
Abstract
Achromatopsia is a congenital, autosomal recessively inherited disorder characterized by a lack of color discrimination, low visual acuity (<0.2), photophobia, and nystagmus. Mutations in the genes for CNGA3, CNGB3, and GNAT2 have been associated with this disorder. Here, we analyzed the spectrum and prevalence of CNGB3 gene mutations in a cohort of 341 independent patients with achromatopsia. In 163 patients, CNGB3 mutations could be identified. A total of 105 achromats carried apparent homozygous mutations, 44 were compound (double) heterozygotes, and 14 patients had only a single mutant allele. The derived CNGB3 mutation spectrum comprises 28 different mutations including 12 nonsense mutations, eight insertions and/or deletions, five putative splice site mutations, and three missense mutations. Thus, the majority of mutations in the CNGB3 gene result in significantly altered and/or truncated polypeptides. Several mutations were found recurrently, in particular a 1 bp deletion, c.1148delC, which accounts for over 70% of all CNGB3 mutant alleles. In conclusion, mutations in the CNGB3 gene are responsible for approximately 50% of all patients with achromatopsia. This indicates that the CNGB3/ACHM3 locus on chromosome 8q21 is the major locus for achromatopsia in patients of European origin or descent.
Similar content being viewed by others

Introduction
Achromatopsia (rod monochromacy/total colorblindness; OMIM 262300, 216900) is a congenital, autosomal recessively inherited disorder characterized by reduced visual acuity, pendular nystagmus, increased sensitivity to light (photophobia), a small central scotoma (which is often difficult to demonstrate), eccentric fixation, and reduced or complete loss of color discrimination. Hyperopia is common. Nystagmus develops during the first few weeks after birth followed by increased sensitivity to bright light. Best visual acuity varies with severity of the disease and is 20/200 or less in complete achromatopsia and may be as high as 20/80 in incomplete achromatopsia. Visual acuity is usually stable over time, but both nystagmus and sensitivity to bright light may improve slightly. Although the fundus is usually normal, macular changes and vessel narrowing may be present in some patients. In electroretinographic recordings, cone function cannot be detected, while rod function has been reported as essentially normal.1
Mutations in the CNGA3 and CNGB3 genes, which encode the α- and the β-subunits of the cyclic nucleotide-gated cation channel (CNG) in cone photoreceptors, and also in the GNAT2 gene, encoding the α-subunit of the cone photoreceptor transducin, have been shown to cause this disorder.2, 3, 4, 5, 6 All three gene products are involved in the cone phototransduction cascade (photopigment → G-protein → phosphodiesterase/cGMP → CNG channel). The CNG channels are located in the cell membrane of the outer segments of the photoreceptor. In the dark, they are kept open by high intracellular levels of cGMP (dark current). Illumination triggers a signal transduction cascade – involving transducin – that results in the decrease of the intracellular cGMP level. This decrease in ligand concentration leads to the closure of the cGMP-gated channels and thus generates a membrane hyperpolarization signal that subsequently modulates glutamate release at the photoreceptor synapse.7
While we recently reported the results of a comprehensive screen for mutations in CNGA3,8 there are as yet only limited data for CNGB3.3, 4, 9, 10 The aim of this study was to determine the prevalence of CNGB3 gene mutations and to establish a comprehensive CNGB3 gene mutation spectrum in a large collection of patients affected by achromatopsia.
Materials and methods
Patients and clinical examination
Patients were informed about the objectives of the studies and consented to participate. The research program followed the tenets of the Declaration of Helsinki. The recruitment and clinical examinations were performed in several ophthalmic centers notably in Germany, the Netherlands, Denmark, Italy, the United States, Sweden, Hungary, France, Switzerland, and Israel. The clinical diagnosis was established by standard clinical examinations (visual acuity, color vision, ophthalmoscopy) and, if applicable, by further electroretinographic and psychophysical testings.
Venous blood was collected from all patients and available family members after informed consent and total genomic DNA was extracted according to standard procedures.
The patient sample comprised of 341 independent subjects with a diagnosis of congenital achromatopsia including patients diagnosed with complete (rod monochromacy) and incomplete achromatopsia.
Patients with blue cone monochromacy, caused by a defect in the red-/green opsin gene cluster, were excluded through prior genetic testing (data not shown).
In all, 80 cases of this patient sample were shown to carry homozygous or compound heterozygous CNGA3 gene mutations2, 9 (unpublished data) and were excluded from screening of the CNGB3 gene.
Clinical details of some of these patients with CNGB3 mutations have recently been reported.11, 12, 13
Mutation screening in achromatopsia
In a first screening series, 70 patients, in which CNGA3 mutations had been excluded, were analyzed by PCR amplification of all 18 exons and flanking intron sequences of the CNGB3 gene from total genomic DNA and subsequent direct DNA sequencing. PCR products were purified using Gibco PCR Purification Columns (GibcoBRL, Life Technologies, Rockville, MD, USA) or treated with ExoSAP-IT (USB Corp., Cleveland, OH, USA) and sequenced employing Big Dye Terminator Chemistry (Applied Biosystems, Weiterstadt, Germany). Sequencing reactions were separated on ABI 377 and ABI 3100 DNA sequencers (PE Biosystems, Weiterstadt, Germany), analyzed by manual inspection and comparative assembly with the SeqMan program (DNASTAR, Madison, WI, USA).
In a second screening series, DNA samples of 67 achromatopsia patients were subjected to whole CNGB3 gene SSCP analysis. For exons 1, 5, 9, and 11–16, PCR products were separated on 10% polyacrylamide gels (37.5:1) without glycerol at 4°C, exons 3, 8, 10, and 18 were run on 10% polyacrylamide gels (37.5:1) without glycerol at room temperature, and exons 4, 6, 7, and 17 were analyzed on 10% polyacrylamide gels (37.5:1) with 10% glycerol at 4°C. All gels were run in 1 × TBE over night at constant voltage (100–300 V) depending on fragment size. Polyacrylamide gels were then silver-stained for visualization of single- and double-stranded DNA fragment bands.
In addition, to account for the most frequent mutations, DNA samples of patients were analyzed for common mutations as follows: c.819–826del8 – PCR with mismatch primers for exon 6 and RFLP with HinfI; c.886–896del11insT – PCR with mismatch primers for exon 7 and RFLP with ApoI; c.991-3T>G – PCR with mismatch primers for intron 8/exon 9 and RFLP with HinfI; c.1006G>T (p.E336X) – standard PCR for exon 9 and RFLP with ApoI; and c.1148delC – standard PCR for exon 10 and RFLP with BfaI. Digested PCR products were separated on 2% agarose gels and visualized by ethidium bromide staining in comparison to a healthy control subject.
Our most recent screening strategy combines these PCR/RFLP assays and subsequent direct sequencing of all coding exons (remaining number of patients).
Control experiments
To evaluate the different identified sequence variations, we analyzed 100 healthy control DNA samples for the following mutations (in parts reported previously):3 c.467C>T (p.S156F), c.819–826del8, c.886–896del11insT, c.926C>T (p.P309L), c.991-3T>G, c.1006G>T (p.E336X), c.1148delC, c.1304C>T (p.S435F), and c.1578+1G>A. None of the mutations were observed in the control panel.
Segregation analysis
Segregation analysis for the presence and independent inheritance of two mutant alleles were performed in all cases (68/163), for whom samples from additional family members were available. Analysis was carried out either by PCR/RFLP analysis, SSCP analysis, or direct sequencing.
Results
Prevalence of CNGB3 mutations in achromatopsia patients
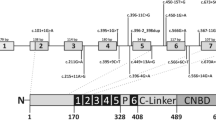
Our original patient sample comprised 341 independent patients with a clinical diagnosis of achromatopsia. Of these patients, 80 were shown to carry mutations in the CNGA3 gene (unpublished data)2, 8 and five patients carried GNAT2 mutations.5 In 163 patients, we were able to identify mutations in the CNGB3 gene. The spectrum of CNGB3 mutations comprised 28 different mutations including three missense mutations, 12 nonsense mutations, five small deletions of 1, 2, 8, 10, or 21 bp, respectively, three 1 bp insertions, and five putative splice site mutations (Table 1). The mutations observed in CNGB3 were spread almost evenly over all coding exons (Figure 1). This is in contrast to the distribution of mutations in CNGA3, which are almost exclusively confined to the three terminal exons encoding the structural and functional domains of the channel α-subunit.8

Distribution of the mutations found in the CNGB3 gene and the CNGB3 polypeptide. The structure of the exon coverage of the CNGB3 gene is displayed compared with the structural and functional CNGB3 channel domains. The mutations are shown in relative position to this arrangement. The different mutation types are displayed as follows: ▵ deletion, ▿ insertion, □ missense mutation, ○ nonsense mutation, ◊ splice site mutation. S1–S6: transmembrane domains; CaM: Ca2+/calmodulin domains.
Among the 163 patients with CNGB3 mutations, both mutant alleles were identified in 149 subjects and only a single heterozygous mutation was detected in 14 subjects (Table 2).
Of the 149 achromatopsia patients with two mutations in the CNGB3 gene, heterozygous mutations were observed in 44 cases and apparent homozygosity was present in 105 cases including 95 patients with homozygosity for the c.1148delC mutation allele. However, for most of the subjects with apparent homozygous mutations, there was no evidence for parental consanguinity. For 68 of the 163 CNGB3 mutation carrying patients, family members were available for segregation analysis, which provided no evidence for in-phase double mutations (ie heterozygous mutations in cis) or hemizygous mutations that mimic homozygosity. In fact, the presence and segregation of two independent mutation alleles could be unequivocally confirmed in 51 cases in which both parents were genotyped (data not shown). A total of 19 patients had affected family members. All affected family members were shown to carry the same genotype as the index patient.
In 14 achromatopsia patients, only a single heterozygous mutation was detected, although all 18 coding exons had been sequenced. In addition, the presence of larger rearrangements and deletions was excluded by means of semiquantitative multiplex PCR and long-distance PCR covering all coding exons in overlapping segments (data not shown).
Discussion
Nature and impact of CNGB3 mutations
A major finding of this study is that the majority of mutations in the CNGB3 gene (25/28) – if expressed – give rise to truncated CNGB3 polypeptides and most probably represent null alleles. Four of the 12 nonsense mutations result in premature translation termination before transmembrane domain S1, and the remaining mutations well before or right at the beginning of the cGMP binding site (Figure 1). Similarly, all but one of the deletion/insertion mutations result in frameshifts that lead to largely truncated polypeptides. It is very likely that these mutations do not contribute to the formation of functional heteromeric cone CNG channels, since all of these mutant CNGB3 polypetides lack the cGMP binding site. The high proportion of protein truncation mutations in CNGB3 contrasts with the mutation spectrum in the CNGA3 gene, where missense mutations predominate.8 The reason for this obvious discrepancy in the prevalence of different types of mutations is unclear. One might speculate that the CNGB3 polypeptide, which is less stringently conserved in evolution than CNGA3, tolerates to a larger extent substitution mutations. However, the presence of only few sequence variants/polymorphisms argues against this.
Five of the detected mutations most probably affect splicing of the CNGB3 pre-mRNA. Three of them (c.1578+1G>A; c.1781+1G>C; c.1781+1G>A) alter the almost perfectly conserved guanosine of the splice donor 5′-terminal -GT- dinucleotide. Such mutations typically lead to a skipping of the preceding exon during transcript processing. The other two splice site mutations c.644-1G>C and c.991-3T>G affect the splice acceptor consensus sequence. The mutation c.644-1G>C alters the last nucleotide of the introns' almost perfectly conserved terminal -AG- dinucleotide. The mutation c.991-3T>G is located three nucleotides upstream of the intron 8/exon 9 boundary. Statistically, this nucleotide position is most frequently occupied by a cytidine or by a thymidine nucleotide. In most cases, mutations at the splice acceptor site lead to a skipping of the downstream exon. Arguments supporting the pathogenicity of the putative splice site mutations are their occurrence in association with a second clearly pathogenic mutation in compound heterozygotes (see Table 2: allele compositions) and the recurrency of the c.991-3T>G mutation in Dutch patients.
The association of the missense mutations with the achromatopsia phenotype is also worthy of discussion. Firstly, all three mutations have been excluded in over 100 healthy controls and shown to segregate with the achromatopsia phenotype in the respective pedigrees.
The missense mutation p.S156F was observed in two independent achromatopsia families in our study and has also been reported by Sundin and co-workers.4 Although it affects an amino residue in the less stringently conserved amino-terminus, it was found as the sole alteration in trans with clear pathogenic mutations (Table 2). Most recent research has shown that p.S156F is located in the vicinity of an amino-terminal Ca2+/calmodulin binding site (aa 132–147).14
The mutation p.P309L affects an amino-acid residue in transmembrane domain S3 that is evolutionarily highly conserved among human and murine CNGB3 and human CNGB1. It was observed in trans with the most frequent mutant allele c.1148delC.
The p.S435F mutation has been reported in prior studies to segregate with ‘Pingelapese Blindness’ (OMIM262300).3, 4 This kind of achromatopsia is very common in the Pingelap islander population of the Eastern Caroline Islands of Micronesia (incidence 5–10%).4, 15 Recent work achieved by heterologous coexpression of p.S435F mutant CNGB3- and wild-type CNGA3-subunits in Xenopus oocytes suggests that the mutation does not disturb subunit assembly or plasma membrane targeting, but rather seems to result in an increase of the affinity for cAMP and cGMP, and changes the pore properties of the channel including decreased single channel conductance and sensitivity to block by L-cis-diltiazem.16
Recurrent mutations
Nine of the 28 different mutations were found recurrently (Table 1). Some of these were confined to specific geographic locations suggesting local founder effects. The mutations c.819–826del8 and c.886–896del11insT that account for 16 and eight disease alleles, respectively, in our sample were found only in patients originating from Germany or the Netherlands. Even more restricted in its geographic distribution is the putative splice site mutation c.991-3T>G in intron 8. Except for one patient, this mutation was exclusively found in patients originating from the Netherlands. Of the 40 patients of Dutch origin, five patients were compound heterozygous and three homozygous for this mutation (11/13 alleles).
Most strikingly, we identified as many as 233 c.1148delC mutant alleles in our total of 163 independent patients with CNGB3 mutations. In all, 95 patients were homozygous (or at least apparently homozygous) for this mutation (190 alleles) and 43 were heterozygous. In seven of the latter patients, the c.1148delC mutation was the sole CNGB3 mutation detected (see below). In comparison, another study reports eight families with CNGB3 mutations, of these five patients were apparently homozygous, one compound heterozygous, and one carried a single c.1148delC allele.10 This means that the c.1148delC mutation accounts for over 70% of all CNGB3 mutant alleles in our achromat sample. This predominance of a single mutation is one of very few examples among retinal diseases such as the p.P23H RHO gene mutation in the US patients with autosomal dominant retinitis pigmentosa,17 the p.R345W EFEMP1 gene mutation in Malattia Leventinese and Doyne honeycomb retinal dystrophy,18 or the c.2588G>C ABCA4 gene mutation in middle and northern European Stargardt disease patients.19 In comparable samples, screening for the c.1148delC mutation will enable the molecular diagnosis of almost 40% of all achromatopsia cases with a single assay.
Singular mutations
In 14 index patients, we could only detect a single heterozygous mutation. The lack of a second mutation in these patients might be explained by several arguments. A general problem in PCR-based screening protocols – as used in this study – is the failure to identify large genomic deletions and rearrangements. We tried to address this problem by performing semiquantitative multiplex and long-distance PCR experiments for the CNGB3 gene in these subjects. Yet, these investigations did not provide evidence for the presence of such deletions/rearrangements in our patients with single heterozygous mutations. Another possibility is that the missing mutations are located within introns (‘deep intron mutations’) or in yet unidentified upstream untranslated sequences critical for CNGB3 splicing or expression.
Finally, given the large number of subjects screened in this study, we have to consider that one or the other of these single heterozygotes might be a carrier for a mutant CNGB3 allele, while the disease is caused by mutations in another gene. Current estimates for the prevalence of achromatopsia are between 1:30 000 and 1:50 000, with about half of them caused by CNGB3 mutations as shown in this study. The expected carrier frequency for CNGB3 mutations therefore ranges between 0.006 and 0.008.
Prevalence of CNGB3 mutations in achromatopsia
Our study demonstrates that mutations in CNGB3 are the most common cause of achromatopsia in patients of European origin or descent. Of 341 patients in our sample, 163 (47%) carry mutations in CNGB3. In contrast, mutations in CNGA3 account for only 23% of cases in this sample (unpublished results).8 Mutations in the third gene known to be associated with achromatopsia, GNAT2, are even less prevalent and are found in less than 2% of achromatopsia patients.5, 6
Although there has been considerable progress in the genetic basis of achromatopsia, and the genetic defect can now be defined in almost three of every four patients within our study sample, there still remains a fair proportion of achromats that tested negative for all three known achromatopsia genes. Discordant marker segregation patterns for all three loci in a few families with multiple affecteds (unpublished data) additionally support the existence of at least one further achromatopsia locus in the human genome.
Phenotypic presentation in human
All patients with CNGB3 mutations initially presented with typical complete achromatopsia characterized by severely reduced visual acuity, pendular nystagmus, photophobia, and loss of color vision. Electrophysiological examination, when possible, regularly showed the absence of cone function. Although typical complete achromatopsia is usually a nonprogressive disease, a preliminary analysis suggests that the symptoms of a few of our patients are consistent with a progressive disease manifesting with decreasing visual acuity and central visual field defects.13 Another case of progressive cone dystrophy associated with mutations in CNGB3 has been described by Michaelides and co-workers.20 However, while complete achromatopsia and progressive cone dystrophies have also been associated with mutations in CNGA3, so far incomplete achromatopsia has only been associated with mutations in CNGA3.8, 11, 21
Pathophysiology and comparison to animal models
Native cone photoreceptor CNG channels most likely represent heterotetramers of two α- and two β-subunits encoded by CNGA3 and CNGB3, respectively.22 In heterologous expression systems, the α-subunit is per se able to form functional cation channels.23 In contrast, the β-subunit alone cannot conduct measurable ion currents.
However, it has been demonstrated for the mouse ortholog cng6 that in coexpression experiments, the cng6 gene product modulates the biophysical and electrophysiological behavior of the functional channel-forming α-subunit. It induces flickering channel gating, a decrease in outward rectification, and sensitivity to the blocking agent L-cis-diltiazem similar to native cone CNG channels.24 Since mutations in CNGB3 cause complete achromatopsia, it must be reasoned that the presence of the β-subunit is indispensable for appropriate channel function in vivo.
Very recent research has shown that autosomal recessive canine cone degeneration (cd) in the Alaskan malamute and the German shorthaired pointer breeds is caused by mutations in the canine CNGB3 gene.25 In the naturally occurring Alaskan malamut, cone-degenerate pups develop dayblindness and photophobia between 8 and 12 weeks postnatal, the age when retinal development is normally completed in dogs. Symptoms are present only in bright light, vision in dim light is normal. Affected dogs remain ophthalmoscopically normal throughout life. Cone function can be detected electroretinographically in very young cd-affected pups (3–6 weeks old), but begins to fail at 6–12 weeks of age and is nonrecordable in mature cd-affected dogs.26 Adult cd-affected retinas lack all cones. Cones degenerate by extrusion of the nucleus into the inner segment and later displacement of the cone nuclei in the interphotoreceptor space.27, 28 The genetic analysis has shown that in the Alaskan malamute, the complete gene is deleted, while in the German shorthaired pointers, the disease is caused by a missense mutation c.784G>A, resulting in the amino-acid substitution p.D262N (Genbank Accession no. AF490511). This amino-acid residue is located in the second transmembrane domain and is known to be conserved between human and dog CNGB3.27
References
Hess RF, Sharpe LT, Nordby K (eds): Night Vision: Basic, Clinical and Applied Aspects. Cambridge: Cambridge University Press, 1990.
Kohl S, Marx T, Giddings I et al: Total colorblindness is caused by mutations in the gene encoding the α-subunit of the cone photoreceptor cGMP-gated cation channel. Nat Genet 1998; 19: 257–259.
Kohl S, Baumann B, Broghammer M et al: Mutations in the CNGB3 gene encoding the β-subunit of the cone photoreceptor cGMP-gated channel are responsible for achromatopsia (ACHM3) linked to chromosome 8q21. Hum Mol Genet 2000; 9: 2107–2116.
Sundin OH, Yang JM, Li Y : Genetic basis of total colourblindness among the Pingelapese islanders. Nat Genet 2000; 25: 289–293.
Kohl S, Baumann B, Rosenberg T et al: Mutations in the cone photoreceptor G-protein alpha-subunit gene GNAT2 in patients with achromatopsia. Am J Hum Genet 2002; 71: 422–425.
Aligianis IA, Forshew T, Johnson S et al: Mapping of a novel locus for achromatopsia (ACHM4) to 1p and identification of a germline mutation in the alpha subunit of cone transducin (GNAT2). J Med Genet 2002; 39: 656–660.
Müller F, Kaupp UB : Signaltransduktion in Sehzellen. Naturwissenschaften 1998; 85: 49–61.
Wissinger B, Gamer D, Jagle H et al: CNGA3 mutations in hereditary cone photoreceptor disorders. Am J Hum Genet 2001; 69: 722–737.
Rojas CV, Maria LS, Santos JL, Cortes F, Alliende MA : A frameshift insertion in the cone cyclic nucleotide gated cation channel causes complete achromatopsia in a consanguineous family from a rural isolate. Eur J Hum Genet 2002; 10: 638–642.
Johnson S, Michaelides M, Aligianis IA et al: Achromatopsia caused by novel mutations in both CNGA3 and CNGB3. J Med Genet 2004; 41: e20.
Jägle H, Kohl S, Apfelstedt-Sylla E, Wissinger B, Sharpe LT : Manifestations of rod monochromacy. Col Res Appl 2001; 26: S96–S99.
Eksandh L, Kohl S, Wissinger B : Clinical features of achromatopsia in Swedish patients with defined genotypes. Ophthal Genet 2002; 23: 109–120.
Kellner U, Wissinger B, Kohl S, Kraus H, Foerster MH : Molecular genetic findings in patients with congenital cone dysfunction: mutations in the CNGA3, CNGB3, or GNAT2 genes. Ophthalmologe 2004; 101: 830–835.
Peng C, Rich ED, Thor CA, Varnum MD : Functionally important calmodulin binding sites in both N- and C-terminal regions of the cone photoreceptor cyclic nucleotide-gated channel CNGB3 subunit. J Biol Chem 2003; 278: 24617–24623.
Sheffield VC : The vision of typhoon lengkieki. Nat Med 2000; 6: 746–747.
Peng C, Rich ED, Varnum MD : Achromatopsia-associated mutation in the human cone photoreceptor cyclic nucleotide-gated channel CNGB3 subunit alters the ligand sensitivity and pore properties of heteromeric channels. J Biol Chem 2003; 278: 34533–34540.
Dryja TP, McGee TL, Reichel E et al: A point mutation of the rhodopsin gene in one form of retinitis pigmentosa. Nature 1990; 343: 364–366.
Stone EM, Lotery AJ, Munier FL et al: A single EFEMP1 mutation associated with both Malattia Leventinese and Doyne honeycomb retinal dystrophy. Nat Genet 1999; 22: 199–202.
Maugeri A, Flothmann K, Hemmrich N et al: The ABCA4 2588G>C Stargardt mutation: single origin and increasing frequency from South-West to North-East Europe. Eur J Hum Genet 2002; 10: 197–203.
Michaelides M, Aligianis IA, Ainsworth JR et al: Progressive cone dystrophy associated with mutation in CNGB3. Invest Ophthalmol Vis Sci 2004; 45: 1975–1982.
Trankner D, Jagle H, Kohl S et al: Molecular basis of an inherited form of incomplete achromatopsia. J Neurosci 2004; 24: 138–147.
Peng C, Rich ED, Varnum MD : Subunit configuration of heteromeric cone cyclic nucleotide-gated channels. Neuron 2004; 42: 401–410.
Wissinger B, Müller F, Weyand I et al: Cloning, chromosomal localization and functional expression of the gene encoding the α-subunit of the cGMP-gated channel in human cone photoreceptors. Eur J Neurosci 1997; 9: 2512–2521.
Gerstner A, Zong X, Hofmann F, Biel M : Molecular cloning and functional characterization of a new modulatory cyclic nucleotide-gated channel subunit from mouse retina. J Neurosci 2000; 20: 1324–1332.
Sidjanin DJ, Lowe JK, McElwee JL et al: Canine CNGB3 mutations establish cone degeneration as orthologous to the human achromatopsia locus ACHM3. Hum Mol Genet 2002; 11: 1823–1833.
Aguirre GD, Rubin LF : The electroretinogram in dogs with inherited cone degeneration. Invest Ophthalmol 1975; 14: 840–847.
Aguirre GD, Rubin LF : Pathology of hemeralopia in the Alaskan malamut dog. Invest Ophthalmol 1974; 13: 231–235.
Gropp KE, Szel A, Huang JC, Acland GM, Farber DB, Aguirre GD : Selective absence of cone outer segment beta 3-transducin immunoreactivity in hereditary cone degeneration (cd). Exp Eye Res 1996; 63: 285–296.
Acknowledgements
We thank all patients and family members for participating in this study, and Manfred Stuhrmann-Spangenberg, Jennifer Kemp, Karmen Trzupek, and Gert Matthijs for supplying us with patients and blood/DNA samples. We also thank Jochen Scholl, Martina Borghammer, Sabine Tippmann, and Eva Weber for excellent technical assistance. This work was supported by the following grants: Grant IB2 and Q1 from the Bundesministerium für Bildung und Forschung (01 KS 9602) and the Interdisziplinäres Zentrum für Klinische Forschung (IZKF) Tübingen to LTS and BW; a grant of the Schilling Stiftung to LTS; Grants SFB430/A5 and W: 1189/6-1 of the Deutsche Forschungsgemeinschaft to BW; Grant 32-065250.01 from the Swiss National Science Foundation to FLM; Grant DFGLo457,1–3 of the Deutsche Forschungsgemeinschaft to BL; a grant from the Foundation Fighting Blindness and Research to Prevent Blindness to RGW. CR: None.
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article
Cite this article
Kohl, S., Varsanyi, B., Antunes, G. et al. CNGB3 mutations account for 50% of all cases with autosomal recessive achromatopsia. Eur J Hum Genet 13, 302–308 (2005). https://doi.org/10.1038/sj.ejhg.5201269
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.ejhg.5201269
Keywords
This article is cited by
-
Structure of the human cone photoreceptor cyclic nucleotide-gated channel
Nature Structural & Molecular Biology (2022)
-
Achromatopsia: Genetics and Gene Therapy
Molecular Diagnosis & Therapy (2022)
-
Molecular and clinical characterization of Thai patients with achromatopsia: identification of three novel disease-associated variants in the CNGA3 and CNGB3 genes
International Ophthalmology (2021)
-
Naturally-occurring myopia and loss of cone function in a sheep model of achromatopsia
Scientific Reports (2020)
-
Molecular genetic analysis using targeted NGS analysis of 677 individuals with retinal dystrophy
Scientific Reports (2019)
