
Abstract
Intraspecific genetic variation in three non-coding chloroplast DNA (cpDNA) regions (trnT-L and trnL-F spacers, and trnL intron) of Japanese beech (Fagus crenata Blume) was investigated. This species is a major constituent of the typical cool–temperate deciduous forests in Japan. Twenty-one F. crenata populations from throughout Japan, and four F. japonica populations, a close relative of F. crenata, were examined. Seven haplotypes were distinguishable in F. crenata based on nucleotide substitutions and indels. Pairwise nucleotide diversities among haplotypes ranged from 0.0000 to 0.0042 for F. crenata, including F. japonica. The geographical distribution of cpDNA haplotypes was found to be highly structured in F. crenata. Four haplotypes predominated: haplotypes FC1 and FC4 are prevalent on the Pacific Ocean coast, haplotype FC6 is prevalent on the Japan sea coast from the San-in district to Hokkaido, whilst haplotype FC3 is restricted to northern Kyushu and the western-most part of Honshu. Two haplotypes (FC5 and FC7) are restricted to single populations and one haplotype (FC2) is a derivative of FC1. Each of these haplotypes, except FC2, are thought to be derived from different glacial refugia. Phylogenetic analysis showed that neither F. crenata nor F. japonica was monophyletic for the haplotypes, suggesting either ancestral polymorphism or ancient introgression between the lineages of these two Fagus species.
Similar content being viewed by others


Introduction
Japanese beech, Fagus crenata Blume, is widely distributed throughout Japan. This species often dominates typical cool–temperate deciduous forests, and together with Japanese oak (Quercus mongolica Fisch. ex Turcz var. grosseserrata (Blume) Rehder & EH Wilson) forms a major vegetation class, the so-called beech-oak forests. The northern-most F. crenata population is found at the Kuromatsunai lowland on the Oshima Peninsula in Hokkaido. It grows at sea level to 900 m on the Oshima Peninsula and to 1400 m in northeastern Honshu. On the other hand, beech forests can be found in mountainous areas at heights of more than 800 m in southwestern Japan and are often isolated in small populations. The southern-most population is at Mt Takakuma on the Osumi Peninsula in Kyushu. Due to the limited use of the wood in the past, many forests still remain in a natural condition, although rapid fragmentation is occurring throughout Japan, especially due to human exploitation.
Two endemic Fagus species occur in Japan, F. crenata and F. japonica. Fagus japonica is located mainly on the Pacific Ocean side of Japan and is distributed at lower altitudes than F. crenata, although these two species sometimes form mixed forests. Intraspecific allozyme variation has been examined in Japanese beech (Takahashi et al, 1994; Tomaru et al, 1997); within-population variation was detected but among-population genetic differentiation was small. Despite low differentiation among populations, significant correlations have been found between the clinal distribution of allele frequencies and both the latitudinal and longitudinal ranges of the species (Tomaru et al, 1997). Much greater genetic differentiation has been revealed by RFLP analysis of mitochondrial DNA (mtDNA), showing that populations are divided into several regional groups throughout Japan (Koike et al, 1998; Tomaru et al, 1998). Palynological studies have suggested that the full glacial distribution of Fagus was restricted to coastal regions of the Japanese Archipelago, south of 38°N (Tsukada, 1982a, b). Rapid expansion of Fagus vegetation was thought to have occurred simultaneously toward the interior and north of Japan about 12000 years ago. Beech forests became established from the San-in district to northern Honshu about 10000 years ago; however, at around the same time, they began to retreat from the lowlands and moved to higher altitude areas. The present distribution of Fagus forests in the northern part of Japan and their altitudinal limits were estimated to have been established about 7000 years ago (Tsukada, 1982b). The phylogeographical distribution of mtDNA during the late-Quaternary period was discussed by Tomaru et al (1998) and Koike et al (1998).
A relatively slow rate of chloroplast DNA (cpDNA) sequence change has been reported in angiosperms (Banks and Birky, 1985; Palmer, 1987; Wolfe et al, 1989). The synonymous substitution rate in the nuclear genes is about four times that in cpDNA (Wolf et al, 1987). However, recent studies in Quercus (Ferris et al, 1997; Petit et al, 1997), Liriodendron (Swell et al, 1996) and alpine plants (Fujii et al, 1995, 1996) have revealed intraspecific cpDNA variation. CpDNA is predominantly maternally inherited in angiosperms (Corriveau and Coleman, 1988), including oaks (Dumolin et al, 1995). Thus greater genetic differentiation among subpopulations is expected because of low gene flow via seeds and the haploid nature of the genome (Ennos, 1994). Analysis of the geographical distribution of cytoplasmic genetic markers in trees has thus enabled the present distribution of forests to be related to postglacial migration routes from different refugia (Newton et al, 1999). Chloroplast genomes are considered to be much more stable than mitochondrial genomes, which frequently show structural rearrangements and celluler heteroplasmy (Newton, 1988). Consequently, cpDNA is considered to be more suitable for phylogeographical analysis. The reported faster evolution of cpDNA, compared with mtDNA, in higher plants at synonymous sites (Wolfe et al, 1989) should also increase resolution in phylogeographical analysis.
In the view of its importance as a major component of the cool–temperate deciduous forest ecosystem in Japan, we intended to clarify the genetic structure of the present populations of F. crenata using cpDNA markers. Length variation in amplified fragments of three non-coding cpDNA regions are examined and the geographical structure of the variation investigated.
Materials and methods
Plant material
Fresh leaves or winter buds were collected from 488 trees in 21 populations of F. crenata. An additional four trees were sampled from four populations of F. japonica (Table 1 and Figure 1). Samples were obtained from trees separated by 30 m, to avoid collecting half sibs, and immediately processed for DNA extraction or frozen at −20°C until needed.

Location of sampled Fagus crenata and F. japonica populations. Numbers correspond to population numbers in Table 1.
DNA extraction
Total genomic DNA was extracted using the CTAB method of Doyle and Doyle (1990) with minor modifications. DNA samples were stored at 4°C until used. Besides samples for DNA extraction, additional leaves and buds were kept for voucher specimens, which are deposited in the herbarium of Forest Resources Department, Ehime University.
PCR amplification
Three non-coding cpDNA regions were amplified using the universal primers reported by Taberlet et al (1991). The primer pairs denoted a-b, c-d and e-f by Taberlet et al (1991) amplify the spacer between the trnT(UGU) and trnL(UAA) 5′ exon (ie the trnT-L spacer), the intron of trnL(UAA) 5′ exon and the trnL(UAA) 3′ exon (ie the trnL intron), and the spacer between the trnL(UAA) 3′ exon and the trnF(GAA) exon (ie the trnL-F spacer), respectively. Five ng genomic DNA was subjected to 30 cycles of amplification (preheating for 2 min at 94°C, cycles of 30 sec at 94°C, 30 sec at 51°C and 1 min at 72°C) using Gene Amp PCR systems 2400 and 9700 (PE Biosystems). The amplified products were subjected to final extension for 5 min at 72°C, and then kept at 4°C. The products were then purified by ethanol precipitation and separated on agarose gels to determine DNA concentration and length variation.
Sequencing of three non-coding regions of cpDNA
PCR products were directly sequenced using the dye terminator method (Murray, 1989) without subcloning. The cycle sequencing reaction was performed using a BigDye terminator cycle sequencing FS kit (PE Biosystems). 50 ng template DNA was subjected to 25 cycles of amplification (preheating for 2 min at 96°C, cycles of 30 sec at 96°C, 30 sec at 50°C and 4 min at 60°C) using Gene Amp PCR systems. The universal primers for amplifying the three non-coding regions were used as the forward and reverse sequencing. Labelled products were purified by ethanol precipitation and then applied to an ABI PRISM 310 Genetic Analyzer (PE Biosystems).
Nucleotide sequence analysis and phylogeny construction
Sequence data were aligned using Clustal X (Thompson et al, 1997) and further corrected using SeqPup v. 0.6 (developed by D Gilbert, University of Indiana and available at http://iubio.bio.indiana.edu/soft/molbio/seqpup). A phylogenetic tree for F. crenata was constructed using nucleotide substitutions and indels. Maximum Parsimony (MP) trees were obtained using a branch-and-bound search with 1000 bootstrap replications using PAUP v. 3.1.1 (Swofford, 1993). Quercus mongolica var. grosseserrata was used as an out group species. American beech, F. grandifolia, and European beech, F. sylvatica were also included (gift from K Kitamura, FFPRI, Japan). Neighbour-joining (NJ) trees (Saitou and Nei, 1987) were constructed using Clustal X based on nucleotide substitutions; 1000 bootstrap replicates were performed to estimate clade robustness. Nucleotide diversity was calculated by pairwise comparison in three non-coding regions using DnaSP version 3.0 (Roza and Roza, 1999). The sequences reported here have been deposited in the DDBJ database under accession numbers AB043709-AB043717, AB043730-AB043738 and AB043751-AB043759 for F. crenata, and AB043718-AB043721, AB043739-AB043742 and AB043760-AB043763 for F. japonica, and AB066496, AB066497 and AB066498 for F. grandifolia, and AB070963, AB070964 and AB070965 for F. sylvatica, and AB043748, AB043753 and AB043758 for Q. mongolica var. grosseserrata for the trnT-L spacer, trnL intron and trnL-F spacer, respectively.
Results
Fragment length variation
For the initial analysis of length variation in the three non-coding regions, one sample was randomly chosen from each F. crenata population. Two length differences (long and short with estimated size of 805 and 747 bp, respectively) in the trnT-L spacer and three length differences (long, medium and short with estimated size of 232, 223 and 200 bp, respectively) in the trnL-F spacer were detected, but no length differences were detected in the trnL intron. Four combinations were identified among the 21 F. crenata individuals and designated as follows: Type I (long trnT-L spacer and long trnL-F spacer), Type II (long trnT-L spacer and medium trnL-F spacer), Type III (short trnT-L spacer and short trnL-F spacer) and Type IV (short trnT-L spacer and long trnL-F spacer). Subsequently, 351 individuals from 21 F. crenata populations were examined in the same way (Table 1). Fragment types are shown to be regionally aggregated (Figure 2) with populations at Mt Izumigadake (population 5), Mt Takami (population 13) and Mt Ishizuchi (population 18) being dimorphic, whilst the other populations were monomorphic. A measure of population differentiation (GST, Nei, 1973) for fragment type was calculated to be 0.95, indicating a strong geographical structure in cpDNA length variation throughout the Japanese Archipelago.

Geographical distribution of four fragment types determined by length variations in two cpDNA spacer regions of Fagus crenata. Fragment Types, I, II, III and IV correspond to long-long, long-medium, short-short and short-long combinations of the trnT-L and trnL-F spacers, respectively. Circle size is proportional to the number of individuals examined. Numbers of individual populations are given in Table 1.
Sequence analysis of non-coding regions
Nucleotide sequences of non-coding regions were determined for 24 individuals from 21 F. crenata populations. One individual was randomly chosen from each monomorphic population and two representative individuals were chosen from populations for polymorphic fragment type. In addition, nucleotide sequences of four F. japonica individuals, one from each of the four populations, were determined. Regions corresponding to exons, determined by alignment with tobacco cpDNA sequence (Shinozaki et al, 1986), were eliminated from the analysis. TrnT-L spacer length varied from 736 bp to 785 bp, whilst trnL-F spacer length varied from 165 bp to 194 bp, and trnL intron length varied from 533 bp to 536 bp. In total, seven nucleotide substitutions, three indels, and five mononucleotide repeat variations were found among the 24 F. crenata individuals. One insertion occurred in the trnT-L spacer, a tandemly repeated 43-bp sequence, and two deletions (9 bp and 28 bp) occurred in the trnL-F spacer. These indels correspond to the fragment length variation described in the previous section; the 43-bp insertion corresponds to the long trnT-L spacer, and 9 bp and 28 bp deletions correspond to the medium and short trnL-F spacers, respectively. Seven cpDNA haplotypes were distinguished based on these mutations; mononucleotide repeats were disregarded since they appeared too variable to classify, and were considered to be subtypes (Table 2). In F. japonica, five nucleotide substitutions, one insertion common to that in F. crenata, and four mononucleotide repeat length variants were found, producing four haplotypes (Table 2).
The geographical distribution of F. crenata and F. japonica haplotypes is shown in Figure 3. Types I, II, III and IV (Figure 2) corresponded to haplotypes FC1, FC3, FC4 and FC6, respectively, except for Type IV in Mt Amagi (population 10; haplotype FC5), Dantouradani (population 11; haplotype FC7) and Mt Ishizuchi (population 18; haplotype FC2). An additional nine sequences were examined for Mt Amagi and Dantouradani; all of these sequences were identical within populations. This, together with the results of fragment type analysis, suggests that most populations, except dimorphic populations (5, 13 and 18), had fixed haplotypes.

Geographical distribution of cpDNA haplotypes based on sequence analysis of three non-coding regions in two Fagus species. Numbers show haplotypes of F. crenata (Table 2). Underlined numbers are F. japonica haplotypes.
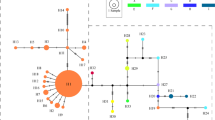
In order to clarify the genetic relationship between these haplotypes, phylogenetic trees were constructed using the MP and NJ methods (Figure 4). Sequence data were combined for the three non-coding regions. Unweighted MP analysis of 11 haplotypes with two references (F. grandifolia and F. sylvatica) and one out group sequence (Q. mongolica var. grosseserrata) resulted in one most parsimonious tree (Figure 4a; length = 86 steps, consistency index (CI) = 0.988, retention index (RI) = 0.929). Two monophyletic groups, one including FC1, FC2 and FC3 and the other including FC5, FC6 and FC7 were suggested although the bootstrap probability is not high. The position of FC4 was not resolved in this tree. Monophyly of the group with FC1, FC2 and FC3 and the group with FC5, FC6 and FC7 is better supported in the NJ tree with bootstrap probabilities 79.8% and 67.7%, respectively. Monophyly of the former group is supported by the homoplasious insertion at position 193 in the trnT-L spacer (Figure 4a). This is also supported by the observation that haplotypes in this group have a similar combination of mononucleotide repeats, A16 (or A15)-A10-A7-A5 (or A6)-T7, located at the positions 344, 638 and 738 in the trnT-L spacer, 149 in the trnL intron and 129 in the trnL-F spcer in this order (Table 2). The grouping with FC6 and FC7 is strongly supported (87% in MP tree and 86% in NJ tree). This grouping is also supported by their having the same combination of mononucleotide repeats, A10-A10-A8-A4-T7 at the positions mentioned above. There are potential problems of homoplasy with these repeats; however, the probability of coincident occurrence in five sites seems very low. FC1 is a prevalent type on the Pacific Ocean coast as well as FC4, whilst FC6 is prevalent on the Japan Sea coast from the San-in district to Hokkaido. FC3 which is closely related to FC1 is restricted in distribution to northern Kyushu and the western-most part of Honshu. Both FC5 and FC7 are phylogentically related to FC6, but are uniquely found on the Pacific Ocean side in Mt Amagi and Dantouradani, respectively. FC2 was found only at Mt Ishizuchi (population 18), a polymorphic population. FC2 has the same sequence as FC1, except for an insertion in the trnT-L spacer. This insertion was probably lost in FC2 (Figure 4a).

Phylogenetic trees constructed by the MP (a) and NJ (b) methods. FC and FJ indicate haplotypes for Fagus crenata and F. japonica, respectively. FG, FS and QM represents haplotypes of Fagus grandifolia, F. sylvatica and Quercus mongolica var. grosseserrata, respectively. Bootstrap values based on 1000 replicates are shown above branches. The vertical bars on the MP tree indicate the expected position of indels. Open bars indicate the insertion at position 193 in the trnT-L spacer, solid bars indicate the deletion at position 10 and the shaded bar indicates the deletion at position 68 in the trnL-F spacer. Checked bar indicates the position of expected loss of the insertion at position 193. Unit length of the branch in MP tree indicates one nucleotide substitution or one indel event. Seventy nucleotide substitutions were found between QM and the common ancestor of F. crenata.
Neither F. crenata nor F. japonica was monophyletic in the MP and NJ trees. For example, haplotypes FC6 and FJ4 have the same nucleotide sequence, except for mononucleotide repeats in the trnT-L spacer and suggested to be closely related. The nucleotide sequences of FC4 and FJ1 were also identical, except for the insertion in FJ1 in the trnT-L spacer, the deletion in FC4 at the position 68 in the trnL-F spacer and three mononucleotide repeats. Interestingly, FJ4 was found in Komenono in the population 19 which is remotely located from the area where FC6 was found. Similarly, FJ1 was found in Mt Osorakan in the population 16 and located distantly from where FC4 was found. This suggests that phylogenetic relationship found in the haplotypes of F. crenata and F. japonica are not correlated with their geographical distribution.
Only the F. sylvatica haplotype was included in the clade of F. crenata and F. japonica in the MP tree, whilst both F. sylvatica and F. grandifolia haplotypes were included in the clade in NJ tree. Therefore the position of these species are not clear.
Pairwise nucleotide diversities among the haplotypes of F. crenata and F. japonica ranged from 0.0000 to 0.0042 (mean 0.00220 ± 0.00026 (s.e.)) in F. crenata and 0.0000 to 0.0042 (mean 0.00195 ± 0.00020 (s.e.)) between F. crenata and F. japonica, indicating that there is no significant differentiation between these species in cpDNA variation.
Discussion
Geographical distribution of cpDNA variation
Seven cpDNA haplotypes were distinguished throughout the Japanese Archipelago and showed a strong geographical structure. Among them, FC6 prevails on the Japan Sea coast, from the San-in district northward to Hokkaido. FC6 has a boundary with FC3 in the southwest part of Japan, somewhere between Mt Hiruzen (population 15) and Mt Osorakan (population 16). The Mt Izumigadake population (population 5) appears located in the region where FC6 merges with FC4 in northeastern Honshu. The geographical distribution of haplotypes indicates that populations which expanded north along the Japan Sea coast 12 000 years ago, as inferred from palynological studies (Tsukada, 1982a, b), had haplotype FC6, whilst populations that expanded along the Pacific Ocean coast had haplotype FC4. Since the Japanese Archipelago is traversed by mountain ranges that are north–south, it may have been difficult for Fagus forests, comprising trees with relatively large seeds, to have migrated across it; rather, Fagus forests may have moved along the coasts. Yasuda (1985) pointed out that the expansion of vegetation may correspond in a change to a moist oceanic postglacial climate, following warming and an elevation in sea level during this period. Haplotypes FC1, FC2 and FC3 comprised one phylogenetically related group. FC3 is the dominant haplotype in northern Kyushu and the western-most part of Honshu, whilst FC1 is the dominant haplotype in southern Kyushu and Shikoku. Consequently, beech forests in Japan are divided into four prevailing regional groups that presumably developed from different refugia, characterised by different cpDNA haplotypes, in the last glacial period. Haplotypes FC5 and FC7 were uniquely found in Mt Amagi (population 10) and Dantouradani (population 11), respectively. These haplotypes are fixed in each population and suggest that they have arisen from independent refugia. Tomaru et al (1998) and Koike et al (1998) also reported the existence of four major population groups, with monophyletic or identical haplotypes, based on mtDNA variation: one in northern Kyushu and the western part of Honshu; one in southern Kyushu and Shikoku; one in central Japan; and the other in Tohoku and Hokkaido. The groupings are concordant with those reported here, although the phylogenetic relationships among groups are somewhat different. The Mt Amagi population also made an independent branch in the phylogeny of Tomaru et al (1998), suggesting that this population may have been derived from an old refugium in Izu Peninsula. In contrast to the cpDNA results, both Tomaru et al (1998) and Koike et al (1998) showed that populations on the Japan Sea coast of Japan from the San-in district northward to Hokkaido are not monomorphic. This suggests that the refugia assumed to have been located along the coastal region below 38°N latitude (Tsukada, 1982a, b) were polymorphic for mtDNA or that these populations originated from closely related multiple refugia, as suggested by Tomaru et al (1998).
Tsukada's palynological study (1982b) also indicated that there were expansions of beech forests 12000 years ago in the coastal regions at Osaka and Ise Bays. This may have been an extension of the forests on the Japan Sea coast or may have developed from refugia located around these areas. The route from Wakasa Bay on the Japan Sea coast to Osaka or Ise Bay, through the lowland of Lake Biwa, is probably the route for migration across Honshu. Migration of plants along this route may have occurred in the interglacial period, especially during the cooling of the climate in the past glacial ages and may have formed genetically related refugia on the Pacific side. There is only one nucleotide substitution between FC6 and FC7 (Table 2). Assuming that cpDNA nucleotide substitution rate in the Fagaceae is 2.36 ± 0.79 × 10−10 per site per year (Frascaria et al, 1993) and considering this value to be a random variable, the lower 95% confidence limit of the branching time with one nucleotide substitution in a 1463 bp sequence is estimated to be 3.7 × 104 years ago with mean divergence time of 1.63 million years ago (upper limit is 6.6 million years ago), following the method of Haubold and Wiehe (2001). This time well exceeded the last glacial-maximum. Thus, the Dantouradani population may not be derived from FC6 during the expansion of vegetation 12000 years ago, rather it originated from an independent refugia established around this area sometime before the last glacial-maximum.
Geographical variation in leaf morphology has been shown in F. crenata. Hagiwara (1977) reported that leaf length and area of F. crenata show a clinal increase from the south to north, with a latitudinal increase throughout its range. Hashizume et al (1997) investigated the variation in leaf shape among transplanted F. crenata provenances. They also found a significant latitudinal cline in leaf morphology. This suggests that the geographical cline in leaf morphology is formulated by adaptive genetic changes during vegetation expansion. However, apparently another factor exists that differentiates leaf morphology on the Japan Sea and the Pacific Ocean coasts; leaves are generally larger and thinner on the Japan Sea coast compared to the Pacific coast. Differentiation in leaf morphology apparently corresponds to the contrasting climates on the Japan Sea and the Pacific Ocean coasts of the Japanese Archipelago (Yasuda, 1985). In this respect, F. crenata has occasionally been classified into two subspecies: F. crenata var. grandifolia Nakai (large-leaf beech) on the Japan Sea coast and F. undulata Buerger ex Miq. (small-leaf beech) on the Pacific Ocean coast from the south of Kinka-zan in the Miyagi Prefecture (Satake et al, 1989). Apparently, the distribution of the large-leaf beech (Hagiwara, 1977) is almost identical to that of FC6. Therefore, the beech bearing FC6 is suggested to be the type adapted to the moist winter, and expanded rapidly northward on the Japan Sea coast, following climate change.
Polyphyly of cpDNA in F. crenata and F. japonica
Phylogenetic analysis showed that F. crenata and F. japonica cpDNA haplotypes formed a polyphyletic clade. FC6 and FJ4 showed exactly the same sequences, except for two mononucleotide repeats. Nucleotide sequences of FC4 and FJ1 were also similar. The geographical distributions of haplotypes (Figure 4) showed that those haplotypes with common sequences were not found at the same location. FJ4 was found on the Pacific coast (population 19), whilst haplotype FC6 was found on the Japan Sea coast. Based on mast peduncles, beech species have often been classified into two groups: long and short mast peduncle groups (Namiki and Okamoto, 1985). Fagus japonica belongs to the first group, whilst F. crenata belongs to the latter. Peduncle morphology is also correlated with different patterns of cotyledon folding (Namiki and Okamoto, 1985). On the other hand, Tanai (1972) discussed the relationships between modern and fossil beeches in the context of leaf morphology, such as the number of secondary veins, leaf index and marginal serration. He classified the extant species into two form-groups; F. grandifolia (American beech) group and the F. sylvatica (European beech) group. Fagus crenata belongs to the latter and F. japonica belongs to the former. He claimed that these two lineages could be found in the fossil records by the middle of Miocene (Tanai, 1974). These observations suggest that an evolutionary relationship between F. crenata and F. japonica is remote. Furthermore, no natural hybrids has been observed between these two species. Thus the possibility of introgression of cpDNA among these two extant species may be discarded. Several Fagus fossils have been recorded in Japan. According to Tanai (1972), the first fossil species described in Japan was F. antipofi, a widely recorded species in the Northern Hemisphere, from the Lower Miocene in Hokkaido. Under the somewhat warmer climate of the Middle Miocene, F. antipofi became more widely recorded in northern Japan, northeastern Korea and Saghalien (Tanai, 1972). In the Late Miocene, F. palaeocrenata appeared abundantly throughout Japan, which is similar to the European beech, F. sylvatica, and also similar to the modern Japanese beech, F. crenata. In place of F. antipofi, F. protojaponica which shows similar characters to F. japonica appeared in the Late Miocene. During a cooling of the climate in the Late Pliocene to Early Pleistocene, modern species prevailed and completely replaced fossil taxa at about 30 × 104 years ago in the Late Quaternary (Tanai, 1972). Although the relationship between extant and fossil species is unclear, occasional hybridisation among the ancestral species may have resulted in cpDNA introgression. However, the relative richness of variation found in the cpDNA non-coding regions suggests ancestral polymorphism to be more likely. In the most remote case of FC2 and FC7 with six nucleotide difference, the mean divergence time is estimated to be 9.8 × 106 years ago with a lower 95% confidence limit of 2.8 × 106 years ago, following Haubold and Wiehe (2001). This could reach to the Early Miocene, when F. antipofi, a proposed common ancestor (Tanai, 1972), prevailed in Japan. The polymorphic insertion at the position of 193 in the trnT-L spacer is found in F. sylvatica, but not found in F. grandifolia (Table 2) suggesting this insertion to have been polymorphic in the common ancestor. In order to clarify this point, further analysis of cpDNA variation including foreign species such as F. grandifolia and F. sylvatica is required.
References
Banks, JA, Birky, CW Jr (1985). Chloroplast DNA diversity is low in a wild plant. Lupinus texensis. Proc Natl Acad Sci USA, 82: 6950–6954.
Corriveau, JL, Coleman, AW (1988). Rapid screening method to detect potential biparental inheritance of plastid DNA and results for over 200 angiosperm species. Am J Bot, 75: 1443–1458.
Doyle, JJ, Doyle, JJ (1990). Isolation of plant DNA from fresh tissue. Focus, 12: 13–15.
Dumolin, S, Demesure, B, Petit, RJ (1995). Inheritance of chloroplast and mitochondrial genomes in pedunculate oak investigated with an efficient PCR method. Theor Appl Genet, 91: 1253–1256.
Ennos, RA (1994). Estimating the relative rate of pollen and seed migration among plant populations. Heredity, 72: 250–259.
Ferris, C, King, AR, Vainola, R, Hewitt, GM (1997). Chloroplast DNA recognizes three refugial sources of European oaks and suggests independent eastern and western immigrations to Finland. Heredity, 80: 584–593.
Frascaria, N, Maggia, L, Michaud, M, Bousquet, J (1993). The rbcL gene sequence from chestnut indicates a slow rate of evolution in Fagaceae. Genome, 36: 668–671.
Fujii, N, Ueda, K, Watano, Y, Shimizu, T (1995). Intraspecific sequence variation in chloroplast DNA of Primula cunefolia Ledeb. J Phytogeogr Taxon, 43: 15–24.
Fujii, N, Ueda, K, Shimizu, T (1996). Intraspecific sequence variation of chloroplast DNA in Japanese alpine plants. J Phyto-geogr Taxon, 44: 72–81.
Hagiwara, S (1977). Clines on leaf size of beech Fagus crenata. Spec Biolog Res, 1: 39–51. (in Japanese).
Hashizume, H, Lee, NH, Yamamoto, F (1997). Variation in the leaf shape of planted trees of Fagus crenata Blume among provenances. Ouyou Shinrin Kenkyu, 6: 115–118. (in Japanese).
Haubold, B, Wiehe, T (2001). Statistics of divergence times. Mol Biol Evol, 18: 1157–1160.
Koike, T, Kato, S, Shimamoto, Y, Kitamura, K, Kawano, S, Ueda, K et al (1998). Mitochondrial DNA variation follows a geographic pattern in Japanese beech species. Bot Acta, 111: 87–92.
Murray, V (1989). Improved double-strand DNA sequencing using the linear polymerase chain reaction. Nucl Acids Res, 17: 251–257.
Namiki, M, Okamoto, M (1985). Evolutionary history of Japanese Beech. In: Umehara T (ed) Bunatai Bunka. Shisaku-Sha: Tokyo. pp 65–88. (in Japanese).
Nei, M (1973). Analysis of gene diversity in subdivided populations. Proc Natl Acad Sci USA, 70: 3321–3323.
Newton, KJ (1988). Plant mitochondrial genomes: organization, expression and variation. Ann Rev Plant Physiol Plant Mol Biol, 39: 503–532.
Newton, AC, Allnutt, TR, Gillies, ACM, Lowe, AJ, Ennos, RA (1999). Molecular phylogeography, intraspecific variation and the conservation of tree species. Trends Ecol Evol, 14: 140–145.
Palmer, JD (1987). Chloroplast DNA evolution and biosystematic uses of chloroplast DNA variation. Am Nat, 130: S6–29.
Petit, RJ, Pineau, E, Demesure, B, Bacilieri, R, Ducousso, A (1997). Chloroplast DNA footprints of post-glacial recolonization by oaks. Proc Natl Acad Sci USA, 94: 9996–10001.
Roza, J, Roza, R (1999). DnaSP version 3: an integrated program for molecular population genetics and molecular evolution analysis. Bioinformatics, 15: 174–175.
Saitou, N, Nei, M (1987). The neighbor joining method: a new method for reconstructing phylogentic trees. Mol Biol Evol, 4: 406–425.
Satake, Y, Hara, H, Watari, S, Tominari, T (1989). Wild Flowers of Japan, Woody Plants. Heibonsha: Tokyo (in Japanese).
Shinozaki, K, Ohme, M, Tanaka, M, Wakasugi, T, Hayasihida, N, Matsubayashi, T et al (1986). The complete nucleotide sequence of the tobacco chloroplast genome: its organization and expression. EMBO J, 5: 2043–2049.
Swell, MM, Parks, CR, Chase, MW (1996). Intraspecific chloroplast DNA variation and biogeography of North American Liriodendron L. (Magnoliaceae). Evolution, 50: 1147–1154.
Swofford, DL (1993). PAUP. Phylogenetic Analysis Using Parsimony Version 311. The Illinois Natural History Survey: Champaign.
Taberlet, P, Gielly, L, Pautou, G, Bouvet, J (1991). Universal primer for amplification of three non-coding regions of chloroplast DNA. Plant Mol Biol, 17: 1105–1109.
Takahashi, M, Tsumura, Y, Nakamura, T, Uchida, K, Ohba, K (1994). Allozyme variation in Fagus crenata in northeastern Japan. Can J Forest Res, 2: 1071–1074.
Tanai, T (1972). Tertiary history of vegetation in Japan. In: Graham A (ed) Floristics and Paleofloristics of Asia and Eastern North America. Elsevier: Amsterdam-London-New York. pp 235–255.
Tanai, T (1974). Evolutionary trend of the genus Fagus around the Northern Pacific Basin. Birbal Sahni Inst Paleobot Spec Publ, 1: 62–83.
Thompson, JD, Gibson, TJ, Plewniak, F, Jeanmougin, F, Higgins, DG (1997). The Clustal X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucl Acids Res, 24: 4876–4882.
Tomaru, N, Mitsutsuji, T, Takahashi, M, Tsumura, Y, Uchida, K, Ohba, K (1997). Genetic diversity in Fagus crenata (Japanese beech): influence of the distributional shift during the late-Quaternary. Heredity, 78: 241–251.
Tomaru, N, Takahashi, M, Tsumura, Y, Takahashi, M, Ohba, K (1998). Intraspecific variation and phylogeographic patterns of Fagus crenata (Fagaceae) mitochondrial DNA. Am J Bot, 85: 629–636.
Tsukada, M (1982a). Late-Quaternary development of Fagus forest in the Japanese Archipelago. Jap J Ecol, 32: 113–118.
Tsukada, M (1982b). Late-Quaternary shift of Fagus distribution. Bot Mag Tokyo, 95: 203–217.
Wolfe, KH, Li, W-H, Sharp, PM (1987). Rates of nucleotide substitution vary greatly among plant mitochondrial, chloroplast, and nuclear DNA. Proc Natl Acad Sci USA, 84: 9054–9058.
Wolfe, KH, Sharp, PM, Li, WH (1989). Rates of synonymous substitution in plant nuclear genes. J Mol Evol, 29: 208–211.
Yasuda, Y (1985). Natural history and civilization of two beech forests in the east and the west. In: Umehara T (ed) Bunatai Bunka. Shisaku-sha: Tokyo. pp 29–63. (in Japanese).
Acknowledgements
We are very grateful to Prof Hidenori Tachida for his valuable comments on our manuscript. We are also grateful to the school forests of the University of Tokyo, Kyushu University, Tottori University and Kyoto University for providing some of the Fagus and Quercus samples. We also thank Keiko Kitamura, FFPRI, Japan for providing samples of F. grandifolia and F. sylvatica. We are grateful to Masato Tani, Yorihito Takayanagi, Tatsuhiko Ohkubo, Toru Hiromi, Reiji Yoneda, Koichi Kamiya, Tomotaka Konishi, Ryosuke Kozuki, Kenzo Tanaka and Seiichi Kashimura for help during sample collection.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Okaura, T., Harada, K. Phylogeographical structure revealed by chloroplast DNA variation in Japanese Beech (Fagus crenata Blume). Heredity 88, 322–329 (2002). https://doi.org/10.1038/sj.hdy.6800048
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.hdy.6800048
Keywords
This article is cited by
-
Can species distribution models and molecular tools help unravel disjunct distribution of Rhododendron arboreum?
Journal of Genetics (2021)
-
Genetic relationships among sympatric varieties of Acer mono in the Chichibu Mountains and Central Hokkaido, Japan
Journal of Forestry Research (2017)
-
Detecting east–west genetic differentiation in Castanopsis (Fagaceae) on the main islands of Japan and north–south on the Ryukyu Islands, based on chloroplast haplotypes
Plant Systematics and Evolution (2016)
-
Towards a better understanding of the Taraxacum evolution (Compositae–Cichorieae) on the basis of nrDNA of sexually reproducing species
Plant Systematics and Evolution (2015)
-
Decline in gene diversity and strong genetic drift in the northward-expanding marginal populations of Fagus crenata
Tree Genetics & Genomes (2015)
