
Abstract
Individual BCL2 family members couple apoptosis regulation and cell cycle control in unique ways. Antiapoptotic BCL2 and BCL-xL are antiproliferative by facilitating G0. BAX is proapoptotic and accelerates S-phase progression. The dual functions in apoptosis and cell cycle are coordinately regulated by the multi-domain BCL2 family members (MCL-1) and suggest that survival is maintained at the expense of proliferation. The role of BH3-only molecules in cell cycle is more variable. BAD antagonizes both the cell cycle and antiapoptotic functions of BCL2 and BCL-xL through BH3 binding. BID has biochemically separable functions in apoptosis and S-phase checkpoint, determined by post-translational modification. p53-induced PUMA is known only to have apoptotic function. Inhibition of apoptosis is oncogenic, whereas promotion of cell cycle arrest is tumor suppressive. Paradoxically, selected BCL2 family members can be both oncogenic and tumor suppressive. Which of the dual functions predominates is lineage specific and context dependent.
Similar content being viewed by others

Introduction
Connections between cell cycle and cell death have long been noted. It has been generally accepted that cycling cells are more susceptible to cell death, whereas quiescent cells are relatively more resistant to killing. One principle of cancer treatment has been to recruit more cells into the generally small growth fraction of the tumor, so that they can be susceptible to chemotherapeutic drugs. The retinoblastoma protein pRB both checks cells in G0/G1 and protects them against apoptosis. Cells undergoing apoptosis often exhibit activation of cell cycle events, such as cdk activation and abortive cell cycle progression.1, 2 Many oncogenes have dual functions of positively regulating proliferation and apoptosis, such as Myc and E2F, whereas tumor suppressors, such as Rb and p53, inhibit cell cycle. BCL2 is an oncogene that inhibits apoptosis but, paradoxically, it is also antiproliferative.
BCL2 enhances G0 and delays G0 to S transition
BCL2's antiapoptosis function was first linked to effects on proliferation by the observation that when deprived of growth factor, BCL2-overexpressing IL-3-dependent FDC-P1 cells were smaller than cells in the presence of IL-3 and were mostly arrested in G0/G1.3 The lack of Myc expression and decreased nuclear size indicated that BCL2 cells arrested in G0 to maintain viability when deprived of growth factor. When the HL60 promyelocytic leukemia cell line was treated with DMSO, a differentiative stimulus that did not involve cell death, cells overexpressing BCL2 decreased RNA content more quickly than controls, suggesting that BCL2 expression facilitated exit to G0.4 These early experiments demonstrated an effect of BCL2 on G0 that appeared to be separate from its antiapoptotic function.
A second observation of BCL2's effect on proliferation was that bone marrow-derived IL-3-dependent BAF3 cells expressing BCL2 were arrested in G1 and protected from apoptosis upon IL-3 removal.5 These cells were refractory to cell cycle re-entry upon IL-3 re-stimulation. A series of papers ensued, including several from the group at the Walter and Eliza Hall Institute, which examined the effect of BCL2 not only on G0/G1 arrest but also on cell cycle progression. Primarily using lymphocytes from BCL2 transgenic mice, these studies found that in T and B cells, and in certain thymocyte subpopulations, BCL2 expression correlated with a higher G0/G1 fraction, lower S-phase fraction, and decreased BrdU incorporation.6, 7, 8 During activation of quiescent T and B cells in culture and serum stimulation of experimentally arrested NIH3T3 cells, BCL2 expression delayed the onset of S phase, indicating inhibition of G0 to S progression. Indeed, expression of not only BCL2, but also its homologs BCL-xL, BCL-w, and E1B19K, similarly retarded progression to S phase, demonstrating that this cell cycle effect of BCL2 is manifested in other antiapoptotic molecules within the BCL2 family, and is not cell type restricted.5, 9
The physiologic relevance of the cell cycle inhibitory effects of BCL2 was first demonstrated by Stan Korsmeyer's laboratory in a systematic study comparing bcl2-deficient, bcl2 heterozygous, wild-type, and transgenic BCL2 T cells.10 The G0 state and the kinetics of cell cycle entry in response to T-cell activation of these genotypes varied progressively from the least to the most arrested. Cell size was largest in resting bcl2−/− T cells and smallest in lck-BCL2 transgenics. Onset of S phase was quickest in activated bcl2−/− T cells and slowest in lck-BCL2 cells. Bcl2−/− T cells produced the most and lck-BCL2 cells produced the least IL-2. Recognizing that the T-cell pools from these mice are not identical, in that the CD8 T-cell population of bcl2−/− mice is relatively smaller, and the lck-BCL2 CD8 T-cell population is relatively larger than wild-type controls, these genotype comparisons nevertheless provided strong evidence that at least in T cells, endogenous BCL2 plays a role in regulating cell cycle entry. Despite initial BrdU- and thymidine-labeling experiments suggesting BCL2 may be generally growth inhibitory, growth rate measurements in conventional and continuous chemostat cultures revealed that in cycling cells, BCL2 does not significantly affect growth rates under optimal conditions, but prolongs G1 in suboptimal conditions.7, 8, 10, 11, 12, 13 It became increasingly clear that the cell cycle delay effect of BCL2 is selective for cell cycle re-entry from G0.
Is G0 arrest distinct from delayed cell cycle progression?
Are BCL2-mediated G0 arrest and BCL2 inhibition of progression to S phase two separate activities or manifestations of the same cell cycle function? The cdk inhibitor p27 is normally upregulated in G0, and prevents the activation of G1 cyclins. Various groups showed that p27, as well as the pRB relative p130, which binds E2F4 in quiescence, was elevated significantly more than usual in BCL2 cells during arrest.10, 14, 15, 16 With stimulation of cell cycle, p27 levels decreased, but still remained higher than in wild-type cells. Activation of cyclinE/cdk2 and cyclinD/cdk4, which defines the restriction point in normal G1 to S progression, was delayed and dampened in BCL2 and BCL-xL cells, owing to persistently high p27 in the cyclin/cdk complexes.16, 17 That p27 is key in mediating the cell cycle function of BCL2 and BCL-xL was supported by the inability of BCL2 transgene to delay activation-induced proliferation in p27−/− mice and the failure of BCL-xL to delay cell cycle in p27−/− MEFs.16, 17 Interestingly, with cell cycle stimulation of G0-arrested cells, early marker events of G1 entry, which include the induction of c-Fos, c-Jun, Myc, cyclin D, all occurred at the same time in BCL2 or BCL-xL cells as in controls cells.10, 17 Thus, in BCL2 or BCL-xL cells, the early signaling events initiating G0 to G1 transition are intact, but the critical step of transition into S phase, that is, cdk2/4 activation, is delayed.
Time-course measurements of cell size and cellular RNA content indicated that BCL2 cells and BCL-xL cells remain small and do not initiate macromolecular synthesis despite the induction of Myc and cyclin D.18 Furthermore, cells sorted for the same size, regardless of BCL2 or BCL-xL expression level, entered cell cycle with similar kinetics, indicating that the main function of BCL2 and BCL-xL is to drive cells into G0.18 Thus, prolonged G0 is mainly responsible for the observed delay in reaching S phase, and the function of BCL2 and BCL-xL is further focused as facilitating G0 arrest (Figure 1).

Ability of BCL2/BCL-xL to drive cells into enhanced G0 arrest leads to delay in G1 progression and G1/S transition
Antiapoptosis and cell cycle inhibition: separate functions or one?
Despite indications that the cell cycle effects of BCL2 and BCL-xL are not simply a result of apoptosis inhibition, mutation analysis of BCL2 and BCL-xL could not consistently identify separate domains for the cell cycle function and for cell survival.5, 18 Mutation at residue Y28 in the BH4 domain was reported to preserve the survival but not the cell cycle function of BCL2.9 Deletion of the non-conserved ‘loop’ region of BCL2 was also reported to facilitate cell proliferation while preserving the antiapoptotic effect.19 However, others could not reproduce the phenotypes of these mutants in cells or in mice.18, 20 It is unclear whether the discrepancy is simply owing to expression level and cell line differences, or other indirect effects, and the divergent phenotypes of the Y28 mutation remain a curious question.
One hypothesis is that the cell cycle function of BCL2 and BCL-xL is dependent on an intact survival function. For example, caspases are involved in cell cycle progression, and BCL2 or BCL-xL may mediate arrest through the inhibition of caspase activation. However, enhancing survival by caspase inhibition does not result in the same cell cycle phenotype as BCL2 and BCL-xL, suggesting that mechanisms in addition to inhibition of the apoptosome are necessary10 (E Yang, unpublished data). p27 has emerged as an essential downstream mediator of the antiproliferative function of BCL2 and BCL-xL. Upregulation of p27 appears to be a direct effect of BCL2 and BCL-xL, most likely at the post-translational level, although the precise mechanism is still unknown. As the BCL2 family members are mitochondrial molecules regulating many aspects of mitochondria physiology, including ATP generation, permeability transition pore, and mitochondrial potential, it is likely that mitochondrial bioenergetics are involved. How mitochondrial signals lead to p27 protein elevation is an interesting circuit to unravel.
BCL2 cell cycle control and tumorigenesis
The role of BCL2's antiproliferative function in lymphomagenesis is complex. Transgenic BCL2 expression in the lymphoid compartment is clearly oncogenic, but lymphomas developed only in a fraction of mice after long latencies.21, 22 The coexistence of the cell cycle inhibitory function with the antiapoptosis function may explain why BCL2 does not induce tumors with higher penetrance. Half of the BCL2 lymphomas were found to harbor Myc rearrangements, illustrating that secondary genetic alterations, which counter the growth inhibitory function of BCL2, are necessary for tumor development.21 The marked synergy between BCL2 and Myc in lymphomagenesis is classically attributed to the ability of BCL2 to inhibit Myc-induced apoptosis, enabling Myc-induced proliferation to proceed unchecked.23, 24, 25 However, cell culture findings on the interaction of BCL2 and Myc were not always consistent. Expression of BCL2 maintained survival of Eμ-Myc bone marrow cells in culture, but the cells proliferated only slowly, suggesting that by arresting cells in G0, BCL2 inhibited Myc-induced proliferation as well as Myc-induced apoptosis.3 In Rat1 MycER cells, BCL2 inhibited the apoptotic function of Myc, but had no effect on cell division when measured by time-lapse microscopy.24 Yet, another group found that both BCL2 and Myc were required for IL-2-stimulated proliferation.26 These different results on the role of BCL2 in Myc-induced proliferation may be due to differences between animal models and cell culture, and whether apoptosis or proliferation plays a dominant role in the particular model system. In Myc-induced lymphoma formation, the antiapoptotic function of BCL2 is clearly dominant over its antiproliferative function, perhaps owing to the strong proliferative function of Myc.
In other tissues, the antiproliferative function of BCL2 translates into tumor suppression. In colon cancer cell lines, BCL2 unexpectedly inhibited proliferation to the same extent as p53, but in a p53-independent manner, and decreased clonogenicity in soft agar.6 In human colon cancer, multiple studies showed that BCL2 expression is correlated with favorable outcome.27 In multi-stage liver carcinogenesis models, BCL2 expression inhibits the growth of early proliferative foci and counteracts hepatic carcinogenesis induced by TGFα and Myc.28, 29 In support of this, induction of BCL2 also delays hepatocyte cell cycle entry in liver regeneration.30 In WAP-TAg and carcinogen-driven mammary tumor models, in which stages of initial proliferation and progression are obvious, BCL2 expression reduces both proliferation and apoptosis early in the process, but the antiproliferative effect is lost as tumors progress to adenocarcinoma.31, 32 Association of BCL2 with differentiated phenotypes and better prognosis is borne out in human breast cancer studies.33 In the classical two-stage skin carcinogenesis model, BCL2 expression in basal epidermal keratinocytes similarly increased the latency and reduced the frequency of papillomas converting to malignant carcinomas.34 In contrast to Myc-induced lymphomas, these solid tumors are characterized by a proliferative pretumor phase during which BCL2's antiproliferative effect could be more consequential than its antiapoptosis activity. Therefore, the balance between the antiapoptotic and the cell cycle effects of BCL2 can be influenced by tumor physiology.
Other antiapoptotic BCL2 family members
Two other antiapoptotic BCL2 family members, BCL-w and myeloid cell leukemia-1 (MCL-1), also have antiproliferative effects, but they are much less studied. In earlier experiments, BCL-w behaved like BCL2 and BCL-xL in delaying cell cycle entry.9 This inhibitory activity on cell cycle was revisited in the developing testis and spermatogenesis.35 Transgenic expression of BCL-w driven by the chicken β-actin promoter resulted in decreased germ cell number and male sterility, which correlated with reduced number of BrdU-positive spermatogonia in the first postnatal week. This single finding is consistent with inhibition of cell cycle reentry or G1 to S transition by BCL-w overexpression, but the physiological significance of this was not substantiated in bcl-w−/− mice.36
MCL-1 was originally identified as an upregulated gene in a human myeloblastic leukemia cell line induced to differentiate in the monocyte lineage.37 Overexpression of this gene in cell lines caused decreased BrdU uptake and slower doubling rate.38, 39 In one study, the antiproliferative function of MCL-1 was clearly linked to its ability to bind proliferating cell nuclear antigen (PCNA), but distinct from its antiapoptotic activity.38 Another report identified a short form of MCL-1 in the nucleus (snMCL-1) that binds and negatively regulates cdk1 activity,39 but its function in cell survival is unclear. In both cases, MCL-1's cell cycle function is in S and G2 phases, not in G0. Although it is also antiproliferative, MCL-1's cell cycle function is very different from BCL2 or BCL-xL. It is interesting to speculate that the cell cycle function of MCL-1 may be responsible for MCL-1's role in implantation, but there are no data to support this.
To date, no role in cell cycle has been identified for A1. In fact, A1 was shown specifically not to have a cell cycle inhibitory effect when expressed as a transgene driven by the lck distal promoter, in that more A1-expressing T cells accumulated in culture after activation than BCL2-expressing T cells.40 It was suggested that this is because A1 rescued T cells from activation-induced cell death and allowed them to cycle, whereas BCL2 saved the cells from apoptosis but also inhibited their proliferation.
Although most of the antiapoptotic BCL2 family members are antiproliferative, all do not have the same activity in cell cycle. BCL2 and BCL-xL clearly have a G0 function. BCL-w may be similar to BCL2 and BCL-xL, but MCL-1's cell cycle activity is in S or G2, whereas A1 has no known cell cycle function.
BAX and the multi-domain proapoptotic molecules
Whereas transgenic BCL2 T cells are delayed in activation-induced cell cycle entry, transgenic BAX T cells enter S phase faster than wild-type counterparts. CD2-BAX and lck-BAX thymi have higher fractions of cells in S phase and exhibit increased BrdU uptake.14, 41 Resting transgenic BAX T cells are larger and their activation is associated with increased p27 degradation and increased cdk2 activation, exactly the opposite of transgenic BCL2 T cells.20, 42 Whereas BCL2 is prosurvival and antiprolifeative, BAX is proapoptotic and proliferative, suggesting that the cell cycle functions of the multi-domain BCL2 family members are directly linked to life or death decisions. Although bax−/− cells do not have an obvious cell cycle phenotype and transgenic BCL2 on bax−/− background still delays cell cycle entry, bax−/−bak−/− double knockout mice have increased hematopoietic progenitors and mature lymphocytes. bax−/−bak−/− lymphocytes are smaller, reminiscent of BCL2 cells, indicating that the absence of Bax and Bak may promote G0.43 It would be of great interest to examine the ability of BCL2 to regulate cell growth in bax−/−bak−/− doubly deficient cells, which should settle the question whether BCL2 exerts its cell cycle effects through BAX and BAK, or whether the effectors for BCL2's cell cycle functions are different from those involved in BCL2's antiapoptosis function.
The effect of BAX expression on tumorigenesis is paradoxical. If BCL2 is oncogenic in the lymphoid lineage, then BAX might be expected to be tumor suppressive. Yet, bax deficiency alone or in combination with p53 deletion was not oncogenic, perhaps because p53 loss already largely abrogated apoptosis.41 In the presence of oncogenes providing strong proliferative drive associated with apoptosis, including T antigen and E1A, bax deficiency did enhance transformation, presumably by blocking apoptosis, resulting in further enhancement of proliferation.44, 45 A tumor suppressive role for the multi-domain proapoptotic molecules was further demonstrated by the cooperation of bax and bak deficiency with p53 inactivation in E1A-mediated tumor formation.46 Surprisingly, lymphomagenesis owing to p53 deficiency was potentiated by lck-BAX. Here, the proliferative effect of BAX was presumably dominant over its proapoptotic activity. Thus, the proapoptotic function of BAX is tumor suppressive and its proliferative function is oncogenic. The relative contribution of each function to the overall effect appears to be influenced by the choice of its oncogene partners.
Overexpression of the BH3-only molecule BAD renders the cell unable to arrest in G0 and persistently activate cdk2.18, 47 This effect is completely dependent on BAD binding to BCL-xL and BCL2; therefore, it is not surprising that deficiency of BAD itself is only minimally oncogenic.48
Proapoptotic BID
Proapoptotic BID was cloned through interaction with BCL2 and BAX,49 and biochemically purified as a protein mediating cytochrome c release from mitochondria following activation of death receptors.50 In vitro studies of mitochondria and recombinant truncated BID indicate that it activates the multi-domain BCL2 family members BAX or BAK, resulting in allosteric conformational change and release of cytochrome c.51, 52 The role of BID in normal development and cellular homeostasis has been characterized using mice in which Bid has been disrupted. These bid-deficient mice are viable and execute developmental cell death normally.53 When challenged with agonistic anti-fas antibody, bid-deficient mice are resistant to the hepatocellular apoptosis that kills wild-type mice, indicating a critical role for BID in this Fas-signaled death. Aging bid-deficient mice spontaneously develop a myeloproliferative disorder with elevated absolute neutrophil counts, and over time, the mice progress to a fatal clonal disorder resembling chronic myelomonocytic leukemia (CMML).54 Myeloid progenitors from Bid-deficient mice exhibit resistance to death receptor-induced apoptosis, and demonstrate a competitive advantage in vivo. These studies indicate an essential role for BID in maintaining myeloid homeostasis and suppressing leukemogenesis.
BID's role in tumorigenesis may be cell type specific. Bid−/− mice demonstrate decreased tumor growth in the liver following treatment with diethylnitrosamine (DEN) in a mouse model of hepatocellular carcinoma.55 Bid−/− hepatocytes display fewer cells in S phase by BrdU incorporation following DEN treatment as well as partial hepatectomy, perhaps suggesting a role for BID in regulating proliferation in the liver, and its absence may slow tumor growth.
BID is unique among the BH3-only BCL2 family members in interconnecting death receptors to the mitochondrial amplification loop of the intrinsic pathway. BID's potent proapoptotic activity and broad expression patterns require that cells carefully regulate its apoptotic activation. Subcellular localization appears to play a role in directing BID's proapoptotic activity. Following death receptor stimulation, BID is activated by caspase-8 cleavage and N-myristoylation to target mitochondria where it activates BAX and BAK, or is alternatively sequestered by antiapoptotic BCL2 members, preventing death.56 Full-length BID is also capable of translocation to the mitochondria in at least one case facilitated by other proteins such as PACS2.57, 58, 59 At the mitochondria, full-length BID has been shown to potentiate cell death following certain apoptotic signals, suggesting that caspase cleavage is not an absolute requirement for activating BID's proapoptotic function.58, 60
Recent studies indicate that activation of BID's prodeath activity may be negatively regulated by phosphorylation. Casein kinases have been implicated in BID phosphorylation, and ATM has been shown to phosphorylate BID following DNA damage.61, 62, 63 Phosphorylated BID is resistant to caspase cleavage in in vitro assays, and MEFs harboring phosphorylation-defective S78A BID are more sensitive to etoposide-induced cell death. The above data are consistent with a role for phosphorylation to inhibit activation of BID's proapoptotic function.61, 63 How might BID be involved in suppressing leukemogenesis? Although the loss of BID could theoretically reset death susceptibility in both intrinsic and extrinsic pathways, it is less obvious why the absence of BID should prove so oncogenic. A striking feature of the bid-deficient CMML is the frequent presence of chromosomal instability, as evidenced by chromosomal translocations seen on spectral karyotype analysis.54 Wild-type hematopoietic cells have a marked propensity for apoptosis in response to DNA damage; yet, in the absence of BID, myeloid cells accumulate mutations, resist apoptosis, and display aspects of unchecked proliferation.62 This suggests that BID itself may play a role in DNA repair, in cell cycle checkpoint responses, or in integrating apoptosis and the DNA repair response.
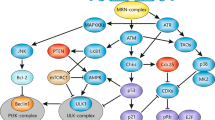
Consistent with the above hypothesis, bid−/− myeloid progenitor cells and primary activated T cells manifest increased chromosomal damage following mitomycin C treatment, with tri- and quadriradial chromosomal figures quantifiable by an increase in the number of chromosomal breaks per cell.62 These abnormal chromosomal structures represent ‘chromatid-type’ errors, resulting from improperly repaired DNA damage accrued during S phase of the cell cycle and are characteristic of cells with a defect in DNA repair, such as those in Fanconi anemia, Bloom's syndrome, and the hereditary breast and ovarian cancer syndromes involving BRCA1. Following replicative stress, BID is localized in the nucleus, positioning it to play a role in integrating the apoptotic and DNA repair responses downstream of DNA damage, or a direct role in DNA repair.62 Bid−/− myeloid progenitor cells and MEFs fail to properly execute the ionizing radiation-induced intra-S-phase checkpoint.62, 63 This S-phase role is mediated through BID phosphorylation at position 78 by the DNA damage kinase ATM, demonstrating a direct link between BID and the DNA damage response. These studies demonstrate that BID plays a novel role in preserving genomic integrity that places BID at an early point in the path to determine the fate of a cell (Figure 2).

Model for the dual function. Following death receptor suimulation, BID initiates a proapoptotic program at the mitochondria. Following DNA damage, BID is phosphorylated in the nucleus and plays a role in cell cycle checkpoint control
The BCL2 family has been shown to play a role in myeloid leukemogenesis. Leukemic cells from most human acute myelogenous leukemias (AMLs) have been found to express elevated levels of BCL2 relative to normal cellular counterparts.64 Transgenic mice overexpressing BCL2 in myeloid cells develop a myeloproliferative disorder, and when crossed with lpr mice harboring a mutation in the Fas receptor, the mice progress to AML, implicating a synergistic role for the Fas pathway and BCL2 in tumor suppression in the myeloid lineage.65 Deletion of BID in myeloid cells promotes myeloid leukemogenesis, demonstrating that this single ‘BH3-only’ protein plays a critical role in maintenance of normal myeloid homeostasis and tumor suppression. A mouse model in which the endogenous BID gene has been replaced with a gene that drives the expression of a BID protein carrying mutations in the ATM phosphorylation sites (BIDS61A/S78A) should be instructive in addressing the issues described above. The ATM phosphorylation site at position 78 is conserved in mouse and human BID.62, 63 Given the importance of the BCL2 family in human myeloid malignancies, and the synergistic role of the Fas pathway in mouse models, a role for BID in human disease, with close attention to phosphorylation status, warrants further study.
p53 in DNA damage and apoptosis
p53 plays a pivotal role in the decision of whether the outcome of DNA damage will be growth arrest or apoptosis. The currently accepted model for this choice is based on the idea that p53 is able to differentially transactivate promoters of ‘growth arrest’ and ‘apoptosis’ genes. This idea was built on the suggestion that promoters of growth arrest genes encompass high-affinity p53-binding sites (e.g., p21), whereas the promoters of apoptotic genes contain low-affinity p53-binding sites (e.g., BAX). Several proteins have been identified that can discriminate in favor of the interaction of p53 with the promoters of apoptotic genes.66, 67 Thus, the presence or levels of such proteins in a given cell may dictate the type of response that this cell will undertake following activation of the p53 pathway. With respect to apoptosis, many proapoptotic genes that carry a p53-responsive element have been reported. The products of these may participate in apoptosis in a number of ways. For example, proapoptotic gene products such as the BH3-only PUMA and Noxa, BAX, and p53AIP1 localize to the mitochondria and promote the loss of mitochondrial membrane potential and cytochrome c release.68 Studies have demonstrated that MEFs lacking PUMA or Noxa are resistant to DNA damage-induced apoptosis, a process known to be mediated by p53.69, 70, 71 Another class of proapoptotic genes that can be regulated by p53, such as Fas or DR5/KILLER, are components of the apoptotic extrinsic pathway.72 Finally, genes that encode redox-regulating enzymes such as the PIGs (p53-induced genes), which are involved in reactive oxygen species production, can damage the mitochondria, leading to apoptosis.73 A recent elegant paper demonstrated that Slug, a transcriptional repressor, ‘saves’ hematopoietic progenitors exposed to DNA damage by antagonizing the ability of p53 to transcriptionally induce proapoptotic PUMA.74 Interestingly, p53 also transcriptionally induces Slug. Thus, in certain cells, such as hematopoietic progenitors, p53 circumvents its own ability to induce apoptosis. As p53 transcriptionally induces several proapoptotic proteins, it is likely that additional Slug-like inhibitors exist that act to circumvent p53-induced apoptosis in hematopoietic as well as in non-hematopoietic cells.
Growing evidence suggests that the transcription activity of p53 can be uncoupled from its apoptotic function. Moreover, several recent reports demonstrate the direct localization of p53 to the mitochondria following DNA damage, where p53 can directly interact with BCL2 family members, leading to cytochrome c release.75 A more recent study showed that, after genotoxic stress, the antiapoptotic BCL-xL protein sequesters cytoplasmic p53, whereas nuclear p53 induces transcription of PUMA. PUMA then binds BCL-xL and displaces p53, thereby allowing p53 to directly activate BAX to induce mitochondrial permeabilization.76 p53 is not the only unexpected factor released from the nucleus to induce apoptosis at the mitochondria. Recent evidence revealed an unexpected role also for the linker histone H1.2 in DNA damage-induced apoptosis. Konishi et al.77 demonstrated that DNA double-strand breaks induce translocation of nuclear H1.2 to the cytoplasm, where it promotes release of cytochrome c from mitochondria by activating proapoptotic BAK.
Is there a functional connection between p53 and BID in the DNA damage response? The p53 protein is a target of ATM and ATR, and its activation by these kinases (which results in its accumulation) can lead to either cell cycle arrest at the G1 phase or apoptosis. As both p53 and BID play a balancing act between life and death, and both seem to act at the nucleus and mitochondria, it is tempting to speculate that these two proteins ‘communicate’ with each other following DNA damage. It is documented that p53 acts upstream of BID as its transcriptional activator,78 and as a transcriptional activator of one of its effectors, BAX. On the other hand, it is possible that in the DNA damage pathway, phosphorylated BID might act upstream of p53 by directly regulating its transcriptional-independent activity at the mitochondria, or its transcriptional-dependent activity in the nucleus.
Relationship between BCL2 family cell cycle and antiapoptosis functions in tumorigenesis
Cumulative data indicate that cell cycle control is linked to cell death regulation. The relationship is complex and context dependent. For the antiapoptosis BCL2 family members such as BCL2 and BCL-xL, the parallel effects of antiapoptosis and cell cycle inhibition suggest that cells may maintain survival at the expense of proliferation. Although it remains to be proven, data up to now indicate that the same biochemical function of BCL2 and BCL-xL mediates both survival and quiescence. The multi-domain proapoptotic molecule BAX seems to be the converse of BCL2 and BCL-xL, in that BAX promotes both cell death and cell cycle, suggesting that proliferation is death prone. To date, there is little indication that the two activities are separable in BAX, although this has not been specifically addressed in published reports.
The antiapoptosis function of BCL2 and its homologs renders them as oncogenes, but their cell cycle function is consistent with tumor suppression. Which function is predominant may be in part determined by the physiology of the cell and tissue type. The hematopoietic system, particularly the lymphoid lineage, is constantly exposed to apoptosis signaling in development and maturation. One reasonable hypothesis would be that the antiapoptosis function of BCL2 exerts a dominant effect over the antiproliferative function in this scenario, and BCL2 emerges as an oncogene. In contrast, in epithelial and mesenchymal tissues, such as breast and liver, the proliferative phases preceding progression to carcinoma may provide an opportunity for the antiproliferative function of BCL2 to be more evident. The overall effect of BCL2 in this context, then, would be more tumor suppressive than oncogenic.
In the lymphoid system, the antiapoptosis molecule BCL2 paradoxically acts as a relatively weak oncogene. By itself, BCL2 promotes tumors at a low but significant rate, but BCL2 is much more remarkable in potentiating other oncogenes, especially c-Myc. This could be explained by BCL2's simultaneous antiproliferative function. If the cell cycle inhibitory function of BCL2 could be abrogated, presumably BCL2 would be a more potent oncogene. Conversely, augmenting the antiproliferative function of BCL2 could decrease tumor aggressiveness.
In contrast to the case of BCL2, where the antiapoptotic function correlates with its oncogenic function, this correlation is less obvious in proapoptotic molecules. Absence of proapoptotic BH3-only molecules results in spontaneous malignancy in mouse models lacking BID and BAD.48, 54 However, the absence of multi-domain BAX or BAK results in prominent inhibition of apoptosis emanating from both intrinsic and extrinsic pathways with resultant abrogation of homeostatic control.79 Despite this potent perturbation of apoptosis, to date, bax−/− or bak mice do not progress to malignancy. BAX deficiency and BAX overexpression can synergize with other oncogenes, and like BCL2, BAX has dual roles to either enhance or inhibit tumorigenesis depending on the genetic context.
Proapoptotic BID possesses an additional function in regulating the ability of cells to stop DNA replication following DNA damage, presumably allowing cells to repair damaged DNA and prevent propagation of potentially harmful mutations. This cell cycle function is independent of the proapoptotic BH3 domain of BID, in contrast to BCL2 and BCL-xL, in which the cell cycle and apoptotic functions are linked. Perturbation of both DNA damage-induced cell cycle checkpoints and apoptosis has the potential to enahnce tumorigenesis in the case of BH3-only BID.
Like BCL2 and BCL-xL, recent data indicate that BID's ability to suppress oncogenesis may be context- and lineage-dependent. Absence of BID results in decreased tumor growth in a mouse model of carcinogen-induced hepatocellular carcinoma, whereas BID-deficient mice develop spontaneous CMML. The hematopoietic system is highly susceptible to DNA damage, and organisms rely on apoptosis for removal of damaged cells. In this context, the proapoptotic function of BID may play a more prominent role.
Conclusion
For the multi-domain anti- and proapoptotic BCL2 members (except MCL1), the cell cycle and apoptosis functions are coordinately regulated. The interplay of cell cycle and apoptosis is more variable for the BH3 molecules, all of which cannot be easily explained by their ability to bind antiapoptotic family members. BAD is basically the antithesis of BCL2 and BCL-xL, consistent with the selective high affinity of BAD for BCL-xL and BCL2.80 For Bid, the choice appears to be either apoptosis or cell cycle. In this case, the cell cycle function is independent of BH3-mediated binding to BAX or BCL2.62 For PUMA, the choice between apoptosis and cell cycle arrest occurs upstream at the level of gene induction, and to date, there is no evidence that this BH3 molecule has any cell cycle function (Figure 3). Perhaps, this is because PUMA binds all of the antiapoptotic BCL2 family members,80 but in that case, one might expect greatly accelerated cell cycling. It is clear that the cell cycle function of certain BCL2 family members is distinct from their apoptosis or survival function.

BCL2 family members couple apoptosis and cell cycle in different ways. BCL2, BCL-xL, and BAX coordinately regulate survival and proliferation; BID functions in either apoptosis or cell cycle; PUMA is only apoptotic. For each molecule, whether the apoptosis or cell cycle function predominates depends on cell type and genetic context
The BCL2 family offers a unique opportunity to study the intersection of two major cellular pathways: regulation of apoptosis and cell cycle control. For the BCL2 family members known to have dual functions, such as BCL2, BCL-xL, MCL-1, BAX, and BID, their relative roles in apoptosis versus cell cycle are highly dependent on cell lineage and genetic context. These variables present challenges in the discovery of cell cycle functions for the other BCL2 family members, such as A1, BIM, PUMA, NOXA. Increased understanding of the BCL2 family proteins in recent years illustrates that individual members couple cell cycle and apoptosis in unique ways. That the mitochondria may be at the center of a cell's decision between survival and proliferation is very intriguing. More research needs to be focused on precisely how the apoptotic regulators fit into known cell cycle signaling cascades. The concept of survival at the expense of proliferation awaits further validation as more mechanistic data come to light.
Dedication
This review is written in honor of Stan Korsmeyer, who was a wonderful mentor. All three authors are grateful for the rigorous post doc training in Stan's lab. He will always remain a guiding force to our science.
Abbreviations
- DMSO:
-
dimethylsulfoxide
- MEF:
-
mouse embryo fibroblasts
- CMML:
-
chronic myelomonocytic leukemia
- DEN:
-
diethylnitrosamine
- AML:
-
acute myelogenous leukemia
References
Pandey S and Wang E (1995) Cells en route to apoptosis are characterized by the upregulation of c-fos, c-myc, c-jun, cdc2, and RB phosphorylation, resembling events of early cell-cycle traverse. J. Cell Biochem. 58: 135–150.
Meikrantz W, Gisselbrecht S, Tam SW and Schlegel R (1994) Activation of cyclin A-dependent protein kinases during apoptosis. Proc. Natl. Acad. Sci. USA 91: 3754–3758.
Vaux DL, Cory S and Adams JM (1988) Bcl-2 gene promotes haemopoietic cell survival and cooperates with c-myc to immortalize pre-B cells. Nature 335: 440–442.
Vairo G, Innes KM and Adams JM (1996) Bcl-2 has a cell cycle inhibitory function separable from its enhancement of cell survival. Oncogene 13: 1511–1519.
Marvel J, Perkins GR, Lopez Rivas A and Collins MK (1994) Growth factor starvation of bcl-2 overexpressing murine bone marrow cells induced refractoriness to IL-3 stimulation of proliferation. Oncogene 9: 1117–1122.
O'Reilly LA, Huang DC and Strasser A (1996) The cell death inhibitor Bcl-2 and its homologues influence control of cell cycle entry. EMBO J. 15: 6979–6990.
O’Reilly LA, Harris AW and Strasser A (1997) bcl-2 transgene expression promotes survival and reduces proliferation of CD3-CD4-CD8- T cell progenitors. Int. Immunol. 9: 1291–1301.
O’Reilly LA, Harris AW, Tarlinton DM, Corcoran LM and Strasser A (1997) Expression of a bcl-2 transgene reduces proliferation and slows turnover of developing B lymphocytes in vivo. J. Immunol. 159: 2301–2311.
Huang DC, O’Reilly LA, Strasser A and Cory S (1997) The anti-apoptosis function of Bcl-2 can be genetically separated from its inhibitory effect on cell cycle entry. EMBO J. 16: 4628–4638.
Linette GP, Li Y, Roth K and Korsmeyer SJ (1996) Cross talk between cell death and cell cycle progression: BCL-2 regulates NFAT-mediated activation. Proc. Natl. Acad. Sci. USA 93: 9545–9552.
Mazel S, Burtrum D and Petrie HT (1996) Regulation of cell division cycle progression by bcl-2 expression: a potential mechanism for inhibition of programmed cell death. J. Exp. Med. 183: 2219–2226.
Borner C (1996) Diminished cell proliferation associated with the death-protective activity of Bcl-2. J. Biol. Chem. 271: 12695–12698.
Simpson NH, Singh RP, Emery AN and Al-Rubeai M (1999) Bcl-2 over-expression reduces growth rate and prolongs G1 phase in continuous chemostat cultures of hybridoma cells. Biotechnol. Bioeng. 64: 174–186.
Brady HJ, Gil-Gomez G, Kirberg J and Berns AJ (1996) Bax alpha perturbs T cell development and affects cell cycle entry of T cells. EMBO J. 15: 6991–7001.
Lind EF, Wayne J, Wang QZ, Staeva T, Stolzer A and Petrie HT (1999) Bcl-2-induced changes in E2F regulatory complexes reveal the potential for integrated cell cycle and cell death functions. J. Immunol. 162: 5374–5379.
Vairo G, Soos TJ, Upton TM, Zalvide J, DeCaprio JA, Ewen ME, Koff A and Adams JM (2000) Bcl-2 retards cell cycle entry through p27(Kip1), pRB relative p130, and altered E2F regulation. Mol. Cell Biol. 20: 4745–4753.
Greider C, Chattopadhyay A, Parkhurst C and Yang E (2002) BCL-x(L) and BCL2 delay Myc-induced cell cycle entry through elevation of p27 and inhibition of G1 cyclin-dependent kinases. Oncogene 21: 7765–7775.
Janumyan YM, Sansam CG, Chattopadhyay A, Cheng N, Soucie EL, Penn LZ, Andrews D, Knudson CM and Yang E (2003) Bcl-xL/Bcl-2 coordinately regulates apoptosis, cell cycle arrest and cell cycle entry. EMBO J. 22: 5459–5470.
Uhlmann EJ, D’Sa-Eipper C, Subramanian T, Wagner AJ, Hay N and Chinnadurai G (1996) Deletion of a nonconserved region of Bcl-2 confers a novel gain of function: suppression of apoptosis with concomitant cell proliferation. Cancer Res. 56: 2506–2509.
Cheng N, Janumyan YM, Didion L, Van Hofwegen C, Yang E and Knudson CM (2004) Bcl-2 inhibition of T-cell proliferation is related to prolonged T-cell survival. Oncogene 23: 3770–3780.
McDonnell TJ and Korsmeyer SJ (1991) Progression from lymphoid hyperplasia to high-grade malignant lymphoma in mice transgenic for the t(14; 18). Nature 349: 254–256.
Linette GP, Hess JL, Sentman CL and Korsmeyer SJ (1995) Peripheral T-cell lymphoma in lckpr-bcl-2 transgenic mice. Blood 86: 1255–1260.
Strasser A, Harris AW, Bath ML and Cory S (1990) Novel primitive lymphoid tumours induced in transgenic mice by cooperation between myc and bcl-2. Nature 348: 331–333.
Fanidi A, Harrington EA and Evan GI (1992) Cooperative interaction between c-myc and bcl-2 proto-oncogenes. Nature 359: 554–556.
Bissonnette RP, Echeverri F, Mahboubi A and Green DR (1992) Apoptotic cell death induced by c-myc is inhibited by bcl-2. Nature 359: 552–554.
Miyazaki T, Liu ZJ, Kawahara A, Minami Y, Yamada K, Tsujimoto Y, Barsoumian EL, Permutter RM and Taniguchi T (1995) Three distinct IL-2 signaling pathways mediated by bcl-2, c-myc, and lck cooperate in hematopoietic cell proliferation. Cell 81: 223–231.
Buglioni S, D’Agnano I, Cosimelli M, Vasselli S, D’Angelo C, Tedesco M, Zupi G and Mottolese M (1999) Evaluation of multiple bio-pathological factors in colorectal adenocarcinomas: independent prognostic role of p53 and bcl-2. Int. J. Cancer 84: 545–552.
Pierce RH, Vail ME, Ralph L, Campbell JS and Fausto N (2002) Bcl-2 expression inhibits liver carcinogenesis and delays the development of proliferating foci. Am. J. Pathol. 160: 1555–1560.
de La Coste A, Mignon A, Fabre M, Gilbert E, Porteu A, Van Dyke T, Kahn A and Perret C (1999) Paradoxical inhibition of c-myc-induced carcinogenesis by Bcl-2 in transgenic mice. Cancer Res. 59: 5017–5022.
Vail ME, Chaisson ML, Thompson J and Fausto N (2002) Bcl-2 expression delays hepatocyte cell cycle progression during liver regeneration. Oncogene 21: 1548–1555.
Furth PA, Bar-Peled U, Li M, Lewis A, Laucirica R, Jager R, Weiher H and Russell RG (1999) Loss of anti-mitotic effects of Bcl-2 with retention of anti-apoptotic activity during tumor progression in a mouse model. Oncogene 18: 6589–6596.
Murphy KL, Kittrell FS, Gay JP, Jager R, Medina D and Rosen JM (1999) Bcl-2 expression delays mammary tumor development in dimethylbenz(a)anthracene-treated transgenic mice. Oncogene 18: 6597–6604.
Zhang GJ, Kimijima I, Tsuchiya A and Abe R (1998) The role of bcl-2 expression in breast carcinomas (Review). Oncol. Rep. 5: 1211–1216.
Rossiter H, Beissert S, Mayer C, Schon MP, Wienrich BG, Tschachler E and Kupper TS (2001) Targeted expression of bcl-2 to murine basal epidermal keratinocytes results in paradoxical retardation of ultraviolet- and chemical-induced tumorigenesis. Cancer Res. 61: 3619–3626.
Yan W, Huang JX, Lax AS, Pelliniemi L, Salminen E, Poutanen M and Toppari J (2003) Overexpression of Bcl-W in the testis disrupts spermatogenesis: revelation of a role of BCL-W in male germ cell cycle control. Mol. Endocrinol. 17: 1868–1879.
Print CG, Loveland KL, Gibson L, Meehan T, Stylianou A, Wreford N, de Kretser D, Metcalf D, Kontgen F, Adams JM and Cory S (1998) Apoptosis regulator bcl-w is essential for spermatogenesis but appears otherwise redundant. Proc. Natl. Acad. Sci. USA 95: 12424–12431.
Kozopas KM, Yang T, Buchan HL, Zhou P and Craig RW (1993) MCL1, a gene expressed in programmed myeloid cell differentiation, has sequence similarity to BCL2. Proc. Natl. Acad. Sci. USA 90: 3516–3520.
Fujise K, Zhang D, Liu J and Yeh ET (2000) Regulation of apoptosis and cell cycle progression by MCL1. Differential role of proliferating cell nuclear antigen. J. Biol. Chem. 275: 39458–39465.
Jamil S, Sobouti R, Hojabrpour P, Raj M, Kast J and Duronio V (2005) A proteolytic fragment of Mcl-1 exhibits nuclear localization and regulates cell growth by interaction with Cdk1. Biochem. J. 387: 659–667.
Gonzalez J, Orlofsky A and Prystowsky MB (2003) A1 is a growth-permissive antiapoptotic factor mediating postactivation survival in T cells. Blood 101: 2679–2685.
Knudson CM, Johnson GM, Lin Y and Korsmeyer SJ (2001) Bax accelerates tumorigenesis in p53-deficient mice. Cancer Res. 61: 659–665.
Gil-Gomez G, Berns A and Brady HJ (1998) A link between cell cycle and cell death: Bax and Bcl-2 modulate Cdk2 activation during thymocyte apoptosis. EMBO J. 17: 7209–7218.
Rathmell JC, Lindsten T, Zong WX, Cinalli RM and Thompson CB (2002) Deficiency in Bak and Bax perturbs thymic selection and lymphoid homeostasis. Nat. Immunol. 3: 932–939.
Yin C, Knudson CM, Korsmeyer SJ and Van Dyke T (1997) Bax suppresses tumorigenesis and stimulates apoptosis in vivo. Nature 385: 637–640.
McCurrach ME, Connor TM, Knudson CM, Korsmeyer SJ and Lowe SW (1997) bax-deficiency promotes drug resistance and oncogenic transformation by attenuating p53-dependent apoptosis. Proc. Natl. Acad. Sci. USA 94: 2345–2349.
Degenhardt K, Chen G, Lindsten T and White E (2002) BAX and BAK mediate p53-independent suppression of tumorigenesis. Cancer Cell 2: 193–203.
Chattopadhyay A, Chiang CW and Yang E (2001) BAD/BCL-[X(L)] heterodimerization leads to bypass of G0/G1 arrest. Oncogene 20: 4507–4518.
Ranger AM, Zha J, Harada H, Datta SR, Danial NN, Gilmore AP, Kutok JL, Le Beau MM, Greenberg ME and Korsmeyer SJ (2003) Bad-deficient mice develop diffuse large B cell lymphoma. Proc. Natl. Acad. Sci. USA 100: 9324–9329.
Wang K, Yin XM, Chao DT, Milliman CL and Korsmeyer SJ (1996) BID: a novel BH3 domain-only death agonist. Genes Dev. 10: 2859–2869.
Luo X, Budihardjo I, Zou H, Slaughter C and Wang X (1998) Bid, a Bcl2 interacting protein, mediates cytochrome c release from mitochondria in response to activation of cell surface death receptors. Cell 94: 481–490.
Desagher S, Osen-Sand A, Nichols A, Eskes R, Montessuit S, Lauper S, Maundrell K, Antonsson B and Martinou JC (1999) Bid-induced conformational change of Bax is responsible for mitochondrial cytochrome c release during apoptosis. J. Cell Biol. 144: 891–901.
Wei MC, Lindsten T, Mootha VK, Weiler S, Gross A, Ashiya M, Thompson CB and Korsmeyer SJ (2000) tBID, a membrane-targeted death ligand, oligomerizes BAK to release cytochrome c. Genes Dev. 14: 2060–2071.
Yin XM, Wang K, Gross A, Zhao Y, Zinkel S, Klocke B, Roth KA and Korsmeyer SJ (1999) Bid-deficient mice are resistant to Fas-induced hepatocellular apoptosis. Nature 400: 886–891.
Zinkel SS, Ong CC, Ferguson DO, Iwasaki H, Akashi K, Bronson RT, Kutok JL, Alt FW and Korsmeyer SJ (2003) Proapoptotic BID is required for myeloid homeostasis and tumor suppression. Genes Dev. 17: 229–239.
Bai L, Ni HM, Chen X, DiFrancesca D and Yin XM (2005) Deletion of Bid impedes cell proliferation and hepatic carcinogenesis. Am. J. Pathol. 166: 1523–1532.
Danial NN and Korsmeyer SJ (2004) Cell death: critical control points. Cell 116: 205–219.
Tafani M, Karpinich NO, Hurster KA, Pastorino JG, Schneider T, Russo MA and Farber JL (2002) Cytochrome c release upon fas receptor activation depends on translocation of full-length bid and the induction of the mitochondrial permeability transition. J. Biol. Chem. 277: 10073–10082.
Valentijn AJ and Gilmore AP (2004) Translocation of full-length Bid to mitochondria during anoikis. J. Biol. Chem. 279: 32848–32857.
Simmen T, Aslan JE, Blagoveshchenskaya AD, Thomas L, Wan L, Xiang Y, Feliciangeli SF, Hung CH, Crump CM and Thomas G (2005) PACS-2 controls endoplasmic reticulum-mitochondria communication and Bid-mediated apoptosis. EMBO J. 24: 717–729.
Sarig R, Zaltsman Y, Marcellus RC, Flavell R, Mak TW and Gross A (2003) BID-D59A is a potent inducer of apoptosis in primary embryonic fibroblasts. J. Biol. Chem. 278: 10707–10715.
Desagher S, Osen-Sand A, Montessuit S, Magnenat E, Vilbois F, Hochmann A, Journot L, Antonsson B and Martinou JC (2001) Phosphorylation of bid by casein kinases I and II regulates its cleavage by caspase 8. Mol. Cell 8: 601–611.
Zinkel SS, Hurov KE, Ong C, Abtahi FM, Gross A and Korsmeyer SJ (2005) A role for proapoptotic BID in the DNA-damage response. Cell 122: 579–591.
Kamer I, Sarig R, Zaltsman Y, Niv H, Oberkovitz G, Regev L, Haimovich G, Lerenthal Y, Marcellus RC and Gross A (2005) Proapoptotic BID is an ATM effector in the DNA-damage response. Cell 122: 593–603.
Bensi L, Longo R, Vecchi A, Messora C, Garagnani L, Bernardi S, Tamassia MG and Sacchi S (1995) Bcl-2 oncoprotein expression in acute myeloid leukemia. Haematologica 80: 98–102.
Traver D, Akashi K, Weissman IL and Lagasse E (1998) Mice defective in two apoptosis pathways in the myeloid lineage develop acute myeloblastic leukemia. Immunity 9: 47–57.
Flores ER, Tsai KY, Crowley D, Sengupta S, Yang A, McKeon F and Jacks T (2002) p63 and p73 are required for p53-dependent apoptosis in response to DNA damage. Nature 416: 560–564.
Samuels-Lev Y, O’Connor DJ, Bergamaschi D, Trigiante G, Hsieh JK, Zhong S, Campargue I, Naumovski L, Crook T and Lu X (2001) ASPP proteins specifically stimulate the apoptotic function of p53. Mol. Cell 8: 781–794.
Matsuda K, Yoshida K, Taya Y, Nakamura K, Nakamura Y and Arakawa H (2002) p53AIP1 regulates the mitochondrial apoptotic pathway. Cancer Res. 62: 2883–2889.
Shibue T, Takeda K, Oda E, Tanaka H, Murasawa H, Takaoka A, Morishita Y, Akira S, Taniguchi T and Tanaka N (2003) Integral role of Noxa in p53-mediated apoptotic response. Genes Dev. 17: 2233–2238.
Jeffers JR, Parganas E, Lee Y, Yang C, Wang J, Brennan J, MacLean KH, Han J, Chittenden T, Ihle JN, McKinnon PJ, Cleveland JL and Zambetti GP (2003) Puma is an essential mediator of p53-dependent and -independent apoptotic pathways. Cancer Cell 4: 321–328.
Villunger A, Michalak EM, Coultas L, Mullauer F, Bock G, Ausserlechner MJ, Adams JM and Strasser A (2003) p53- and drug-induced apoptotic responses mediated by BH3-only proteins puma and noxa. Science 302: 1036–1038.
Takimoto R and El-Deiry WS (2000) Wild-type p53 transactivates the KILLER/DR5 gene through an intronic sequence-specific DNA-binding site. Oncogene 19: 1735–1743.
Polyak K, Xia Y, Zweier JL, Kinzler KW and Vogelstein B (1997) A model for p53-induced apoptosis. Nature 389: 300–305.
Wu WS, Heinrichs S, Xu D, Garrison SP, Zambetti GP, Adams JM and Look AT (2005) Slug antagonizes p53-mediated apoptosis of hematopoietic progenitors by repressing Puma. Cell 123: 641–653.
Chipuk JE, Kuwana T, Bouchier-Hayes L, Droin NM, Newmeyer DD, Schuler M and Green DR (2004) Direct activation of Bax by p53 mediates mitochondrial membrane permeabilization and apoptosis. Science 303: 1010–1014.
Chipuk JE, Bouchier-Hayes L, Kuwana T, Newmeyer DD and Green DR (2005) PUMA couples the nuclear and cytoplasmic proapoptotic function of p53. Science 309: 1732–1735.
Konishi A, Shimizu S, Hirota J, Takao T, Fan Y, Matsuoka Y, Zhang L, Yoneda Y, Fujii Y, Skoultchi AI and Tsujimoto Y (2003) Involvement of histone H1.2 in apoptosis induced by DNA double-strand breaks. Cell 114: 673–688.
Sax JK, Fei P, Murphy ME, Bernhard E, Korsmeyer SJ and El-Deiry WS (2002) BID regulation by p53 contributes to chemosensitivity. Nat. Cell. Biol. 4: 842–849.
Lindsten T, Ross AJ, King A, Zong WX, Rathmell JC, Shiels HA, Ulrich E, Waymire KG, Mahar P, Frauwirth K, Chen Y, Wei M, Eng VM, Adelman DM, Simon MC, Ma A, Golden JA, Evan G, Korsmeyer SJ, MacGregor GR and Thompson CB (2000) The combined functions of proapoptotic Bcl-2 family members bak and bax are essential for normal development of multiple tissues. Mol. Cell 6: 1389–1399.
Chen L, Willis SN, Wei A, Smith BJ, Fletcher JI, Hinds MG, Colman PM, Day CL, Adams JM and Huang DC (2005) Differential targeting of prosurvival Bcl-2 proteins by their BH3-only ligands allows complementary apoptotic function. Mol. Cell 17: 393–403.
Acknowledgements
We thank Mike Knudson for comments.
Author information
Authors and Affiliations
Corresponding author
Additional information
Edited by C Borner
Rights and permissions
About this article
Cite this article
Zinkel, S., Gross, A. & Yang, E. BCL2 family in DNA damage and cell cycle control. Cell Death Differ 13, 1351–1359 (2006). https://doi.org/10.1038/sj.cdd.4401987
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.cdd.4401987
Keywords
This article is cited by
-
Joint Analysis of Genome-wide DNA Methylation and Transcription Sequencing Identifies the Role of BAX Gene in Heat Stress–Induced-Sertoli Cells Apoptosis
Reproductive Sciences (2024)
-
Anti-apoptotic protein Bcl-2 contributes to the determination of reserve cells during myogenic differentiation of C2C12 cells
In Vitro Cellular & Developmental Biology - Animal (2024)
-
Mild heat treatment in vitro potentiates human adipose stem cells: delayed aging and improved quality for long term culture
Biomaterials Research (2023)
-
Synthetic lethality of drug-induced polyploidy and BCL-2 inhibition in lymphoma
Nature Communications (2023)
-
Single-cell morphological and transcriptome analysis unveil inhibitors of polyploid giant breast cancer cells in vitro
Communications Biology (2023)
