
Abstract
Rett syndrome (RTT) is one of the most common neurodevelopmental disorders in females. The disease is caused by mutations in the methyl-CpG-binding protein 2 gene (MECP2), and various mutations have been reported. The phenotypic spectrum in both female and male patients is diverse, ranging from very mild to congenital encephalopathy and prenatal lethality. In this study, the question was addressed as to whether implementation of systematic screening of MECP2 in patients with an unexplained mental retardation in DNA diagnostics would be reasonable, and the spectrum of phenotypes resulting from mutations in this gene was further explored. Mutational analysis of MECP2 was performed in mentally retarded female patients who were negative for FMR1 CGG repeat expansion, in male and female patients with clinical features suggestive of either Angelman or Prader-Willi syndrome without methylation defects on chromosome 15q11–q13. In the cohort of females negative for the molecular Fragile-X studies (N=92), one nonsense mutation (p.Q406X) was found. In the cohort of Angelman-negative patients (N=63), two missense mutations (p.R133C in a female patient and a mosaic p.T158M in a male patient) were found, which have been reported many times in patients with classical RTT syndrome. In the Prader-Willi-negative group (N=98), no pathogenic mutations were found.
The results support testing of patients with features suggestive of Angelman syndrome, but without methylation defects on chromosome 15q11–q13 for mutations in MECP2. In the remaining patients with unexplained mental retardation, additional clinical features should determine whether analysis of MECP2 is indicated.
Similar content being viewed by others


Introduction
Heterozygous mutations in the X-linked MECP2 gene have been first reported in Rett syndrome (RTT), a progressive neurologic developmental disorder, occurring almost exclusively in females.1 In approximately 95% of patients, these mutations occur de novo, and it has been shown that in most cases they are of paternal origin.2
Recent studies indicate that females with RTT appear to represent a more heterogeneous phenotype than was first realized.3,4,5 There are cases with a very mild phenotype, with preserved speech variant or with a congenital, early seizure onset. It is hypothesized that these differences are mainly due to the genotype, variation in X-inactivation patterns and probably other polygenic modifiers.3,6
Unlike previous thoughts, MECP2 mutations are not necessarily prenatally lethal in males, and are the cause of a variable phenotype, ranging from lethal congenital encephalopathy to Angelman-like phenotype and mild nonspecific X-linked mental retardation (MRX).7,8,9,10,11,12 Since the complete phenotypic spectrum of MECP2 mutations in both male and female patients is presently unknown, more studies are required. Previously, we reported on MECP2 mutation analysis in a cohort of 475 mentally retarded males who were negative for FMR1 CGG repeat expansion, and found one mutation that correlated with a Prader-Willi-like phenotype.13,14 It was suggested that MECP2 might have a role in regulating genes involved in body habitus. Since some patients with features of Angelman syndrome also had mutations in the MECP2 gene,10,11 genes in the region 15q11–q13 could be target genes downstream of MECP2. Therefore, it was suggested that it was worthwhile to evaluate the prevalence of MECP2 mutations among patients with features suggestive of the Prader-Willi syndrome, but without the characteristic molecular abnormalities on chromosome 15q.
To answer the question whether implementation of systematic screening of MECP2 in patients with an unexplained mental retardation in DNA diagnostics would be reasonable, and to explore further the spectrum of phenotypes resulting from MECP2 mutations, we performed mutational analysis in three additional patient groups: in mentally retarded female patients (N=92) who were negative for FMR1 CGG repeat expansion, in male and female patients with a phenotype suggestive of Angelman syndrome (N=63) without methylation defects in chromosome 15q11–q13 and in male and female patients with a phenotype suggestive of Prader-Willi syndrome (N=98), also without methylation abnormalities on chromosome 15q11–q13.
Subjects and methods
Patients
The patients had been referred to our DNA diagnostic division during the last 5 years. The patients were subdivided in three panels according to their previous request for DNA testing for Fragile-X syndrome, Angelman syndrome or Prader-Willi syndrome. The panels were called Fragile-X-negative (92 female patients), Angelman-negative (30 females and 33 males) or Prader-Willi-negative (41 females and 57 males). If the patient's DNA had been analysed for Angelman syndrome or Prader-Willi syndrome and Fragile-X syndrome, the patient was included in the Angelman- negative or Prader-Willi-negative panel. For all patients, informed consent was obtained.
DNA analysis
DNA was extracted from peripheral blood as described previously.15 The three coding exons of MECP2 were amplified using PCR and genomic DNA as a template. Both strands were sequenced. Sequencing reactions were run on an Applied Biosystems 3730 automated sequencer.
X-inactivation studies
X-inactivation studies were carried out on DNA isolated from lymphocytes from the p.Q406X patient, as described by Allen et al.16 However, the analysis was performed on an automated sequencer (ABI 3100 Prism) instead of Southern hybridisation.
Results
DNA studies
The results of mutation analysis of all the three panels are shown in Table 1a and b. In the Fragile-X-negative panel, two sequence changes were found, one known polymorphism c.1126C → T (p.P376S) and one nonsense mutation c.1216C → T (p.Q406X). The mutation was not found in the parents. X-inactivation studies in the patient lymphocytes showed a 100% skewed pattern.
In the Angelman-negative group, two known pathogenic missense mutations were found: c.397C → T (p.R133C) in a female patient and a mosaic c.473G → T (p.T158M) in a male patient, and one novel change c.1497G → C in a female patient. The c.1497G → C is in the 3′ UTR of the gene, and was also found in the mother of the patient. The known polymorphism c.1126C → T (p.P376S) was also found in this cohort in a female patient and in her father. The p.R133C was not found in the mother. The mother of the male patient with the mosaic p.T158M mutation did not have this mutation.
In the cohort of Prader-Willi-like patients one novel silent change c.393C → G (p.A131A) was found.
Clinical studies of patients with a MECP2 mutation
p.Q406X
This girl was born at the term after an uneventful pregnancy and delivery with a birth weight of 2550 g. During early life she was a passive baby. Psychomotor development was delayed; she walked at 3 years and spoke her first words at the age of 5 years. She had no EEG abnormalities.
At the age of 16 years, she attended an institute for child psychiatry because of behavioral problems. The degree of mental retardation was moderate. Chromosomal analysis and DNA studies of the Fragile-X syndrome were normal.
At the age of 22 years, she was re-examined Her height was 160 cm (−1.5 SD), her weight was 57 kg (0 SD), and her skull circumference was 56 cm (0.5 SD). She had a face with a child-like appearance and a strabismus of the left eye. She spoke with short sentences. Her gait was slightly unsteady.
p.R133C
This girl was born at the term after an uneventful pregnancy and delivery, with a birth weight of 3500 g. After 6 months of normal development, there was developmental stagnation, and only a slow further progression. No developmental regression was noted. She walked at 24 months and spoke her first words at 5 years of age.
On investigation at the age of 7 years, she had normal height and head circumference, and was slightly obese. She was a severely retarded, friendly and happy person, and spoke with two-words sentences. She showed teeth grounding and had a wide-based gait. She was insensitive to pain. Since the age of 7 years, there were severe sleep disturbances. Daytime EEG studies showed epileptic activity in less than 1%, whereas during the night more than 75% disturbed EEG patterns were observed. In spite of EEG abnormalities, no convulsions had been observed.
Routine chromosomal studies and molecular studies of the Angelman syndrome were negative.
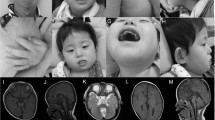
At the age of 14, she was examined again (Figure 1). She was a friendly person with growth retardation (height of 137 cm (<−2.5 SD)), generalised obesity (weight of 60 kg (>+2 SD)) and hypotonia. Head circumference was 53.5 cm (−1 SD). She revealed the same signs as discussed above. Moreover, thoracic scoliosis, cold extremities and tapering of the fingers were noticed. Her gait was unsteady and wide-based, but she could walk and climb stairs unaided. In addition, although with some spilling, she was able to eat and drink.

Female patient with the p.R133C mutation in MECP2 at the age of 14 years.
p.T158M
This boy was born after an uneventful pregnancy and delivery, with a birth weight of 3440 g. During the first year of life, he developed completely normal. Thereafter, his development stagnated followed by a period of regression. He lost his capability of purposeful handuse and walking along the table. At the age of 2 years and 9 months, he was a passive boy with autistic features and hardly any speech. Stereotypic movements like handclapping and teeth grounding were noted. There were convulsions and diffuse EEG abnormalities. Later on, he developed periods of yelling and shouting and sleeping disturbances. No metabolic defects were found. Routine chromosomal studies and DNA studies of the Angelman and Fragile-X syndrome were normal.
At the age of 11 years, he was re-examined (Figure 2). He had a weight of 11 kg (−2 SD) and a skull circumference of 51.5 cm (−2 SD). His height was not measured. In addition to the previous features, he was wheelchairbound, and a severe thoracic scoliosis was observed. Feet were small and cold.

Male patient with the p.T158M mosaic mutation in MECP2 at the age of 11 years. He shows the classical Rett syndrome phenotype.
Discussion
We report on three large patient cohorts in which MECP2 mutation analysis was performed to ascertain whether routine screening of this gene in patients with unexplained MR in a DNA diagnostics setting is indicated, and to further explore the phenotypic spectrum of MECP2 mutations.
In the cohort of females negative for the molecular Fragile-X studies, we found two nucleotide changes. The nonsense p.Q406X mutation that was found in this cohort has been reported once in a family where both the maternal uncle and son had mental retardation and spasticity.12 The two carrier females had a borderline intelligence, but no other symptoms. X inactivation in these two carriers showed random patterns. In our patient, however, the mutation was de novo. She was moderately retarded, had EEG abnormalities, behavioural problems and autistic features without spasticity. Differences in these two phenotypes might be explained by differences in the X-inactivation patterns, or by currently unknown genetic modifiers. X-inactivation analysis in this patient showed completely skewed X inactivation. Unfortunately, it was not possible to analyse which allele was the active or inactive one, but most likely the normal allele was inactivated. This might explain why heterozygosity of the p.Q406X in our patient leads to a severe phenotype, whereas in the two other individuals it results in a nearly normal phenotype. Since X inactivation is performed in lymphocytes, it will not be conclusive for X inactivation in brain cells. Therefore, the question still remains as to whether the differences in the two phenotypes are explained by X-inactivation differences. The other nucleotide change that was found in this cohort (p.P376S) has been reported as a polymorphism.5
In the cohort of 63 Angelman-negative patients, we found three nucleotide changes. Two changes were known missense mutations, one p.R133C and one mosaic p.T158M. The de novo p.R133C mutation has been reported many times in patients with classical RTT syndrome (http://www.ed.ac.uk~skirmis). Though our patient did meet several criteria for classical RTT syndrome, there was no history of regression; she did not have the acquired microcephaly and stereotypic handmovements. In addition, she showed extreme obesity, while most RTT patients had decreased body fat. Owing to this atypical appearance of Rett syndrome, this clinical diagnosis was not considered initially.
The p.T158M mutation found in mosaic form in a severely retarded male patient is a frequently reported mutation in classic RTT syndrome as well. The percentage of mutant versus wild-type allele was estimated to be 25%. Since this is analysed in lymphocytes, it is uncertain whether in brain tissue this percentage will be equal. In males, this mutation has been reported in a Klinefelter syndrome patient17 and post mortem in two brothers with congenital encephalopathy.7 Males with other mosaic MECP2 mutations have been reported as well.18,19 Since they all exhibit features of classic RTT syndrome, it is suggested that an MECP2 mutation associated with RTT syndrome in females could lead to a similar phenotype in males as a result of somatic mosaicism. The clinical diagnosis of Rett syndrome was considered previously, but it was believed at that time that this syndrome did not occur in males.
The c.1497G → C that has not been reported so far, was in the 3′ UTR of the gene, and was also found in the mother of the patient. Therefore, this change is considered a polymorphism.
Finally, in the panel of Prader-Willi-negative patients, one silent change in the coding sequence was found. Although this change has not been reported so far, it is unlikely to have a pathogenic effect.
In female patients with unexplained MR but without features of classic RTT syndrome, the mutation detection rate found here is 1/92. It is believed that the X-inactivation patterns observed in patients with RTT syndrome show a standard distribution. The percentage of possible cases with a MECP2 mutation on both tails of the distribution (neonatal death versus mild neurologic/cognitive defects) is expected to be very low and therefore difficult to find.3 A routine search for MECP2 mutations in female patients with unexplained MR without features of classical Rett syndrome, therefore, might not be worthwhile.
In the Angelman-negative cohort, our mutation rate (2/63) is lower than the ones found by Imessaoudene et al (6/78)10 and Watson et al (5/47).11 Although the patient cohorts in these studies were possibly clinically better defined than in the present study, our results still indicate that MECP2 mutational analysis in patients with a clinical diagnosis of Angelman syndrome but without molecular abnormalities on chromosome 15q11–q13, is worthwhile.
Recently, we suggested thatMECP2 might have a role in regulating genes involved in body habitus.14 Since some patients with features of Angelman syndrome also had mutations in the MECP2 gene,10,11 genes in the region 15q11–q13 might be target genes downstream of MECP2. Therefore, we evaluated the prevalence of MECP2 mutations among patients with features suggestive of the Prader-Willi syndrome, but without the characteristic molecular abnormalities on chromosome 15q11–q13. However, based on the current studies, routine screening of MECP2 in Prader-Willi-like patients might not be worthwhile, since we only found one silent change in a panel of 98 patients.
References
Amir RE, Van den Veyver IB, Wan M, Tran CQ, Francke U, Zoghbi HY : Rett syndrome is caused by mutations in X-linked MECP2, encoding the methyl-CpG-binding protein 2. Nat Genet 1999; 23: 185–188.
Trappe R, Laccone F, Cobilanschi J et al: MECP2 mutations in sporadic cases of Rett syndrome are almost exclusively of paternal origin. Am J Hum Genet 2001; 68: 1093–1101.
Wan M, Lee SS, Zhang X et al: Rett syndrome and beyond: recurrent spontaneous and familial MECP2 mutations at CpG hotspots. Am J Hum Genet 1999; 65: 1520–1529.
Hoffbuhr K, Devaney JM, LaFleur B et al: MECP2 mutations in children with and without the phenotype of Rett syndrome. Neurology 2001; 56: 1486–1495.
Sung Jae Lee S, Wan M, Francke U : Spectrum of MECP2 mutations in Rett syndrome. Brain Dev 2001; 23: 138–143.
Hoffbuhr KC, Moses LM, Jerdonek MA, Naidu S, Hoffman EP : Associations between MECP2 mutations, X-chromosome inativation and phenotype. Ment Retard Dev Disabil Res Rev 2002; 8: 99–105.
Villard L, Kpebe A, Cardoso C, Chelly J, Tardieu M, Fontes M : Two affected boys in a Rett syndrome family: clinical and molecular findings. Neurology 2000; 55: 1188–1193.
Orrico A, Lam CW, Galli L et al: MECP2 mutation in male patients with non-specific X-linked mental retardation. FEBS Lett 2000; 481: 285–288.
Couvert P, Bienvenu T, Aquaviva C et al: MECP2 is highly mutated in X-linked mental retardation. Hum Mol Genet 2001; 10: 941–946.
Imessaoudene B, Bonnefont JP, Royer G et al: MECP2 mutation in non-fatal, non-progressive encephalopathy in a male. J Med Genet 2001; 38: 171–174.
Watson P, Black G, Ramsden S et al: Angelman syndrome phenotype associated with mutations in MECP2, a gene encoding a methyl CpG binding protein. J Med Genet 2001; 38: 224–228.
Meloni I, Bruttini M, Longo I et al: A mutation in the Rett syndrome gene, MECP2, causes X-linked mental retardation and progressive spasticity in males. Am J Hum Genet 2000; 67: 982–985.
Yntema HG, Kleefstra T, Oudakker AR et al: Low frequency of MECP2 mutations in mentally retarded males. Eur J Hum Genet 2002; 10: 487–490.
Kleefstra T, Yntema HG, Oudakker AR et al: De novo MECP2 frameshift mutation in a boy with moderate mental retardation, obesity and gynaecomastia. Clin Genet 2002; 61: 359–362.
Miller SA, Dykes DD, Polesky HF : A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Res 1988; 16: 1215.
Allen RC, Zoghbi HY, Moseley AB, Rosenblatt HM, Belmont JW : Methylation of HpaII and HhaI sites near the polymorphic CAG repeat in the human androgen-receptor gene correlates with X chromosome inactivation. Am J Hum Genet 1992; S1: 1229–1239.
Leonard H, Silberstein J, Falk R et al: Occurence of Rett syndrome in boys. J Child Neurol 2001; 16: 33–38.
Topçu M, Cemaliye A, Sayi A et al: Somatic mosaicism for a MECP2 mutation associated with classic Rett syndrome in a boy. Eur J Hum Genet 2002; 10: 77–81.
Clayton-Smith I, Watson P, Ramsden S, Black GCM : Somatic mutation in MECP2 as a non-fatal neurodevelopmental disorder in males. Lancet 2000; 356: 830–832.
Acknowledgements
This work was supported by grants from ZonMw.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Kleefstra, T., Yntema, H., Nillesen, W. et al. MECP2 analysis in mentally retarded patients: implications for routine DNA diagnostics. Eur J Hum Genet 12, 24–28 (2004). https://doi.org/10.1038/sj.ejhg.5201080
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.ejhg.5201080
Keywords
This article is cited by
-
Progress in Rett Syndrome: from discovery to clinical trials
Wiener Medizinische Wochenschrift (2016)
-
Genetic Approach to Diagnosis of Intellectual Disability
The Indian Journal of Pediatrics (2016)
-
Brief Report: MECP2 Mutations in People Without Rett Syndrome
Journal of Autism and Developmental Disorders (2014)
-
Molecular genetics of Rett syndrome: when DNA methylation goes unrecognized
Nature Reviews Genetics (2006)
-
Mechanisms of Disease: neurogenetics of MeCP2 deficiency
Nature Clinical Practice Neurology (2006)
