
Abstract
Gross deletions of the NF1 gene at 17q11.2 belong to the group of ‘genomic disorders’ characterized by local sequence architecture that predisposes to genomic rearrangements. Segmental duplications within regions associated with genomic disorders are prone to non-allelic homologous recombination (NAHR), which mediates gross rearrangements. Copy number variants (CNVs) without obvious phenotypic consequences also occur frequently in regions of genomic disorders. In the NF1 gene region, putative CNVs have been reportedly detected by array comparative genomic hybridization (array CGH). These variants include duplications and deletions within the NF1 gene itself (CNV1) and a duplication that encompasses the SUZ12 gene, the distal NF1-REPc repeat and the RHOT1 gene (CNV2). To explore the possibility that these CNVs could have played a role in promoting deletion mutagenesis in type-1 deletions (the most common type of gross NF1 deletion), non-affected transmitting parents of patients with type-1 NF1 deletions were investigated by multiplex ligation-dependent probe amplification (MLPA). However, neither CNV1 nor CNV2 were detected. This would appear to exclude these variants as frequent mediators of NAHR giving rise to type-1 deletions. Using MLPA, we were also unable to confirm CNV1 in healthy controls as previously reported. We conclude that locus-specific techniques should be used to independently confirm putative CNVs, originally detected by array CGH, to avoid false-positive results. In one patient with an atypical deletion, a duplication in the region of CNV2 was noted. This duplication could have occurred concomitantly with the deletion as part of a complex rearrangement or may alternatively have preceded the deletion.
Similar content being viewed by others

Introduction
Copy number variants (CNVs), manifesting as duplications, insertions and deletions of specific genomic segments, contribute significantly to human genome diversity.1, 2, 3, 4, 5, 6, 7 The genome-wide distribution of CNVs has been revealed by a variety of different array-based techniques. Thus, using matrix array comparative genomic hybridization (array CGH), CNVs involving from 50 kb up to several megabases have been detected.8, 9, 10, 11, 12, 13, 14, 15 In tandem, SNP genotyping arrays, oligonucleotide arrays and PCR-based genotyping have proved to be excellent tools to identify smaller CNVs.14, 16, 17, 18, 19, 20 In addition, non-array-based techniques, such as the comparison of different human genome assemblies by direct sequence alignments, have been used to confirm the abundance of CNVs in different size ranges.13, 21
The most extensive study on CNVs performed to date was designed so as to allow the construction of a human genome-wide copy number variation map.14 These authors employed array CGH with genomic clones and SNP array analysis to identify some 1447 regions harbouring CNVs in a total of 270 individuals taken from four different human populations (the HapMap collection).14 Some 24% of these 1447 CNV regions were found to be located in regions of segmental duplication. That CNVs occur frequently not only within regions of segmental duplication involved in ‘genomic disorders’, but also in the respective rearranged regions suggests that both the segmental duplications and the CNVs within these regions predispose to the rearrangements associated with genomic disorders.14
Redon et al14 also noted polymorphic copy number variation in the NF1 gene region at 17q11.2. Approximately 5% of NF1 patients exhibit gross NF1 gene deletions,22, 23, 24 which are almost invariably mediated by non-allelic homologous recombination (NAHR) between segmental duplications.25, 26, 27, 28, 29, 30, 31 Thus, gross NF1 deletions may also be considered to belong to the group of genomic disorders. Two distinct types of recurrent NF1 gene deletion have been documented: the first of these, type-1 deletions, span 1.4 Mb and are characterized by breakpoints, which cluster within the NF1-REPs in two regions, ∼15 kb apart, termed PRS1 and PRS2 (paralogous recombination sites 1 and 2).27, 30, 31 By contrast, type-2 NF1 deletions encompass 1.2 Mb with breakpoints in the SUZ12 gene and its pseudogene both of which are located in close proximity to the NF1-REPs (Figure 1).32, 33, 34 Less frequent than the type-1 and type-2 deletions are the so-called atypical NF1 deletions with non-recurring breakpoints.35, 36, 37

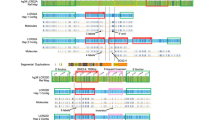
Schematic drawing of the NF1 gene region at 17q11.2. Proximal NF1-REPa and distal NF1-REPc are denoted by grey rectangles. The transcriptional orientation of the various genes within the region is indicated by black arrows. The positions of CNV1 and CNV2 are given, together with the relative positions of the MLPA probes (red arrows). The locus designation for the CNVs is in accordance with the Human Genome Segmental Duplication Database (http://www.projects.tcag.ca/humandup/).
The CNV identified by Redon et al14 encompasses the SUZ12 gene, the LRRC37B gene located in the distal NF1-REPc repeat and the RHOT1 gene. This region was found to be duplicated in one of 270 individuals from the HapMap collection.14 A second region of CNV in the vicinity putatively involves the NF1 gene itself; indeed, Wong et al15 recently claimed to have identified deletions involving the NF1 gene in five individuals and a duplication in 1 out of 95 human DNAs investigated. The details of these CNVs are summarized in Table 1. These NF1 CNVs were originally detected on the basis of deviant fluorescence intensity ratios for BAC RP11-518B17, which spans the distal part of the NF1 gene. However, Khaja et al13 also noted these CNVs within the NF1 gene region by means of direct alignment of the human genomic reference sequence with that of the Celera assembly.
Since CNVs have been suggested to trigger NAHR in regions characterized by genomic disorders, we wondered whether the CNVs in the NF1 gene region could have facilitated the formation of the gross NF1 deletions. To this end, we investigated whether the CNVs within the NF1 gene region occur at increased frequency in the transmitting parents of patients with type-1 deletions, the most common type of gross NF1 deletion. These are constitutional deletions, the vast majority of which occur during maternal meiosis via interchromosomal NAHR.39 In parallel, we also investigated 27 healthy controls, 18 patients with type-2 or atypical deletions and 9 of their parents for the presence of CNVs in the NF1 gene region.
Materials and methods
DNA samples from patients and their parents
Genomic DNA was isolated from peripheral blood samples of patients and their parents, together with healthy donors, using the Qiamp kit (Qiagen, Valencia, CA, USA) or by standard salt precipitation. The type-1 deletions in the respective patients were confirmed by two independent methods: FISH (fluorescent in situ hybridization) and breakpoint junction PCR to amplify across the recurrent breakpoints, PRS1 and PRS2. The methodology and primer pairs used to amplify across PRS2 were as described by Lopez-Correa et al,27 while primer pairs used to amplify across PRS1 were reported by Forbes et al.30 The parents of patients with type-1 deletions (and breakpoints in either PRS1 or PRS2) investigated in this study are listed in Table 2. This study was approved by the Local Institutional Review boards of the participating centres and informed consent was obtained from all patients and their relatives.
Haplotype analysis to determine the origin of the deletions
Analysis of polymorphic markers on chromosome 17 was performed with 6FAM-labelled primers and capillary electrophoresis on an ABI Prism 3100 Genetic Analyzer (Applied Biosystems, Foster City, CA, USA). The respective markers and genotyping data are listed in Supplementary Figure 1 for type-1 deletion patients 270, 450, 801, 344, 752, 1333, 800, 1277, 1547 and for patient 1860 with the atypical NF1 deletion. The parental origin of the type-1 deletions in patients ZL-1 to -9 was previously determined.27, 31
Multiplex ligation-dependent probe amplification
The multiplex ligation-dependent probe amplification (MLPA) assay SALSA P122 NF1 area (version 01, 05-02-2005; MRC Holland, Amsterdam, The Netherlands) was used to screen for CNVs in DNA derived from the parents of patients with type-1, type-2 and atypical deletions. This assay included five probes located within the NF1 gene, and, additionally, seven probes from NF1-flanking regions as summarized in Supplementary Table 1. The DNA samples were analysed by MLPA according to the manufacturer's instructions using 200 ng genomic DNA. After hybridization, ligation and amplification, the PCR products were separated on an ABI Prism 3100 Genetic Analyzer (Applied Biosystems) by capillary electrophoresis. Data analysis was accomplished by exporting the peak area to an Excel file. The relative probe signal was then determined by a normalization procedure as described.40 For sequences present in two copies in a given sample, these calculations were expected to yield a value of 1.0. Any decrease or increase in the peak area values to <0.8 or >1.2, respectively, was considered to be indicative of a deletion or a duplication, respectively, according to the instructions provided by MRC Holland.
Characterization of the deletion in patient 1860
Fluorescent in situ hybridization analysis using BAC RP11-142O6 was performed as previously described.33 The deletion breakpoints were identified in the first instance by CGH using the HG18 CHR17 FT arrays (NimbleGen Systems Inc., Madison, WI, USA). These oligonucleotide arrays are human chromosome 17-specific fine-tiling arrays with a median probe spacing of 160 bp and an isothermal probe design of 50- to 75-mer oligomers. Sample labelling, array manufacturing, hybridization, scanning data extraction and primary data analysis were performed by NimbleGen. After normalization, the data sets were prepared for DNA segmentation analysis using an averaging step in which adjacent windows are averaged. The circular binary segmentation algorithm form was applied to segment the averaged log 2 ratio data into 4-kb windows (Supplementary Table 2). To confirm the deletion boundaries as determined by array CGH, the deletion breakpoints were analysed by PCR with primers listed in Supplementary Tables 3 and 4 using DNA from a somatic cell hybrid containing only the deleted chromosome 17 of the patient. The presence or absence of PCR products was indicative of the location of the deletion boundaries.
Clinical investigation of patient 1860
The female patient was 28 years old at the time of investigation. She presented with learning disabilities and developmental delay but had been able to complete her general school education. Skin manifestations included axillary and inguinal freckling as well as ∼1000 cutaneous and subcutaneous neurofibromas. She had Lisch nodules but no facial dysmorphism and no abnormal joint flexibility. A brain MRI scan failed to indicate any abnormality but whole-body MRI revealed internal tumours, which were confined to the brachial plexus and lumbar region. Neurological and clinical investigation showed no additional abnormal findings.
Results
The CNVs previously reported to occur within the NF1 gene region (schematically indicated in Figure 1) have until now not been independently validated. Wong et al15 detected a deletion in five individuals and a duplication in 1 out of 95 donor samples investigated. The latter CNV appears to encompass the 3′ end of the NF1 gene and includes the OMG, EVI2A, EVI2B and RAB11FIP4 loci. In what follows, we shall refer to these loss-and-gain variants at the NF1 locus as CNV1. The second reported CNV in the NF1 gene region was a duplication observed in 1 out of 270 individuals from the HapMap collection.14 This variant was termed CNV2; it occurs distal to the NF1 gene and encompasses the SUZ12, LRRC37B and RHOT1 genes (Figures 1 and 2). The characteristics of both CNV1 and CNV2 are summarized in Table 1.

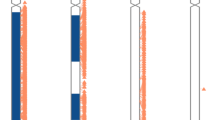
Schematic representation of the genomic region at 17q11.2 indicating in (a) the extent of the CNV2 duplication according to Redon et al,14 (b) the position of the MLPA probes and whether they were deleted or duplicated in patient 1860 and (c) the position of the distal deletion boundary in patient 1860 as determined by PCR analysis using DNA from a somatic cell hybrid containing only the deleted chromosome 17 of the patient. The extent of the duplication in patient 1860 is also indicated in (c) according to the array CGH results. These numbers are in accordance with the nucleotide numbering system of chromosome 17 in the hg18 assembly of the human genome. The BAC clones indicated in (a) are part of the 30K TPA clone set investigated by Redon et al.14 BACs Chr17tp-11E3 and Chr17tp-10C1 exhibited deviant log 2 ratios by array CGH but the overlapping BACs did not.14
Multiplex ligation-dependent probe amplification analysis was performed on DNA derived from the African man, who was positive for CNV2 according to Redon et al14 (DNA sample GM18501; Coriell Cell Repository, Camden, NJ, USA) and the duplication was confirmed (Supplementary Table 5).
MLPA analysis of the transmitting parents of patients with type-1 deletions
To investigate the presence of CNVs within the NF1 gene region, we employed MLPA using probes located in different parts of the NF1 gene region (Figure 1). In total, 18 transmitting (but unaffected) parents of patients with constitutional type-1 deletions as well as 9 non-transmitting parents were analysed (summarized in Table 2). In two cases (patients 1180 and 1872), DNA was available only from the patient's mother and not from the father. Since the vast majority of type-1 deletions are known to originate by NAHR during maternal meiosis,39 it is very likely that in these two cases it was the transmitting parent that was investigated. Notwithstanding that in none of these 29 parents was any deletion or duplication detected in the region of either CNV1 or CNV2.
MLPA analysis of patients with type-2 deletions and their parents
While type-1 NF1 deletions occurring during maternal meiosis usually originate via interchromosomal non-homologous recombination,39 type-2 deletions occur predominantly as mosaic deletions.32, 33, 34 In this study, we investigated 13 NF1 patients with type-2 deletions (Table 3). All exhibited somatic mosaicism, with >90% of peripheral blood cells displaying the deletion.33, 34 Once again, none of the deletion patients possessed either CNV1 or CNV2 on their normal chromosomes 17. In four of the type-2 deletion cases, we also investigated the parent on whose chromosomes the deletion had occurred (Table 3). However, none exhibited copy number variations in the NF1 gene region.
MLPA analysis of patients with atypical deletions
DNA from five patients with atypical NF1 deletions were also analysed by MLPA (Table 3). The breakpoints of four of these deletions have been reported previously,35, 36, 37 while the deletion in patient 1860 was characterized here for the first time (see paragraph below). Copy number variations were not detected in either patients 552, 619, 442 and BUD or the transmitting parents. However, in the peripheral blood of patient 1860, MLPA revealed not only a large NF1 deletion but also a duplication within the regions overlapping CNV2. MLPA analysis of peripheral blood from the mother of patient 1860 indicated that she possessed the same deletion/duplication rearrangement. Thus, we infer that both the deletion and the duplication of the adjacent segment are located on the same chromosome. Genotyping of polymorphic markers using DNA isolated from peripheral blood was not suggestive of somatic mosaicism in the mother (Supplementary Figure 1). The mother of patient 1860 was not available for investigation so we were unable to determine whether she had signs of NF1 or not.
Characterization of the deletion and duplication in patient 1860
Fluorescent in situ hybridization analysis with BAC RP11-142O6 indicated a large deletion in 100% of the cultured blood cells (N=50) from patient 1860. The extent of this deletion was determined by array CGH. The log 2 intensity ratios averaged in 4-kb windows are given in Supplementary Table 2. Accordingly, the proximal deletion breakpoint mapped somewhere between positions 25 966 000 and 25 974 000, while the distal deletion breakpoint was localized between 27 278 000 and 27 282 000 (nucleotide numbering according to the hg18 assembly of chromosome 17, NCBI build 36). To refine the deletion breakpoint positions, PCR was performed with the primers listed in Supplementary Tables 3 and 4 using DNA from a somatic cell hybrid containing only the deleted chromosome 17 from the patient. The presence or absence of the resulting PCR products served to indicate whether the respective regions tagged by these PCRs were deleted or not. Sequencing of the PCR products from the proximal breakpoint region confirmed their origin. The proximal breakpoint was localized to within a 239-bp region (between positions 25 972 442 and 25 972 681). The distal breakpoint was mapped to a 922-bp segment (between positions 27 279 573 and 27 280 495) (Supplementary Tables 3 and 4).
In addition to the deletion, array CGH and MLPA both indicated a duplication in patient 1860 adjacent to the deletion (Figure 2; Supplementary Table 2). According to the array CGH results, the duplication in patient 1860 spans ∼96 kb encompassing the region between nucleotide positions 27 294 000–27 298 000 and 27 390 000–27 394 000 (Supplementary Table 2). Remarkably, the duplication in patient 1860 overlaps with the CNV2 region.
Using Whole Genome TilePath arrays comprising 26 574 large-insert clones (30K TPA clone set), Redon et al14 detected CNV2 in only one African man from the Yoruba tribe among 270 HapMap individuals (DNA sample ID: GM18501). We confirmed the duplication (CNV2) in this Yoruba sample by MLPA (Supplementary Table 5). However, the precise extent of the duplication in this individual could not be determined by MLPA. To obtain more information about the size of the CNV2, we reexamined the array CGH results of Redon et al.14 Increased log 2 ratios indicative of a duplication were noted for two BAC clones, Chr17tp-11E3 and Chr17tp-10C1. According to the positions of these BACs, CNV2 should maximally extend from nucleotide positions 27 245 834 to 27 562 095 (Figure 2). However, CNV2 could be considerably smaller since the overlapping BACs (Chr17tp-11H4 and Chr17tp-3G8), which are also part of the 30K TPA clone set, did not indicate a duplication. Since BAC Chr17tp-10C1 exhibited increased log 2 ratios, CNV2 should extend at least to position ∼27 450 000. The distal duplication boundary in patient 1860 is, however, located between positions 27 390 000 and 27 394 000. Thus, the distal duplication boundary of CNV2 and the duplication identified in patient 1860 may be separated by ∼50 kb.
Frequency of CNV1 and CNV2
In addition to the patients with gross NF1 gene deletions and their parents, we analysed genomic DNA samples from 36 healthy donors of West European origin by MLPA. However, no deletions or duplications suggestive of the presence of CNV1 or CNV2 were apparent (Table 4). Wong et al15 observed CNV1 in 6 out of 95 individuals investigated, a frequency of 3% (95% CI: 1.2–6.7%) (6 CNVs per 190 chromosomes). However, using MLPA, we screened a total of 167 chromosomes (Table 4) for CNV1 and failed to detect any.
Discussion
Non-allelic homologous recombination between segmental duplications can give rise to either the deletion or duplication of the region between the repeats. Owing to a combination of their high degree of homology and the local chromatin organization, such segmental duplications are prone to aberrant recombination. If dosage-sensitive genes are encompassed by the consequent rearrangements, genomic disorders can arise.42, 43, 44, 45 Other apparently homology-independent mechanisms like non-homologous end-joining (NHEJ) also operate in regions predisposing to genomic disorders and can give rise to deletions or duplications with non-recurrent breakpoints.46, 47 Importantly, many segmental duplications flanking the regions rearranged in genomic disorders have a complex structure. These SDs are not simple directly contiguous repeats but instead comprise several sub-repeats with non-duplicated sequences interspersed between different repeated sequences.29, 30, 48, 49, 50, 51, 52 This modular structure is not simply restricted to those segmental duplications involved in genomic disorders but is rather a common feature of segmental duplications in general. It would appear that many segmental duplications constitute unstable genomic regions formed by frequent sequence transfer during recent primate/human genome evolution.51, 53 This evolutionary plasticity is consistent with the observation that segmental duplications are frequent sites of copy number variation.14 Remarkably, the breakpoint regions for 12 of 25 loci involved in genomic disorders, including DiGeorge, Smith–Magenis, Williams–Beuren and Prader–Willi/Angelman syndromes have been found to be highly polymorphic.14 Furthermore, CNVs have also been identified within the regions rearranged in these genomic disorders. Taken together, these findings imply that structural polymorphic variation in regions involved in genomic disorders facilitates NAHR. There are several examples of polymorphic inversions, which predispose to genomic disorders, the former being found at increased frequency in the transmitting parents of patients with Williams–Beuren syndrome, Angelman syndrome and Sotos syndrome as compared with the frequency of the respective inversions in the general population.54, 55, 56 Mispairing during parental meiosis mediated by these inversions in heterozygous form is considered to be responsible for triggering deletion formation.
By analogy with inversions, heterozygously occurring CNVs might also have the potential to give rise to unpaired chromosomal regions. Alternatively, the putatively inherent instability of some CNVs might facilitate genomic rearrangements. In the NF1 gene region, two different sites of CNV have been reported: CNV1 involves the NF1 gene itself whereas CNV2 encompasses the SUZ12 gene, the LRRC37B gene in the distal NF1-REPc repeat and the RHOT1 gene (Table 1; Figures 1 and 2). We used MLPA to ascertain the frequency of these CNVs in patients with NF1 deletions, unaffected controls and transmitting but unaffected parents of patients with type-1 deletions, the most common of the gross NF1 gene deletions.25, 26, 27, 31, 33
CNV2
In none of the 20 transmitting parents of patients with type-1 deletions was CNV2 detected (Table 2). It would therefore seem rather unlikely that CNV2, which encompasses the distal NF1-REPc repeat, could be a frequent trigger of NAHR leading to type-1 deletions. CNV2 was also not detected in 9 non-transmitting parents and 36 healthy donors (Table 4). Finally, we investigated patients with type-2 and atypical NF1 deletions as well as their transmitting parents (Table 3). Only in the atypical deletion patient (1860) did we observe a duplication in the region of CNV2. Patient 1860 possesses a 1.3-Mb deletion, whose proximal breakpoint is located within the NF1-REPa repeat, whereas the distal deletion breakpoint is located 7–8 kb proximal to the SUZ12 gene. Interestingly, the deletion is directly adjacent to the duplication and appears to have occurred on the same chromosome, since the deletion and duplication were both noted in the mother of patient 1860 and hence must have been co-inherited.
Since the duplication observed in patient 1860 and her mother is at least 50 kb smaller than the CNV2 observed by Redon et al,14 it may well be that the duplication and deletion events occurred simultaneously as part of the same complex rearrangement. However, we cannot exclude the possibility that CNV2 was present prior to the deletion in one of the maternal grandparents and could subsequently have facilitated deletion formation.
CNV1
According to the data presented by Wong et al,15 the NF1 gene should constitute a region of fairly frequent copy number variation (CNV1; Figure 1). Investigating 95 normal individuals by array CGH (N=190 chromosomes), they reportedly observed five losses and one gain, suggesting a frequency of 3% CNV1 per chromosome.
However, employing MLPA, we failed to observe any gains or losses in the CNV1 region in a total of 167 chromosomes investigated (P=0.03, two-tailed Fisher's exact test) (Table 4). Thus, our study did not confirm the existence of frequent copy number variation within the NF1 gene region. One explanation for these discrepant findings could be that CNV1 was artefactual in origin. Indeed, it is possible that the numerous pseudogenes of the NF1 gene, in particular those in the pericentromeric region of chromosome 15, which are already known to be polymorphic in copy number,57 could have been responsible for spurious/false-positive array CGH results. However, the screening of a larger number of healthy probands is necessary to exclude unequivocally the existence of CNV1 as a rare variant. Furthermore, it could be that the duplication underlying CNV1 is a highly divergent copy of the NF1 gene, and that this copy was undetectable by MLPA as a consequence of the presence of paralogous sequence variants at sites bound by the MLPA oligonucleotides. Currently, some 6559 CNVs are registered in the Human Genome Segmental Duplication Database (http://www.projects.tcag.ca/humandup/), one among them is CNV1. Our findings suggest that great care should be taken with regard to potential false positives among CNVs detected exclusively by array CGH without confirmation by a second technique. Since it is estimated that many of the CNVs are rare variants rather than common polymorphisms,20 it would appear that the systematic and independent validation of all CNVs hitherto reported is urgently required. A quantitative locus-specific analysis, such as that performed in this study using MLPA, is critically important to discriminate between bona fide CNVs and false-positive CNVs arising due to interference from paralogous loci.
References
Eichler EE : Widening the spectrum of human genetic variation. Nat Genet 2006; 38: 9–11.
Feuk L, Carson AR, Scherer SW : Structural variation in the human genome. Nat Rev Genet 2006; 7: 85–97.
Freeman JL, Perry GH, Feuk L et al: Copy number variation: new insights in genome diversity. Genome Res 2006; 16: 949–961.
Shianna KV, Willard HF : Human genomics: in search of normality. Nature 2006; 444: 428–429.
Beckmann JS, Estivill X, Antonarakis SE : Copy number variants and genetic traits: closer to the resolution of phenotypic to genotypic variability. Nat Rev Genet 2007; 8: 639–646.
Kehrer-Sawatzki H : What a difference copy number variation makes. BioEssays 2007; 29: 311–313.
Shelling AN, Ferguson LR : Genetic variation in human disease and a new role for copy number variants. Mutat Res 2007; 622: 33–41.
Iafrate AJ, Feuk L, Rivera MN et al: Detection of large-scale variation in the human genome. Nat Genet 2004; 36: 949–951.
Sebat J, Lakshmi B, Troge J et al: Large-scale copy number polymorphism in the human genome. Science 2004; 305: 525–528.
Sharp AJ, Locke DP, McGrath SD et al: Segmental duplications and copy number variation in the human genome. Am J Hum Genet 2005; 77: 78–88.
Goidts V, Cooper DN, Armengol L et al: Complex patterns of copy number variation at sites of segmental duplications: an important category of structural variation in the human genome. Hum Genet 2006; 120: 270–284.
Fiegler H, Redon R, Andrews D : Accurate and reliable high-throughput detection of copy number variation in the human genome. Genome Res 2006; 16: 1566–1574.
Khaja R, Zhang J, Macdonald JR et al: Genome assembly comparison identifies structural variants in the human genome. Nat Genet 2006; 38: 1413–1418.
Redon R, Ishikawa S, Fitch KR et al: Global variation in copy number in the human genome. Nature 2006; 444: 444–454.
Wong KK, Deleeuw RJ, Dosanjh NS et al: A comprehensive analysis of common copy-number variations in the human genome. Am J Hum Genet 2007; 80: 91–104.
Conrad DF, Andrews TD, Carter NP, Hurles ME, Pritchard JK : A high-resolution survey of deletion polymorphism in the human genome. Nat Genet 2006; 38: 75–81.
Hinds DA, Kloek AP, Jen M, Chen X, Frazer KA : Common deletions and SNPs are in linkage disequilibrium in the human genome. Nat Genet 2006; 38: 82–85.
McCarroll SA, Hadnott TN, Perry GH, et al, The International HapMap Consortium: Common deletion polymorphisms in the human genome. Nat Genet 2006; 38: 86–92.
Smith AJ, Tsalenko A, Sampas N et al: Array CGH analysis of copy number variation identifies 1284 new genes variant in healthy white males: implications for association studies of complex diseases. Hum Mol Genet 2007; 16: 2783–2794.
Zogopoulos G, Ha KC, Naqib F et al: Germ-line DNA copy number variation frequencies in a large North American population. Hum Genet 2007; 122: 345–353.
Tuzun E, Sharp AJ, Bailey JA et al: Fine-scale structural variation of the human genome. Nat Genet 2005; 37: 727–732.
Cnossen MH, van der Est MN, Breuning MH et al: Deletions spanning the neurofibromatosis type 1 gene: implications for genotype–phenotype correlations in neurofibromatosis type 1? Hum Mutat 1997; 9: 458–464.
Rasmussen SA, Colman SD, Ho VT et al: Constitutional and mosaic large NF1 gene deletions in neurofibromatosis type 1. J Med Genet 1998; 35: 468–471.
Kluwe L, Siebert R, Gesk S et al: Screening of 500 unselected neurofibromatosis 1 patients for deletions of the NF1 gene. Hum Mutat 2004; 23: 111–116.
Dorschner MO, Sybert VP, Weaver M, Pletcher BA, Stephens K : NF1 microdeletion breakpoints are clustered at flanking repetitive sequences. Hum Mol Genet 2000; 9: 35–46.
Jenne DE, Tinschert S, Reimann H et al: Molecular characterization and gene content of breakpoint boundaries in patients with neurofibromatosis type 1 with 17q11.2 microdeletions. Am J Hum Genet 2001; 69: 516–527.
López-Correa C, Dorschner M, Brems H et al: Recombination hotspot in NF1 microdeletion patients. Hum Mol Genet 2001; 10: 1387–1392.
Jenne DE, Tinschert S, Dorschner MO, Hameister H, Stephens K, Kehrer-Sawatzki H : Complete physical map and gene content of the human NF1 tumor suppressor region in human and mouse. Genes Chromosomes Cancer 2003; 37: 111–120.
De Raedt T, Brems H, Lopez-Correa C, Vermeesch JR, Marynen P, Legius E : Genomic organization and evolution of the NF1 microdeletion region. Genomics 2004; 84: 346–360.
Forbes SH, Dorschner MO, Le R, Stephens K : Genomic context of paralogous recombination hotspots mediating recurrent NF1 region microdeletion. Genes Chromosomes Cancer 2004; 41: 12–25.
De Raedt T, Stephens M, Heyns I : Conservation of hotspots for recombination in low-copy repeats associated with the NF1 microdeletion. Nat Genet 2006; 38: 1419–1423.
Petek E, Jenne DE, Smolle J et al: Mitotic recombination mediated by the JJAZF1 (KIAA0160) gene causing somatic mosaicism and a new type of constitutional NF1 microdeletion in two children of a mosaic female with only few manifestations. J Med Genet 2003; 40: 520–525.
Kehrer-Sawatzki H, Kluwe L, Sandig C et al: High frequency of mosaicism among patients with neurofibromatosis type 1 (NF1) with microdeletions caused by somatic recombination of the JJAZ1 gene. Am J Hum Genet 2004; 75: 410–423.
Steinmann K, Cooper DN, Kluwe L et al: Type-2 NF1 deletions are highly unusual by virtue of the absence of non-allelic homologous recombination hotspots and an apparent preference for female mitotic recombination. Am J Hum Genet 2007; 81: 1201–1220.
Kehrer-Sawatzki H, Kluwe L, Funsterer C, Mautner VF : Extensively high load of internal tumors determined by whole body MRI scanning in a patient with neurofibromatosis type 1 and a non-LCR-mediated 2-Mb deletion in 17q11.2. Hum Genet 2005; 116: 466–475.
Kehrer-Sawatzki H, Schmid E, Fünsterer C, Kluwe L, Mautner VF : Absence of cutaneous neurofibromas in an NF1 patient with an atypical deletion partially overlapping the classical 1.4 Mb microdeleted region. Am J Med Genet 2007, (in press).
Mantripragada KK, Thuresson AC, Piotrowski A et al: Identification of novel deletion breakpoints bordered by segmental duplications in the NF1 locus using high resolution array-CGH. J Med Genet 2006; 43: 28–38.
Lander ES, Linton LM, Birren B, et al, International Human Genome Sequencing Consortium: Initial sequencing and analysis of the human genome. Nature 2001; 409: 860–921.
López-Correa C, Brems H, Lazaro C, Marynen P, Legius E : Unequal meiotic crossover: a frequent cause of NF1 microdeletions. Am J Hum Genet 2000; 66: 1969–1974.
Wimmer K, Yao S, Claes K et al: Spectrum of single- and multiexon NF1 copy number changes in a cohort of 1100 unselected NF1 patients. Genes Chromosomes Cancer 2006; 45: 265–276.
Kehrer-Sawatzki H, Tinschert S, Jenne DE : Heterogeneity of breakpoints in non-LCR-mediated large constitutional deletions of the 17q11.2 NF1 tumour suppressor region. J Med Genet 2003; 40: E116.
Inoue K, Lupski JR : Molecular mechanisms for genomic disorders. Annu Rev Genomics Hum Genet 2002; 3: 199–242.
Stankiewicz P, Inoue K, Bi W et al: Genomic disorders: genome architecture results in susceptibility to DNA rearrangements causing common human traits. Cold Spring Harb Symp Quant Biol 2003; 68: 445–454.
Stankiewicz P, Lupski JR : Genome architecture, rearrangements and genomic disorders. Trends Genet 2002; 18: 74–82.
Lupski JR : Hotspots of homologous recombination in the human genome: not all homologous sequences are equal. Genome Biol 2004; 5: 242.
Shaw CJ, Lupski JR : Implications of human genome architecture for rearrangement-based disorders: the genomic basis of disease. Hum Mol Genet 2004; 13 (Spec no 1): R57–R64.
Lupski JR, Stankiewicz P : Genomic disorders: molecular mechanisms for rearrangements and conveyed phenotypes. PLoS Genet 2005; 1: E49.
Amos-Landgraf JM, Ji Y, Gottlieb W et al: Chromosome breakage in the Prader–Willi and Angelman syndromes involves recombination between large, transcribed repeats at proximal and distal breakpoints. Am J Hum Genet 1999; 65: 370–386.
Shaikh TH, Kurahashi H, Saitta SC et al: Chromosome 22-specific low copy repeats and the 22q11.2 deletion syndrome: genomic organization and deletion endpoint analysis. Hum Mol Genet 2000; 9: 489–501.
Park SS, Stankiewicz P, Bi W et al: Structure and evolution of the Smith–Magenis syndrome repeat gene clusters, SMS-REPs. Genome Res 2002; 12: 729–738.
Antonell A, de Luis O, Domingo-Roura X, Perez-Jurado LA : Evolutionary mechanisms shaping the genomic structure of the Williams–Beuren syndrome chromosomal region at human 7q11.23. Genome Res 2005; 15: 1179–1188.
Kurotaki N, Stankiewicz P, Wakui K, Niikawa N, Lupski JR : Sotos syndrome common deletion is mediated by directly oriented subunits within inverted Sos-REP low-copy repeats. Hum Mol Genet 2005; 14: 535–542.
Samonte RV, Eichler EE : Segmental duplications and the evolution of the primate genome. Nat Rev Genet 2002; 3: 65–72.
Osborne LR, Li M, Pober B et al: A 1.5 million-base pair inversion polymorphism in families with Williams–Beuren syndrome. Nat Genet 2001; 29: 321–325.
Gimelli G, Pujana MA, Patricelli MG et al: Genomic inversions of human chromosome 15q11-q13 in mothers of Angelman syndrome patients with class II (BP2/3) deletions. Hum Mol Genet 2003; 12: 849–858.
Visser R, Shimokawa O, Harada N et al: Identification of a 3.0-kb major recombination hotspot in patients with Sotos syndrome who carry a common 1.9-Mb microdeletion. Am J Hum Genet 2005; 76: 52–67.
Barber JC, Cross IE, Douglas F, Nicholson JC, Moore KJ, Browne CE : Neurofibromatosis pseudogene amplification underlies euchromatic cytogenetic duplications and triplications of proximal 15q. Hum Genet 1998; 103: 600–607.
Acknowledgements
This work was supported by the Deutsche Krebshilfe Grant no. 106982.
Author information
Authors and Affiliations
Corresponding author
Additional information
Supplementary Information accompanies the paper on European Journal of Human Genetics website (http://www.nature.com/ejhg)
Rights and permissions
About this article
Cite this article
Steinmann, K., Kluwe, L., Cooper, D. et al. Copy number variations in the NF1 gene region are infrequent and do not predispose to recurrent type-1 deletions. Eur J Hum Genet 16, 572–580 (2008). https://doi.org/10.1038/sj.ejhg.5202002
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.ejhg.5202002
Keywords
This article is cited by
-
Sex-specific recombination patterns predict parent of origin for recurrent genomic disorders
BMC Medical Genomics (2021)
-
Identification of an atypical microdeletion generating the RNF135-SUZ12 chimeric gene and causing a position effect in an NF1 patient with overgrowth
Human Genetics (2017)
-
Emerging genotype–phenotype relationships in patients with large NF1 deletions
Human Genetics (2017)
-
Non-coding RNA ANRIL and the number of plexiform neurofibromas in patients with NF1microdeletions
BMC Medical Genetics (2012)
-
Unambiguous molecular detections with multiple genetic approach for the complicated chromosome 22q11 deletion syndrome
BMC Medical Genetics (2009)
