
Abstract
The Coffin-Lowry syndrome (CLS) is a syndromic form of X-linked mental retardation characterised in male patients by psychomotor and growth retardation, and various skeletal anomalies. CLS is caused by mutations in a gene located in Xp22.2 and encoding RSK2, a growth-factor regulated protein kinase. Mutations are extremely heterogeneous and lead to premature termination of translation and/or to loss of phosphotransferase activity. No correlation between the type and location of mutation and the clinical phenotype is evident. However, in one family (MRX19), a missense mutation was associated solely with mild mental retardation and no other clinical feature. Screening for RSK2 mutations is essential in most cases to confirm the diagnosis as well as for genetic counseling.
Similar content being viewed by others
Clinical definition
Coffin-Lowry syndrome (CLS) is an X-linked disorder characterised by psychomotor retardation, hypotonia and characteristic facial, hand and skeletal malformations. Typically male patients are of short stature and exhibit a characteristic coarse face with a prominent forehead, microcephaly, hypertelorism, downslanting palpebral fissures, ptosis, prominent supraorbital ridges, epicanthic folds, thick lips, a thick nose septum with anteverted nares, and irregular or missing teeth. Their large and soft hands with lax skin and joints, tapering fingers and hypothenar crease, are usually a strong diagnostic feature. Tufting of the terminal phalanges on hand X-ray is also very suggestive. Skeletal malformations appear progressively in most patients, and may include delayed bone development, spinal kyphosis and/or scoliosis and pectus carinatum or excavatum. Some patients present with additional features, including sensorineural deafness (30%), seizures (30%), drop episodes (10–20%) cardiac problems, corpus callosus agenesis or ventricular dilatation, and myelopathy.1,2 Cognitive function is usually moderately impaired, with IQ values often below 55, and the language can usually be acquired. We want to stress that mental retardation is not as severely impaired as previously said, because of better early intervention methods. Furthermore, patients showing only mild psychomotor retardation and/or mild or moderate skeletal anomalies have been documented.3,4 Heterozygote detection in most carrier females is possible based usually on the presence of soft fleshy hands with thick tapering fingers and slight obesity. Furthermore, a coarse face with prominent brow, hypertelorism and thick nasal tissues, can be present. Cognitive function in female heterozygotes is normal or mildly impaired.
Diagnosis
Is based on clinical examination. Since many clinical features are non-specific, it is not always easy to distinguish CLS from other mental retardation syndromes, in particular in young children. In infancy the first diagnostic clues are the neonatal hypotonia together with the facial gestalt including hypertelorism, ptosis, frontal bossing, delayed closure of the anterior fontanel, thick lips and postnatal growth retardation. The typical aspect of the hands develops progressively during early childhood. In addition most cases are sporadic. Mutation detection is, thus, essential to confirm diagnosis of CLS and to offer molecular prenatal or carrier diagnosis to families. Immunoblot and RSK2 kinase assays can be used as rapid (one day) diagnostic tests for CLS.5 A Western blot can be performed on lymphocyte protein extracts prepared directly from fresh (less than 24 h) blood samples. The kinase assay necessitates either a fibroblast or a lymphoblast cell line but remains a diagnostic method of choice as it detects all classes of mutations and also provides information on a possible residual enzyme activity. These assays can be performed on cultured amniocytes for prenatal diagnosis. Figure 1

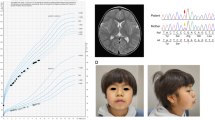
A 9-year-old patient carrying a frameshift mutation in the RSK2 gene. (a) Note the hypertelorism, the broad based nose, the downwards slanting of the palpetral fissures and the large mouth with full lips and missing and abnormal shaped teeth. (b) Note the thickened tapered digits.
Differential diagnoses
In young male patients, physical characteristics of the Coffin-Lowry syndrome are usually mild and can be confused with other syndromes, most notably α-thalassaemia with mental retardation syndrome (ATR-X, MIM 300032), but also with Fragile X syndrome (MIM 309550), Sotos syndrome (MIM 117550), Williams syndrome (MIM 194050) and lysosomal storage disease.6
Disease frequency
CLS has been reported in various ethnic groups. The incidence is unknown but lies probably in the range of 1 in 50–100 000 males.
Gene
The CLS gene was identified in 1996 by positional cloning in the Xp22.2 region.7 It encodes two transcripts of 3.5 and 8.4-kb (alternative polyadenylation), both ubiquitously expressed. The open reading frame is split into 22 exons8 and encodes a protein of 740 amino acids, RSK2, containing two non-identical kinase catalytic domains.9 The gene belongs to a family comprising, in human, four very closely related members (80–85% amino acid sequence identity), RSK1 to RSK4, and homologs have been identified in vertebrate and invertebrate (C elegans, Drosophila) genomes.
Genetic heterogeneity
No genetic heterogeneity has been documented.
Function of the protein
RSKs are serine-threonine protein kinases, acting in the Ras-Mitogen-Activated Protein Kinase (MAPK) signalling pathway. RSKs are directly phosphorylated and activated by ERK1 and 2 (Extracellular Signal-Regulated Kinases) in response to a broad range of cellular perturbations, including stimulation with insulin and growth factors, neurotransmiters, oncogenic transformation and UV-irradiation. Activation of RSKs is accompanied by the phosphorylation of four residues, one in each kinase domain (Ser227 and Thr577) and two in the linker region (Ser369 and Ser386). The N-terminal kinase catalytic domain phosphorylates the substrates of RSK and its activity is regulated by the C-terminal kinase catalytic domain, the linker region and 3-phosphoinositide-dependent protein kinase-1 (PDK1). RSKs have been implicated in several important cellular events, including proliferation and differentiation, cellular stress response and apoptosis. CREB, histone H3, and c-Fos have been demonstrated to be in vivo targets of RSK2. Its induction is therefore thought to influence gene expression.10 (NB: although the ribosomal S6 protein is a good substrate in vitro – their name, Ribosomal S6 Kinase, is due to this property – it has recently been shown that it is not a substrate in vivo).
Animal model
An RSK2 knockout mouse has recently been reported. Mutant mice show no obvious skeletal abnormality but weigh 10% less and are 14% shorter than wild-type littermates. Their glycogen metabolism in skeletal muscle is altered. They also exhibit impaired learning and coordination.11
Mutations
A systematic SSCP screening of the entire coding sequence and the splice sites in 250 unrelated patients with clinical features suggestive of Coffin-Lowry syndrome, revealed 71 distinct mutations in 86 (34%) of these patients.12 Nine mutations have been found in female probands ascertained through learning disability and mild but suggestive physical phenotype, but who have no affected male relatives. Thirty-eight per cent of mutations are missense changes, 20% nonsense, 18% splicing errors and 21% are short deletion or insertion events. The vast majority can be predicted to cause loss of function of the mutant allele. About 57% of mutations result in premature translation termination. Most of the missense mutations affect residues conserved in all known RSK family members from human to C elegans, supporting their pathogenic significance. Functional studies have revealed decreased or absent kinase activity for some of these mutant proteins. Mutations are distributed throughout the RSK2 gene, but five exons contain 50% of the detected mutations (exons 5, 9, 17, 18 and 22). There is no obvious correlation between location of mutations and clinical phenotype. Some missense mutations are associated with milder phenotypes. In one family (MRX19), a missense mutation was associated solely with mild mental retardation.4 In a total of 44 families, for which samples were available from the mothers of the probands, 25 (57%) mutations arose de novo in the proband, a higher frequence than expected. Germinal mosaicism has been reported in some families.13 Information about RSK2 and a continuously updated overview of all RSK2 mutations are available on a Web Page (http://www-ulpmed.u-strasbg.fr/chimbio/diag/coffin). Figure 2

Mutation spectrum of the RSK2 gene. The mutations so far detected are shown above and below a schematic representation of the RSK2 transcripts. NTK and CTK: N-terminal and C-terminal kinase domains respectively. ATP binding sites (ATP BS) and activation loops are coloured light grey and the ERK docking site is striped. Positions of four important regulatory phosphorylation sites (P) are indicated.
Treatment
There is no specific treatment. It is worth noting that the frequency of drop episodes seems to diminish under benzodiazepine medication.
References
Gilgenkrantz S, Mujica P, Gruet P et al. Coffin-Lowry syndrome: a multicenter study Clin Genet 1998 34: 230–245
Young ID . The Coffin-Lowry syndrome J Med Genet 1988 25: 344–348
Manouvrier-Hanu S, Amiel J, Jacquot S et al. Unreported RSK2 missense mutation in two male siblings with an unusually mild form of Coffin-Lowry syndrome J Med Genet 1999 36: 775–778
Merienne K, Jacquot S, Pannetier S et al. A missense mutation in RPS6KA3 (RSK2) responsible for non-specific mental retardation Nature Genet 1999 22: 13–14
Merienne K, Jacquot S, Trivier E et al. Rapid immunoblot and kinase assay tests for a syndromal form of X-linked mental retardation: Coffin-Lowry syndrome J Med Genet 1998 35: 890–894
Plomp AS, De Die-Smulders CEM, Meinecke P, Ypma-Verhulst JM, Lissone DA, Fryns JP . Coffin-Lowry syndrome: clinical aspects at different ages and symptoms in female carriers Gen Counsel 1995 6: 259–268
Trivier E, De Cesare D, Jacquot S et al. Mutations in the kinase Rsk-2 associated with Coffin-Lowry syndrome Nature 1996 384: 567–570
Jacquot S, Merienne K, De Cesare D et al. Mutation analysis of the RSK2 gene in Coffin-Lowry patients: Extensive allelic heterogeneity and a high rate of de novo mutations Am J Hum Genet 1998 63: 1631–1640
Bjorbaek C, Vik TA, Echwald SM et al. Cloning of a human insulin-stimulated protein kinase (ISPK-1) gene and analysis of coding regions and mRNA levels of the ISPK-1 and the protein phosphatase-1 genes in muscle from NIDDM patients Diabetes 1995 44: 90–97
Frodin M, Gammeltoft S . Role and regulation of 90 kDa ribosomal S6 kinase (RSK) in signal transduction Mol Cell Endocrinol 1999 25: 65–77
Dufresne SD, Bjorbaek C, El-Haschimi K et al. Altered extracellular signal-regulated kinase signaling and glycogen metabolism in skeletal muscle from p90 ribosomal S6 kinase 2 knockout mice Mol Cell Biol 2001 21: 81–87
Delaunoy JP, Abidi F, Zeniou M et al. Mutations in the X-linked RSK2 gene (RPS6KA3) in patients with Coffin-Lowry syndrome Hum Mutation 2001 17: 103–116
Jacquot S, Merienne K, Pannetier S, Blumenfeld S, Schinzel A, Hanauer A . Germline mosaicism in Coffin-Lowry syndrome Eur J Hum Genet 1998 6: 578–582
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Jacquot, S., Zeniou, M., Touraine, R. et al. X-linked Coffin-Lowry syndrome (CLS, MIM 303600, RPS6KA3 gene, protein product known under various names: pp90rsk2, RSK2, ISPK, MAPKAP1). Eur J Hum Genet 10, 2–5 (2002). https://doi.org/10.1038/sj.ejhg.5200738
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.ejhg.5200738
Keywords
This article is cited by
-
Spectrum of X-linked intellectual disabilities and psychiatric symptoms in a family harbouring a Xp22.12 microduplication encompassing the RPS6KA3 gene
Journal of Genetics (2019)
-
The role of genetics in the establishment and maintenance of the epigenome
Cellular and Molecular Life Sciences (2013)
-
Implication of abnormal epigenetic patterns for human diseases
European Journal of Human Genetics (2007)
