mammals, the MAPKKs MKK4 and 7 regulate the Jnk pathway. In C. elegans, a lack of the mkk-4 homolog results in an egg-laying defect in adult hermaphrodites.20 MKK7 has two homologs in C. elegans, JKK-1 and MEK-1.20, 21, 22 Disruption of mek-1 results in hypersensitivity to heavy metals and starvation,22 a link suggesting that this gene might be a good candidate for mediating stress-induced apoptosis. When tested in our germ cell apoptosis analysis, mek-1 (ks54); ced-1(RNAi) mutants showed slightly lower levels of stress-induced apoptosis in response to salt, paraquat, heat shock and starvation treatments (1.4-, 1.5-, 1.6- and 2.1-fold increase, respectively) (Figure 4, Supplementary Figure S5f–j and Table 1). These results suggest that mek-1 plays a small role in regulating stress-induced germ cell apoptosis.

The MAPKKs, MEK-1 and SEK-1, and the p53 antagonist protein ABL-1 are essential for germ cell apoptosis induced by several types of stress. Animals from the indicated genetic backgrounds were subjected to stress-inducing conditions as indicated in Materials and Methods. Fold increased germ cell apoptosis are shown in the graph. Picture depicting representative gonads for each genetic background are shown in Supplementary material (Supplementary Figure S5)
We also tested whether stress-induced germ cell apoptosis involves the p38 pathway. In C. elegans, the MAPKK SEK-1, the ortholog of mammalian MKK3 and MKK6, is required for p38 pathway signaling. p38 signaling through SEK-1 protects C. elegans from Salmonella enterica pathogenesis, a condition that triggers germ cell apoptosis in the gonad by an EGL-1 dependent pathway.10, 23, 24 In C. elegans the p38 signalling pathway is also activated by oxidative stress, and is required for oxidative stress resistance.25 We tested sek-1(RNAi); ced-1(e1735) animals for their responses to different types of stress and found that without this MAPK kinase induction of apoptosis by oxidative and heat shock stress is moderately reduced (Figure 4, Supplementary Figure S5k–o and Table 1).
Since it has been shown that the MEK-1 and SEK-1 signaling pathway interact with each other,26, 27 we decided to study stress-induced germ cell apoptosis in the absence of both pathways. We observed that animals lacking both mek-1 and sek-1 genes (mek-1(ks54), sek-1(RNAi); ced-1(e1735)) were unable to induce apoptosis in response to osmotic, oxidative and heat shock stress (Figure 4, Supplementary Figure S5p–s and Table 1). These results suggest that stress-induced germ cell apoptosis is regulated by these pathways. Unexpectedly, even in the absence of MEK-1 and SEK-1, we still observed starvation-induced germ cell apoptosis, suggesting that a different pathway might regulate this response (Figure 4, Supplementary Figure S5t and Table 1).
Over the course of our experiments, we have noted that MAPKK mutants also show lower rates of basal physiological germ cell apoptosis (Figure 5a). We also observed that these mutants produced fewer offspring than wild-type animals (Figure 5b), an outcome that could not be explained by embryo lethality. To test whether the low brood sizes among MAPKK mutants are due to proliferation defects, we stained one- and two-day-old animals with a phospho-histone H3 antibody. We found no difference between the proliferation rates of MAPKK mutants and those of wild-type animals under regular growing conditions (Figure 5c). Therefore, the low rates of basal physiological germ cell apoptosis and reduced offspring production observed in MAPKK mutants cannot be attributed to defects in proliferation.

Mutants in the MAPKK pathway have reduced physiological germ cell apoptosis. (a) Animals of the indicated backgrounds were grown at 20°C and scored for germ cell apoptosis under Nomarski microscopy at L4 stage, and at 1-, 2- and 3-days into adult development. Averages of at least two independent experiments, with a minimum of fifteen animals each, are shown with S.E.M. (b) Animals of the indicated backgrounds were individually selected as L4 specimens, then transferred to new plates every 24 h over the course of 3 days and incubated at 20°C. Plates were scored for dead embryos, and surviving offspring. Offspring percentages from two independent experiments performed with at least 15 animals each are shown. mek-1(ks54); ced-1(RNAi) and sek-1(RNAi), mek-1(ks54); ced-1(e1735) recorded up to a 1.4% occurrence of dead embryos compared to wild type (0%), sek-1(RNAi); ced-1(e1735) animals showed only a 0.2% rate of embryo lethality. Some mek-1(ks54) and mek-1(ks54), sek-1(RNAi) animals were often sterile, in which case they were not scored for this experiment. (c) Animals of the indicated backgrounds were fixed and stained to detect proliferation with a phospho-histone H3 antibody. The proliferation percentages shown were derived from at least two independent experiments conducted with 30 animals each. We did not observe any significant differences between offspring production and proliferation rates when using N2 or ced-1(RNAi); sek-1(RNAi) or sek-1(km4); and mek-1(ks54), sek-1(RNAi); ced-1(e1735) or mek-1(ks54), sek-1(km4); ced-1(RNAi) backgrounds
We also studied how germ cell proliferation was affected by stress. Wild-type animals showed a constant decrease in proliferation under all tested stress conditions (Table 3). The mek-1(ks54) mutant showed almost a 50% reduction in proliferation under heat shock but was unaffected by osmotic stress, whereas in the sek-1(km4) mutant we observed an almost 50% proliferation reduction under osmotic stress but normal proliferation rates after heat shock (Table 3). The mek-1, sek-1 double mutant showed an approximately 80% reduction proliferation after either osmotic or heat shock treatments, indicating that both mek-1 and sek-1 influence the rate of germ cell proliferation in response to osmotic stress and heat shock. In contrast, oxidative stress (paraquat) did not decrease proliferation rates in mek-1(ks54);sek-1(km4) animals (Table 3). Thus, our results demonstrate that the decrease in oxidative stress-induced germ cell apoptosis seen in mek-1(ks54);sek-1(km4) animals does not involve effects on germ cell proliferation, although we cannot exclude this possibility for salt- and heat-induced cell death (Table 3). In contrast, under starvation stress we observed increased germ cell death and dramatically reduced proliferation in all genetic backgrounds tested (Table 3), supporting our model that a MAPKK-independent pathway regulates responses of germ cells to starvation conditions.
The mammalian non-receptor tyrosine kinase, cAbl, can function in either pro- or antiapoptotic pathways in response to DNA damage and oxidative stress and in C. elegans animals have higher basal levels of physiological apoptosis.28, 29 We investigated whether C. elegans ABL-1 influences induction of germline apoptosis by stress. We found that abl-1(ok171); ced-1(e1735) mutant animals showed no increase in their rates of germ cell apoptosis in response to oxidative, osmotic and heat shock conditions, behaving similarly to the mek-1(ks54), sek-1(RNAi) double mutant (Figure 4 and Supplementary Figure S5u–x). Similar to MAPKK mutants, abl-1(ok171); ced-1(e1735) animals were still able to induce some level of apoptosis under starvation conditions (Figure 4 and Supplementary Figure S5y). These results suggested that the MAPKKs MEK-1 and SEK-1 might induce apoptosis through ABL-1 in response to certain stress conditions.
Discussion
Stress-induced germ cell apoptosis is regulated by a p53 independent pathway that also differs from somatic apoptosis
DNA damage induces germ cell apoptosis through the genotoxic pathway, which is dependent upon p53 and involves the proteins EGL-1 and CED-13.3, 4, 8, 9 High levels of germ cell apoptosis are also induced during pathogen infection by an EGL-1 dependent mechanism.10 During oogenesis, 50 percent of germ cells die through the so-called physiological pathway by means of an EGL-1 independent mechanism.2 Here, we show that germ cell apoptosis is induced independently of p53 and EGL-1 in response to oxidative, osmotic, heat shock or starvation stress. We have named this type of response stress-induced germ cell apoptosis to distinguish it from DNA damage- and bacterial-induced apoptosis, which are regulated differently. In wild-type animals, we were able to observe increased numbers of germ cell corpses by Nomarski optics only after paraquat exposure, but analyses with the more sensitive assay of Plim-7ced-1∷GFP expression revealed significant increases in germ cell death also in response to osmotic, heat shock and starvation stress. It is possible that these last stresses reduced cell death to an extent that was subtle enough to be compensated for by the normal rate of engulfment, or might have accelerated engulfment. In either case, our results obtained by scoring ced-1∷GFP expression, which detects corpses at an earlier stage than can be detected by Nomarski optics, validates our observation that apoptotic responses to osmotic, heat shock and starvation were also uncovered in analyses of engulfment-defective strains.
In mammals and other organisms, environmental stress is known to influence germ cell survival. For example, starvation in Drosophila also triggers apoptosis among germ cells.30 Our results show that the C. elegans germline will also be a valuable system for studying germ cell responses to these environmental or nutritional stresses.
Oxidative, osmotic and heat shock stress required the MAPK pathway to induce germ cell apoptosis through ABL-1
MAPK cascades are highly conserved in all eukaryotes and orchestrate diverse cellular activities including apoptosis and stress response.19, 31 In C. elegans, MAPKs are also involved in the animals' defenses against pathogens.23, 24, 32 Virulence factors trigger unknown somatic signals that induce germ cell apoptosis, which in turn enhances pathogen resistance through unknown mechanisms.10 The MAPKK SEK-1 is required for induction of this pathogen-induced germline apoptosis.24 In addition, SEK-1 and p38 signaling in the intestine is important for survival under oxidative stress conditions, because this pathway is required for nuclear localization of the stress-protective transcription factor SKN-1.25 The MAPK, PMK-3, also plays a role in protection from physiological germ cell apoptosis.7
Here, we showed that MEK-1 and SEK-1 are essential for germ cell apoptosis induced by heat shock, oxidative and osmotic stress. Although infection by pathogens and the various stresses both induce apoptosis in the germline, and while both involve the MAPK pathway, it is clear that the downstream activator of apoptosis differs in each case. The induction of apoptosis in response to pathogens is dependent upon the BH3-only domain protein EGL-1.10 In contrast, we found that stress-induced germ cell apoptosis occurs independently of this protein. In yet another variation, germ cell apoptosis induced by nutrient starvation is only partially dependent upon the MEK-1 and SEK-1 pathways. MEK-1 and SEK-1 are necessary for activating at least two types of apoptosis in C. elegans; it will be interesting to dissect the downstream details of the MAPK pathway and to learn how these proteins can mediate apoptosis through different mechanisms.
In C. elegans, an interaction between the MEK-1 and SEK-1 pathways in metal resistance and pathogenesis has been described.26, 27 It is known that MEK-1 can activate both the Jnk (KGB-1) and p38 (PMK-1) pathways. Here, we demonstrated that mutations in mek-1 or sek-1 genes have overlapping essential functions in stress-induced germ cell apoptosis, so that oxidative, osmotic and heat shock stress all fail to induce apoptosis when both pathways have been interrupted. These data are consistent with a model in which these two pathways interact, or else suggest that stimulation of both pathways is required to enact protective stress-induced apoptosis.
It was recently described that the protein ABL-1 functions as an antiapoptotic factor and a negative regulator of p53, preventing DNA damage-induced germ cell apoptosis.29 In contrast, we found that ABL-1 actually promotes apoptosis in response to osmotic, oxidative and heat shock stress. It is interesting that, as in mammals, the C. elegans ABL-1 protein can function either as an anti-29 or a proapoptotic factor (this work) in various contexts. Our results thus provide a genetic system for studying the functions of the ABL kinase in stress responses.
The damaging effects of reactive oxygen species (ROS) on DNA, and their role in pathological processes and aging have been well established.33 Recently, ROS were also described as secondary messengers controlling different cellular functions.34, 35 We speculate that stress conditions tested in this paper would not be strong enough to induce DNA damage, but were sufficient to trigger redox signaling pathways that induce apoptosis through the physiological mechanism. It will be interesting to determine whether these stresses actually cause ROS production in the germline and if higher levels of ROS are able to induce germ cell apoptosis through EGL-1 and p53. It will also be instructive to study the possible relationships between stress and germ cell development, by investigating whether signals that involve ROS play a role in normal regulation of physiological apoptosis and homeostatic maintenance of oocyte production.
Materials and Methods
Strains
Maintenance and genetic manipulation of C. elegans were carried out according to standard procedures. All strains were grown at 20°C except for MD701, which carries a Plim-7ced-1∷gfp fusion and was grown at 24°C (for better GFP germline expression). The following strains have been described previously36 or in the following references: wild-type N2 bristol, ced-1(e1735), ced-3(n717), ced-4(n1162), ced-5(n1812) and ced-9(n1950gf). him-7(e1480); hus-1(op236);8 mrt-2(e2663);16 rad-5(mn159),18 egl-1(n1084 n3082);37 cep-1(w40);3 cep-1(gk138),7 mek-1 (ks54),22 mek-1(ks54), sek-1(km4),27 abl-1(ok171)29 and (MD701) Plim-7ced-1∷gfp.9
Cell corpse assays
Animals were mounted on agar pads that contained a drop of anesthetizing 40 mM NaN3 dissolved in M9. They were observed using Nomarski optics to visualize cell corpses.2 Usually, only one gonad arm per animal was accessible for visualization. On the ced-5(n1812) strain, in which distal tip cell (DTC) migration is frequently affected, only animals showing normal DTC migration were used for our studies. Excel (Microsoft) was used to calculate average standard deviations (S.E.M.) and P-values (by Student's t-test). Images were obtained using a Nikon Eclipse E600 equipped with an AxioCam digital camera (Zeiss); photographs were then processed in Photoshop 7 (Adobe). For time-lapse microscopy, animals were treated with paraquat for 1 h, mounted on agar pads that contained a drop of 3 mM levamisole as described by Gumienny et al.2 and then photograph every 15 min until their corpses had degraded.
As an alternative method to detect germ cell death, we used the vital dye acridine orange, which is used to visualize the highly condensed DNA characteristically present in apoptotic cells.5
Growth conditions prior to treatments
Animals were grown for 3 days at 20°C on NGM plates seeded with Escherichia coli of either the OP50-1 or HT115 strains carrying the corresponding RNAi feeding vector. L4 animals were moved to NGM plates seeded with the indicated food source, where their incubation continued for another 24 h. Following such a period of growth, animals were referred as 1-day old. A standard synchronization of animals was avoided since the bleach and starvation treatments conventionally used to bring animal populations to the same age are stress conditions that would have altered the results. During the course of our experiments, we noticed that animals that were starved, contaminated, exposed at high temperatures or bleached, showed increased cell corpse numbers. We strongly recommend allowing animals to grow under nourishing conditions for at least two generations before trying any stress treatment.
Osmotic and oxidative stress
In order to choose appropriate stress-inducing doses for each of the experimental agents, we initially tested different concentrations of sodium chloride (35–175 mM) and paraquat (1–15 mM). Over the course of these trials, we determined suitable dosage amounts that would induce cell death in the ced-1(e1735) animals without killing them.
One-day old adult animals (∼30) were placed in 200 μl of M9 (control), M9-containing 70 mM NaCl (osmotic stress) or 10 mM paraquat (oxidative stress) for 1 h at 20°C After the incubation period, 1 ml of M9 was added to dilute the solutions; animals were then recovered by pipeting and placed on E. coli-seeded NGM plates. The lids of the plates were left off for 5 min to allow a full draining of excess liquid, the dishes were then covered and animals were allowed to recover for another 55 min more at 20°C. It was important not to leave the lids off for the entire recovery period since the incubator airflow might have caused dehydration in the animals. Finally, the animals were mounted and observed under the microscope (as described previously) to determine cell corpse numbers.
Heat shock and starvation stress
We initially subjected ced-1(e1735) animals to a range of heat shock temperatures, from 30 to 33°C, and found that 33°C induced germ cell apoptosis in a higher proportion of animals without killing them. Likewise, we tested a range of possible times to find the most suitable duration for starvation treatments. With starvation periods lasting longer than 6 h, we observed a higher accumulation of corpses in the animals, but their gonads were dramatically altered in appearance. Therefore, we starved animals for only 6 h since this period would let the animals' gonads remain healthy-looking but still result in increased germ cell apoptosis.
To induce heat shock, several 1-day adult animals were placed in NGM plates seeded with the appropriate strain. Plates were sealed with parafilm and floated directly in a 33°C water bath for 1 h. For starvation experiments, several animals were placed in NGM plates without food source and incubated at 20°C for 6 h. For these stress conditions, control animals were incubated in seeded-NGM plates for the same period of time at 20°C. Immediately after each treatment, animals were mounted and observed in the microscope as previously described.
RNAi studies
To facilitate visualization of cell corpses, we took advantage of the ced-1 mutation. Compared to wild type, ced-1 animals retain apoptotic cells for longer periods of time due to defects in their corpse degradation machinery. We used RNA interference (RNAi) in the ced-1 gene to analyze germ cell apoptosis in several strains. The following primers were used: 5′-CGGAATTCATGCGTCTCATTCTCC-3′ and 5′-CGGAATTCCACTTGTACAGATAC-3′. A fragment containing the first 550 base pairs of ced-1 (starting from ATG) was isolated from the C. elegans cDNA by PCR. The ced-1 product was digested with EcoR1, cloned into the feeding vector pPD129.36, and the resulting plasmid was transformed into the E. coli strain HT115.38
A fragment of ∼600 base pairs from the sek-1 gene was cloned, running from exon 4–exon 6 inclusive, using cDNA from C. elegans. Primers were: 5′-CGGAATTCGAATCAAATCATCAATC-3′ and 5′-CGGAATTCTCATTGATAAACCGAGC-3′. This sek-1 PCR fragment was cloned, as previously described for ced-1, into the feeding vector to effect RNAi. For mek-1 gene cloning, a fragment of ∼980 kb was amplified by PCR using C. elegans cDNA (primers: 5′-GGAATTCATGGAGAGAGACTTCGAC-3′ and 5′-CGAATTCGGTTCCGGCGTATG-3′). PCR product was cut with EcoRI and a band of ∼480 bp was cloned into the feeding vector as described above. To perform RNAi in the abl-1 gene, we used a clone from the C. elegans RNAi v1.1 feeding library (OpenBiosystems, Alabama).
To induce the production of double-stranded RNA in the feeding bacteria, we followed standard procedures.38 Briefly, bacterial cultures were allowed to grow to OD 0.6 at 37°C; on reaching this density, 0.4 mM IPTG was added to the cultures and incubation continued for 2 h at same temperature. Bacteria were collected and resuspended (1 ml for each 10 ml of original culture) in LB containing antibiotics (50 μg/ml ampicillin and 12.5 μg/ml tetracycline), 0.4 mM IPTG and 25% glycerol. One milliliter aliquots were immediately frozen in liquid nitrogen and stored at −70°C. When needed for experiments, aliquots were thawed, centrifuged, and their pellets resuspended in 100 μl LB containing antibiotics and IPTG. Fifty microliters of bacteria were seeded onto each 60 mm plate of NGM containing antibiotics. Plates were stored at 4°C for up to one week.
Apoptosis assay by fluorescence microscopy using a ced-1∷gfp strain
One-day old ced-1∷gfp animals grown at 24°C were treated as previously described to induce germ cell apoptosis under oxidative, osmotic and starvation stress. We found that heat shock affected GFP expression, so in order to improve expression we needed to allow animals to recover for 2.5 h at 24°C following treatment. Animals were observed under fluorescence microscopy to detect GFP expression.
Immunofluorescent staining to detect germ cell proliferation
One-day old animals treated with stress-inducing conditions were dissected, fixed and stained to detect germ cell proliferation using a phospho-histone H3 antibody (Upstate), according to conditions described previously by Kadyk and Kimble.39 Phospho-histone H3 positive cells were counted, as visualized under fluorescence microscopy.
Abbreviations
- MAPKKs:
-
mitogen-activated kinase kinases
References
Schedl T (1997) Developmental genetics of the germ line In C. elegans II Riddle DL, Blumenthal T, Meyer BJ, Priess JR eds (Cold Spring Harbor, NY: Cold Spring Harbor Press) pp. 170–241.
Gumienny TL, Lambie E, Hartwieg E, Horvitz HR and Hengartner MO (1999) Genetic control of programmed cell death in the Caenorhabditis elegans hermaphrodite germline. Development 126: 1011–1022.
Derry WB, Putzke AP and Rothman JH (2001) Caenorhabditis elegans p53: role in apoptosis, meiosis, and stress resistance. Science 294: 591–595.
Schumacher B, Hofmann K, Boulton S and Gartner A (2001) The C. elegans homolog of the p53 tumor suppressor is required for DNA damage-induced apoptosis. Curr. Biol. 11: 1722–1727.
Navarro RE, Shim EY, Kohara Y, Singson A and Blackwell TK (2001) cgh-1, a conserved predicted RNA helicase required for gametogenesis and protection from physiological germline apoptosis in C. elegans. Development 128: 3221–3232.
Boag PR, Nakamura A and Blackwell TK (2005) A conserved RNA–protein complex component involved in physiological germline apoptosis regulation in C. elegans. Development 132: 4975–4986.
Lettre G, Kritikou EA, Jaeggi M, Calixto A, Fraser AG, Kamath RS, Ahringer J and Hengartner MO (2004) Genome-wide RNAi identifies p53-dependent and -independent regulators of germ cell apoptosis in C. elegans. Cell Death Differ. 11: 1198–1203.
Gartner A, Milstein S, Ahmed S, Hodgkin J and Hengartner MO (2000) A conserved checkpoint pathway mediates DNA damage-induced apoptosis and cell cycle arrest in C. elegans. Mol. Cell 5: 435–443.
Schumacher B, Schertel C, Wittenburg N, Tuck S, Mitani S, Gartner A, Conradt B and Shaham S (2005) C. elegans ced-13 can promote apoptosis and is induced in response to DNA damage. Cell Death Differ. 12: 153–161.
Aballay A and Ausubel FM (2001) Programmed cell death mediated by ced-3 and ced-4 protects Caenorhabditis elegans from Salmonella typhimurium-mediated killing. Proc. Natl. Acad. Sci. USA 98: 2735–2739.
Zhou Z, Hartwieg E and Horvitz HR (2001) CED-1 is a transmembrane receptor that mediates cell corpse engulfment in C. elegans. Cell 104: 43–56.
Wu YC and Horvitz HR (1998) C. elegans phagocytosis and cell-migration protein CED-5 is similar to human DOCK180. Nature 392: 501–504.
del Peso L, Gonzalez VM, Inohara N, Ellis RE and Nunez G (2000) Disruption of the CED-9.CED-4 complex by EGL-1 is a critical step for programmed cell death in Caenorhabditis elegans. J. Biol. Chem. 275: 27205–27211.
Lettre G and Hengartner MO (2006) Developmental apoptosis in C. elegans: a complex CEDnario. Nat. Rev. Mol. Cell Biol. 7: 97–108.
Song YH (2005) Drosophila melanogaster: a model for the study of DNA damage checkpoint response. Mol. Cells 19: 167–179.
Ahmed S and Hodgkin J (2000) MRT-2 checkpoint protein is required for germline immortality and telomere replication in C. elegans. Nature 403: 159–164.
Hofmann ER, Milstein S, Boulton SJ, Ye M, Hofmann JJ, Stergiou L, Gartner A, Vidal M and Hengartner MO (2002) Caenorhabditis elegans HUS-1 is a DNA damage checkpoint protein required for genome stability and EGL-1-mediated apoptosis. Curr. Biol. 12: 1908–1918.
Ahmed S, Alpi A, Hengartner MO and Gartner A (2001) C. elegans RAD-5/CLK-2 defines a new DNA damage checkpoint protein. Curr. Biol. 11: 1934–1944.
Pearson G, Robinson F, Beers Gibson T, Xu BE, Karandikar M, Berman K and Cobb MH (2001) Mitogen-activated protein (MAP) kinase pathways: regulation and physiological functions. Endocr. Rev. 22: 153–183.
Villanueva A, Lozano J, Morales A, Lin X, Deng X, Hengartner MO and Kolesnick RN (2001) jkk-1 and mek-1 regulate body movement coordination and response to heavy metals through jnk-1 in Caenorhabditis elegans. EMBO J. 20: 5114–5128.
Kawasaki M, Hisamoto N, Iino Y, Yamamoto M, Ninomiya-Tsuji J and Matsumoto K (1999) A Caenorhabditis elegans JNK signal transduction pathway regulates coordinated movement via type-D GABAergic motor neurons. EMBO J. 18: 3604–3615.
Koga M, Zwaal R, Guan KL, Avery L and Ohshima Y (2000) A Caenorhabditis elegans MAP kinase kinase, MEK-1, is involved in stress responses. EMBO J. 19: 5148–5156.
Kim DH, Feinbaum R, Alloing G, Emerson FE, Garsin DA, Inoue H, Tanaka-Hino M, Hisamoto N, Matsumoto K, Tan MW and Ausubel FM (2002) A conserved p38 MAP kinase pathway in Caenorhabditis elegans innate immunity. Science 297: 623–626.
Aballay A, Drenkard E, Hilbun LR and Ausubel FM (2003) Caenorhabditis elegans innate immune response triggered by Salmonella enterica requires intact LPS and is mediated by a MAPK signaling pathway. Curr. Biol. 13: 47–52.
Inoue H, Hisamoto N, An JH, Oliveira RP, Nishida E, Blackwell TK and Matsumoto K (2005) The C. elegans p38 MAPK pathway regulates nuclear localization of the transcription factor SKN-1 in oxidative stress response. Genes Dev. 19: 2278–2283.
Mizuno T, Hisamoto N, Terada T, Kondo T, Adachi M, Nishida E, Kim DH, Ausubel FM and Matsumoto K (2004) The Caenorhabditis elegans MAPK phosphatase VHP-1 mediates a novel JNK-like signaling pathway in stress response. EMBO J. 23: 2226–2234.
Kim DH, Liberati NT, Mizuno T, Inoue H, Hisamoto N, Matsumoto K and Ausubel FM (2004) Integration of Caenorhabditis elegans MAPK pathways mediating immunity and stress resistance by MEK-1 MAPK kinase and VHP-1 MAPK phosphatase. Proc. Natl. Acad. Sci. USA 101: 10990–10994.
Yoshida K, Yamaguchi T, Natsume T, Kufe D and Miki Y (2005) JNK phosphorylation of 14-3-3 proteins regulates nuclear targeting of c-Abl in the apoptotic response to DNA damage. Nat. Cell Biol. 7: 278–285.
Deng X, Hofmann ER, Villanueva A, Hobert O, Capodieci P, Veach DR, Yin X, Campodonico L, Glekas A, Cordon-Cardo C, Clarkson B, Bornmann WG, Fuks Z, Hengartner MO and Kolesnick R (2004) Caenorhabditis elegans ABL-1 antagonizes p53-mediated germline apoptosis after ionizing irradiation. Nat. Genet. 36: 906–912.
Drummond-Barbosa D and Spradling AC (2001) Stem cells and their progeny respond to nutritional changes during Drosophila oogenesis. Dev. Biol. 231: 265–278.
Wada T and Penninger JM (2004) Mitogen-activated protein kinases in apoptosis regulation. Oncogene 23: 2838–2849.
Huffman DL, Abrami L, Sasik R, Corbeil J, van der Goot FG and Aroian RV (2004) Mitogen-activated protein kinase pathways defend against bacterial pore-forming toxins. Proc. Natl. Acad. Sci. USA 101: 10995–11000.
Halliwell B and Gutteridge JMC (1989) Free Radicals in Biology and Medicine. (London: Clarenton Press, Oxford University Press).
Finkel T (2003) Oxidant signals and oxidative stress. Curr. Opin. Cell Biol. 15: 247–254.
Aguirre J, Rios-Momberg M, Hewitt D and Hansberg W (2005) Reactive oxygen species and development in microbial eukaryotes. Trends Microbiol. 13: 111–118.
Riddle DL, Blumenthal T, Meyer BJ and Priess JR (eds) (1997 C. elegans II. (Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press).
Conradt B and Horvitz HR (1998) The C. elegans protein EGL-1 is required for programmed cell death and interacts with the Bcl-2-like protein CED-9. Cell 93: 519–529.
Timmons L, Court DL and Fire A (2001) Ingestion of bacterially expressed dsRNAs can produce specific and potent genetic interference in Caenorhabditis elegans. Gene 263: 103–112.
Kadyk LC and Kimble J (1998) Genetic regulation of entry into meiosis in Caenorhabditis elegans. Development 125: 1803–1813.
Acknowledgements
We thank Drs. Jesús Aguirre, Geraldine Seydoux and Keith Blackwell for kindly editing and critically reading this manuscript and also for their helpful discussions. We also thank Dr. Claudia Kent and Leslie Tu for editing the manuscript. We are indebted to anonymous reviewers for helpful suggestions and manuscript editing. We are grateful to the C. elegans Genetics Center, which is supported by NIH's National Center for Research Resources, for providing strains. We would like to express gratitude to Drs. K. Matsumoto and T. Mizuno for kindly providing the mek-1(ks54), sek-1(km4) double mutant. This work was supported by grants from UNAM-PAPIIT (IN217102-3) to REN and EM, CONACYT (41518-Q) to REN and CONACYT (41587Q) to EM.
Author information
Authors and Affiliations
Corresponding author
Additional information
Edited by M Hengartner
Supplementary Information accompanies the paper on Cell Death and Differentiation website (http://www.nature.com/cdd)
Supplementary information
Rights and permissions
About this article
Cite this article
Salinas, L., Maldonado, E. & Navarro, R. Stress-induced germ cell apoptosis by a p53 independent pathway in Caenorhabditis elegans. Cell Death Differ 13, 2129–2139 (2006). https://doi.org/10.1038/sj.cdd.4401976
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.cdd.4401976
Keywords
This article is cited by
-
Functional recovery of the germ line following splicing collapse
Cell Death & Differentiation (2022)
-
Fatal decision made under pressure
Nature Physics (2021)
-
Isoflurane impairs oogenesis through germ cell apoptosis in C. elegans
Scientific Reports (2021)
-
BRAP-2 promotes DNA damage induced germline apoptosis in C. elegans through the regulation of SKN-1 and AKT-1
Cell Death & Differentiation (2018)
-
N-(3-oxo-acyl) homoserine lactone induced germ cell apoptosis and suppressed the over-activated RAS/MAPK tumorigenesis via mitochondrial-dependent ROS in C. elegans
Apoptosis (2018)
