
Abstract
Although glycogen synthase kinase-3 (GSK-3) might act as a tumor suppressor since its inhibition is expected to mimic the activation of Wnt-signaling pathway, GSK-3β may contribute to NF-κB activation in cancer cells leading to increased cancer cell proliferation and survival. Here we report that GSK-3β activity was involved in the proliferation of human ovarian cancer cell both in vitro and in vivo. Inhibition of GSK-3 activity by pharmacological inhibitors suppressed proliferation of the ovarian cancer cells. Overexpressing constitutively active form of GSK-3β induced entry into the S phase, increased cyclin D1 expression and facilitated the proliferation of ovarian cancer cells. Furthermore, GSK-3 inhibition prevented the formation of the tumor in nude mice generated by the inoculation of human ovarian cancer cells. Our findings thus suggest that GSK-3β activity is important for the proliferation of ovarian cancer cells, implicating this kinase as a potential therapeutic target in ovarian cancer.
Similar content being viewed by others


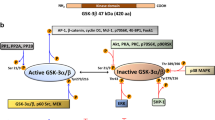
Introduction
Ovarian cancer is a leading cause of death from gynecological malignancies 1. However, the molecular basis of ovarian carcinogenesis remains poorly understood. It is therefore important to explore the molecular mechanism of the development of ovarian cancer.
Recent work in colorectal cancer, pancreatic cancer and hepatocellular carcinoma 2, 3, 4 demonstrates that glycogen synthase kinase-3β (GSK-3β) is involved in the process of tumorigenesis. Inhibition of the expression and activity of GSK-3β attenuates cell proliferation and causes apoptosis in colorectal and pancreatic cancer cells 2, 3. In contrast, activation of GSK-3β by LY294002 sensitizes hepatoma cells to chemotherapy-induced apoptosis 4. GSK-3 is an evolutionarily conserved and ubiquitously expressed serine/threonine kinase and has two homologous mammalian isoforms encoded by different genes (GSK-3α and GSK-3β; Doble BW and Woodgett 5). In addition to glycogen synthesis, from which it takes name, the GSK-3β isoform phosphorylates a number of substrates including metabolic and signaling proteins, structural proteins and transcription factors that regulate cell survival 5, 6. The regulation of GSK-3β itself is dual. Phosphorylation of human GSK-3β at tyrosine 216 is critical for efficient kinase activity, whereas GSK-3β kinase activity is inhibited through phosphorylation of serine 9 by protein kinase A, protein kinase B or protein kinase C 7, 8, 9, 10
Several reports suggest that GSK-3β is a part of a tumor suppressor complex, since it directly phosphorylates the oncoprotein β-catenin and targets it for degradation 5, 11. However, there are also reports that GSK-3β participates in the NF-κB-mediated gene transcription, which predicts that GSK-3β inactivation would decrease cell proliferation 3, 12, 13. Together, these reports prompted us to investigate the role of GSK-3β in ovarian cancer. Our findings demonstrated that GSK-3β was critical in controlling the proliferation of human ovarian cancer cells.
Materials and Methods
Materials
The antibodies against bromodeoxyuridine (BrdU) and GAPDH were from Sigma (St Louis, MO, USA), against GSK-3β, pGSK-3βSer9 and GFP from Cell Signaling Technology (Beverly, MA, USA), against cyclin D1 from Santa Cruz Biotechnology (Santa Cruz, CA, USA). The fluorescence-labeled goat anti-mouse (546) or anti-rabbit (488) IgGs were from Molecular Probes. The anti-rabbit or anti-mouse IgGs conjugated with horseradish peroxidase (HRP) were from Amersham. All other chemicals were obtained from Sigma (St Louis, MO, USA).
Cell culture and transfection
SKOV3 and ES-2 cells, two human ovarian cancer cell lines obtained from ATCC were cultured in RPMI 1640 (Gibco-BRL) supplemented with 10% heat-inactivated fetal calf serum at 37 °C in a humidified atmosphere of 5% CO2 in air. Transfection of SKOV3 was carried out by electroporation using the Amaxa Nucleofector device. Testing plasmids and pCS2, an empty vector, were cotransfected with GFP at a ratio of 3:1, to assess transfection efficiency (typically 50–80%). For stable transfection, SKVO3 cells were cultured overnight after transfection followed by the addition of G418. The G418-resistant cells were pooled 2 weeks later and cultured in the above media supplemented with G418.
Cell growth assay
Cells were seeded into a six-well plate at a density of 1×105 cells/well, followed by treatment of the cells with NaCl, dimethyl sulfoxide (DMSO), lithium chloride (LiCl) or SB216763. After 24, 48 and 72 h of treatment, cells were trypsinized and counted using a cell counter (Beckman Coulter, USA).
BrdU incorporation assay
SKOV3 cells transfected were plated onto coverslips at a density of 2×104cells/coverslip in the medium containing 10 mM BrdU. The cells were then fixed in 4% paraformaldehyde for 20 min at room temperature and rinsed with PBS before being incubated in 2 N HCl at 37 °C for 1 h. Cells were then washed three times with PBS for 5 min. After treatment with a blocking solution (PBS containing 10% goat serum) for 1 h, the cells were incubated with anti-BrdU monoclonal antibody and anti-GFP polyclonal antibody at a 1:500 dilution overnight at 4 °C and washed with PBS containing 0.1% Tween-20 for three times, followed by incubation in the dark with goat anti-mouse IgG 546 and goat anti-rabbit IgG 488 at room temperature for 2 h. The cells were then washed and mounted for observation. The cells were observed under a fluorescence microscope (Olympus). Five independent areas were imaged and used for the calculation of mean values of BrdU-incorporated cells in each condition.
Colony formation assay
SKOV3 cells transfected were incubated with 0.8 mg/ml of G418 for 7 days and 0.4 mg/ml for another 7 days. The cells were then replated at 1 000 cells/well into six-well culture dishes and left to form colonies over a period of 14 days. Cultures were stained with 0.1% crystal violet and the number of colonies in a 2×2 cm grid (on the culture plates) was scored to determine the colony-forming ability of the cells. Colonies containing >50 cells were counted.
Cell cycle analysis
Cell seeded in a 60-mm dish at a density of 1×106 cells/dish were trypsinized and washed twice with PBS. The cells were then fixed with 4% paraformaldehyde followed by the incubation in the fresh nuclei staining buffer (100 mg/ml RNase, and 50 mg/ml PI in PBS) in the dark for 1 h at 37 °C. Cell cycle histograms were generated after analysis of PI-stained cells by fluorescence-activated cell sorting (FACS) with a FACScan (Becton Dickinson). For each sample, at least 1×104events were recorded.
Western blot analysis
Cells were lysed on ice for 30 min in RIPA buffer ((in mM) 150 NaCl, 100 Tris (pH 8.0), 1% Triton X-100, 1% deoxycholic acid, 0.1% SDS, 5 EDTA, 10 NaF, 1 sodium vanadate, 2 leupeptin, 2 aprotinin, 1 phenylmethylsulfonyl fluoride and 1 dithiothreitol). The extracts were clarified by centrifugation and protein concentrations of supernatants were determined using a Bio-Rad protein assay (Bio-Rad). The proteins were separated by SDS-10% polyacrylamide gel electrophoresis, transferred to a polyvinylidene difluoride membrane and probed with the primary antibodies. The secondary antibody conjugated with HRP was then added. The protein bands were visualized with the ECLplus system (Amersham). The density of the bands was normalized relative to the controls.
In vivo experiments
Female athymic Balb/c nude mice (Slac laboratory animal Co. Ltd, Shanghai, China) aged 3-4 weeks were used in the tumor implantation model and housed in IVC cages of isolated ventilation. Experiments were approved by the Ethics Committee for Animal Experimentation of the Institute of Neuroscience according to institutional guidelines. Exponentially growing SKOV3 cells together with NaCl or LiCl (10 mM) were subcutaneously injected into the right flank of athymic nude mice (5×106 cells/mouse). Experiments were conducted in groups of three mice. Three weeks after implantation, mice were euthanized in keeping with the policy of the humane treatment of tumor-bearing animals. Tumor growth was measured by tumor diameters with a vernier caliper and by tumor weight. Tumor volume was calculated according to the formula: TV (mm3)=d2×D/2, where d and D are the shortest and the longest diameter, respectively 14.
Statistical analysis
The paired Student's t-test was used to determine the differences between groups.
Results
Expression of GSK-3β in ovarian cancer cell lines
Levels of GSK-3β and its inactive form pGSK-3βSer9 were investigated in SKOV3 and ES-2, two human ovarian cancer cell lines. SKOV3 is derived from human ovarian adenocarcinoma and ES-2 from ovarian clear cell carcinoma. As shown in Figure 1, both SKOV3 and ES-2 expressed GSK-3β. Interestingly, although the level of GSK-3β was almost identical in the two cell lines, the level of pGSK-3βSer9 in SKOV3 was lower than that in ES-2, indicating that SKOV3 may have more active form of GSK-3β, we thus used SKOV3 as a model for further studies. GSK-3β has been frequently detected in high levels in ovarian cancer tissues than in normal ovary 15, which suggests that GSK-3β may play some role in ovarian cancer development. We thus conducted the following experiments.

Expression of GSK-3β in ovarian cancer cell lines. Total protein extracts from SKOV3 and ES-2 cells were western-blotted using the antibodies against phospho-GSK-3βSer9 (inactive form of GSK-3β) and GSK-3β. GAPDH served as a loading control.
Pharmacological inhibitors of GSK-3β suppressed ovarian cancer cell growth
To assess whether GSK-3β affects the proliferation of ovarian cancer cells, we examined the effect of LiCl, a known inhibitor of GSK-3, on the growth of two human ovarian tumor cell lines, SKOV3 and ES-2. As shown in Figure 2A, the number of the cells was slightly decreased 24 h after treatment with LiCl. However, after 48 and 72 h of treatment, the growth of the cells was significantly inhibited by LiCl, whereas NaCl did not have any effect. Since lithium, frequently used as a non-competitive inhibitor of GSK-3, is not specific and displays a number of other activities 16, we then treated the cells with SB216763, another GSK-3 inhibitor 17, and examined its effect on the cell growth. Similarly, SB216763 in a dose-dependent manner dramatically inhibited SKOV3 and ES-2 cell growth (Figure 2B). These results suggested that although ES-2 cells expressed pGSK-3βSer9 in a relative high level, they did have GSK-3 activity including GSK-3β, which is sensitive to the inhibitors of GSK-3.


Inhibition of GSK-3β suppressed growth of ovarian cancer cells. Effect of LiCl (10 mM) (A) or SB216763 (B) on SKOV3 and ES-2 cell growth over the designated times was assayed by its action on the cell number. Results are means±S.D. from three independent experiments in quadruplicate. *P<0.05 and **P<0.01 versus NaCl or DMSO. (C) Effect of LiCl or SB216763 (50 μM) for 24 h on GSK-3β in SKOV3 cells was western-blotted using the indicated antibodies.
We then asked whether these inhibitors alter the expression and phosphorylation state of GSK-3β. Levels of expression and phosphorylation state of GSK-3β were detected in SKOV3 cells after treatment with each inhibitor for 24 h. As represented in Figure 2C, LiCl, but not SB216763, increased the expression level of the pGSK-3β at Ser9, whereas the level of expression of GSK-3β remained unchanged. The fact that SB216763 did not change the phosphorylation pattern of GSK-3β at Ser9 is consistent with the notion that this compound inhibits GSK-3β by competitive binding to the ATP binding site 17. We thus suggest that LiCl indeed inhibited GSK-3β activity in SKOV3 cells, possibly by phosphorylation of the kinase, as reported previously 18, 19. Taken together, our results support the idea that inhibition of GSK-3β suppressed ovarian cancer cell growth.
GSK-3β activity modulated SKOV3 cell proliferation
To further demonstrate a functional role of GSK-3β in ovarian cancer cell proliferation, we used GSK-3βS9A, a constitutively active form of GSK-3β, in which Ser9 was replaced with alanine, preventing phosphorylation and inactivation of the kinase. SKOV3 cells were cotransfected with a control GFP and GSK-3βS9A. As exemplified in Figure 3A, GSK-3βS9A significantly increased BrdU incorporation in SKOV3 cells compared with control vector. The number of BrdU-positive cells was 44.9% in SKOV3 cells transfected with GSK-3βS9A, whereas 28.5% in SKOV3 cells transfected with control vector (Figure 3B). We further transfected the SKOV3 cells with GID5-6, a peptide inhibitor of GSK-3β derived from the GSK-3β interaction domain of axin 20. The control GID5-6LP contains a single amino acid mutation rendering it unable to interact with GSK-3β 20, 21. Transfection with GID5-6 led to decrease of BrdU intake in SKOV3 cells (20% positive), compared with cells transfected with GID5-6LP (32.4% positive). These results indicated that increasing GSK-3β activity promoted proliferation, conversely, inhibition of GSK-3β activity suppressed proliferation in SKOV3 cells.

GSK-3β regulated the proliferation of ovarian cancer cells. (A) Effect of inhibition of GSK-3β on BrdU incorporation in SKOV3 cells transfected with indicated constructs. BrdU incorporation was measured by double staining of GFP (green) and BrdU (red) under the same microscopic magnification in the GFP-expressing cells. (B) Quantitative analysis of the results as shown in (A). Data are means±S.D. from three independent experiments in triplicate. *P<0.05 versus control. (C) Representative images of inhibition of GSK-3β on clonogenic formation of SKOV3 cells. SKOV3 cells transfected with the indicated plasmids were treated with G418 and stably transfected SKOV3 cells were trypsinized and plated at 1000 cells/well into six-well culture dishes and allowed to form colonies for 14 days, after which they were stained with crystal violet. (D) Quantitative analysis of the results as shown in (C). Data are means±S.D. from three independent experiments in triplicate. *P<0.05 versus ctrl. **P<0.01 versus GID5-6LP.
To evaluate the long-term effects of GSK-3β activity on the growth of SKOV3 cells, we performed colony formation assays. SKOV3 cells were transfected with the above plasmids and cultured under G418 selection. As shown in Figure 3C, GSK-3βS9A enhanced the clonogenic potential of SKOV3 cells, increasing colony formation by 2.2-fold compared with the control vector (Figure 3D). Moreover, GID5-6 led to a 2.6-fold reduction in colony formation compared with GID5-6LP (Figure 3D). These results provided additional evidence that GSK-3β activity was critical for ovarian cancer cell growth.
GSK-3β regulated G1/S phase progression accompanied by cyclin D1 expression change
The above results led us to investigate whether the inhibition or promotion of GSK-3β activity affects cell cycle progression. A typical histogram was shown in Figure 4A. The percentage of the cell population at the S phase in SKOV3 cells transfected with GSK-3βS9A was 44.9%, whereas 34.4% in SKOV3 cells transfected with GFP vector (Figure 4A). Similarly, inhibition of GSK-3β activity by GID5-6 decreased the percentage of SKOV3 cells at the S phase (Figure 4A and 4B). These results indicated that GSK-3β was important for the cell cycle entry from the G1 to S phase, and the alterations in the cell population at the S phase due to the change of GSK-3β activity was in accordance with the BrdU incorporation results.

GSK-3β controlled SKOV3 cell cycle progression. (A) Representative cell cycle histograms recorded in SKOV3 cells transfected for 72 h with the indicated plasmids. The cells expressing GFP proteins were separated and analyzed by FACS analysis. (B) Quantitative analysis of the cells at S phases as shown in (A). Data are means±S.D. from three independent experiments in triplicate. *P < 0.05 versus control. (C) Western blot analysis of cyclin D1 in SKOV3 cells transfected with the indicated plasmids. Relative level of cyclin D1 was made by the ratio of cyclin D1 to GAPDH and normalized to control.
Activation of cyclin/CDK is required for cell cycle progression and G1/S transition. The expression of cyclin D and CDK4/6 in G1 cell cycle acts as the primary sensors of positive and negative environmental signals 22, 23. To explore the mechanism by which the regulation of GSK-3β activity induced the cell cycle changes in SKOV3 cells, we analyzed the cyclin D1 expression during the process. The expression levels of cyclin D1 were inhibited by the transfection of cells with GID5-6 and increased by GSK-3βS9A, compared with that in the controls (Figure 4C). These results indicated that GSK-3β regulated cell cycle progression in ovarian cancer, which may be mediated by change in cyclin D1 expression.
LiCl inhibited ovarian cancer growth in vivo
To determine whether GSK-3 affects ovarian tumor growth in vivo, we used an ectopic xenograft model of ovarian cancer generated by SKOV3 cells. Figure 5A showed the representative image of tumor. LiCl inhibited tumor growth compared to vehicle control. Three weeks after injection, the average tumor volume in control animals was 605.5 mm3, whereas 201.6 mm3 in the LiCl group (P<0.05). The average tumor weight in control group was 405.8 mg compared to 183.7 mg in the LiCl group (Figure 5B). These data indicated that GSK-3 controlled the tumor growth of human ovarian cancer in nude mice.

LiCl inhibited tumour growth generated by human ovarian cancer xenografts. (A) Representative image of tumor specimens dissected from the nude mice xenografted with human ovarian cancer cells. (B) Quantitative analysis of the results as shown in (A). Data are means±S.D. from three independent experiments in triplicate. *P<0.05 versus control.
Discussion
We have assessed the role of GSK-3β in the regulation of cell proliferation in ovarian cancer. The results described above identify a novel role for GSK-3β in regulating ovarian cancer cell proliferation via control of cell cycle progression and possibly cyclin D1 expression. Our results demonstrate that GSK-3β plays an important role in ovarian cancer cell proliferation. Our data are in agreement with recent results for the regulation of cell proliferation by GSK-3β through an NF-κB-dependent pathway in pancreatic tumor 3. It is interesting since NF-κB has been suggested to be the substrate of GSK-3β. It remains to be determined whether GSK-3β affects cell proliferation by NF-κB-mediated gene transcription in ovarian cancer.
One function of GSK-3β is to phosphorylate and inhibit glycogen synthase activity, resulting in a relative increase of glucose metabolism 5, 24. It is well known that malignant cells have accelerated metabolism and high glucose requirements 25, and that tumors consistently rely on anaerobic pathways to convert glucose to ATP even in the presence of abundant oxygen 26, and that tumor cells maintain ATP production by increasing glucose influx to fuel the energy requirements of unrestricted proliferation 25. Thus, activated GSK-3β in ovarian cancer may contribute to high consumption of glucose through inhibition of glycogen synthesis, thereby promoting ovarian cancer cell proliferation.
One of the toughest questions when dealing with ovarian cancer is its resistance to chemotherapy through multiple mechanisms including activation of NF-κB, which might result in lower cell killing and drug resistance 27, 28, 29. Further, increased expression of cyclin D1 has also been reported to contribute to chemotherapy resistance 30. Our results that GSK-3β increased cyclin D1 expression in ovarian cancer cells support a possibility that GSK-3β is involved in chemotherapy resistance. Therefore, it is possible that combination of traditional chemotherapy and GSK-3β inhibitor would benefit ovarian cancer patient survival.
In conclusion, our results show that GSK-3β promotes ovarian cancer cell proliferation, and that cyclin D1 may be involved in the regulation; however, the precise mechanisms still remain unclear. Further studies are needed to determine the molecular pathways that contribute to the regulation, which may provide us novel strategies targeting this kinase for the treatment of ovarian cancer.
References
Barnes MN, Grizzle WE, Grubbs CJ, Partridge EE . Paradigms for primary prevention of ovarian carcinoma. CA Cancer J Clin 2002; 52:216–225.
Shakoori A, Ougolkov A, Yu ZW, et al. Deregulated GSK3β activity in colorectal cancer: its association with tumor cell survival and proliferation. Biochem Biophys Res Commun 2005; 334:1365–1373.
Ougolkov AV, Fernandez-Zapico ME, Savoy DN, Urrutia RA, Billadeau DD . Glycogen synthase kinase-3β participates in nuclear factor κB-mediated gene transcription and cell survival in pancreatic cancer cells. Cancer Res 2005; 65:2076–2081.
Beurel E, Kornprobst M, Blivet-Van Eggelpoel MJ, et al. GSK-3beta reactivation with LY294002 sensitizes hepatoma cells to chemotherapy-induced apoptosis. Int J Oncol 2005; 27:215–222.
Doble BW, Woodgett JR . GSK-3: tricks of the trade for a multitasking kinase. J Cell Sci 2003; 116:1175–1186.
Eldar-Finkelman H . Glycogen synthase kinase 3: an emerging therapeutic target. Trends Mol Med 2002; 8:126–132.
Fang X, Yu S, Lu Y, et al. Phosphorylation and inactivation of glycogen synthase kinase 3 by protein kinase A. Proc Natl Acad Sci U S A 2000; 97:11960–11965.
Li M, Wang X, Meintzer MK, et al. Cyclic AMP promotes neuronal survival by phosphorylation of glycogen synthase kinase 3β. Mol Cell Biol 2000; 20:9356–9363.
Fang X, Yu S, Tanyi JL, et al. Convergence of multiple signaling cascades at glycogen synthase kinase 3: edg receptor-mediated phosphorylation and inactivation by lysophosphatidic acid through a protein kinase C-dependent intracellular pathway. Mol Cell Biol 2002; 22:2099–2110.
Goode N, Hughes K, Woodgett JR, Parker PJ . Differential regulation of glycogen synthase kinase-3 beta by protein kinase C isotypes. J Biol Chem 1992; 267:16878–16882.
Barker N, Clevers H . Catenins, Wnt signaling and cancer. Bioessays 2000; 22:961–965.
Hoeflich KP, Luo J, Rubie EA, et al. Requirement for glycogen synthase kinase-3β in cell survival and NF-κB activation. Nature 2000; 406:86–90.
Takada Y, Fang X, Jamaluddin MS, Boyd DD, Aggarwal BB . Genetic deletion of glycogen synthase kinase-3β abrogates activation of InBa kinase, JNK, Akt, and p44/p42 MAPK but potentiates apoptosis induced by TNF. J Biol Chem 2004; 279:39541–39554.
Cesare DM, Perego P, Righetti SC, et al. Enhanced antitumour efficacy of gimatecan in combination with Bcl-2 antisense oligonucleotide in human melanoma xenografts. Eur J Cancer 2005; 41:1213–1222.
Rask K, Nilsson A, Brännström M, et al. Wnt-signalling pathway in ovarian epithelial tumours: increased expression of β-catenin and GSK3β. Br J Cancer 2003; 89:1298–1304.
Meijer L, Flajolet M, Greengard P . Pharmacological inhibitors of glycogen synthase kinase 3. Trends Pharmacol Sci 2004; 25:471–480.
Coghlan MP, Culbert AA, Cross DA, et al. Selective small molecule inhibitors of glycogen synthase kinase-3 modulate glycogen metabolism and gene transcription. Chem Biol 2000; 7:793–803.
Klein PS, Melton DA . A molecular mechanism for the effect of lithium on development. Proc Natl Acad Sci USA 1996; 93:8455–8459.
Li BG, Hasselgren PO, Fang CH . Insulin-like growth factor-I inhibits dexamethasone-induced proteolysis in cultured L6 myotubes through PI3K/Akt/GSK-3β and PI3K/Akt/mTOR-dependent mechanisms. Int J Biochem Cell Biol 2005; 37:2207–2216.
Hedgepeth CM, Deardorff MA, Rankin K, Klein PS . Regulation of glycogen synthase kinase 3beta and downstream Wnt signaling by axin. Mol Cell Biol 1999; 19:7147–7157.
Zhang F, Phiel CJ, Spece L, Gurvich N, Klein PS . Inhibitory phosphorylation of glycogen synthase kinase-3 (GSK-3) in response to lithium. Evidence for autoregulation of GSK-3. J Biol Chem 2003; 278:33067–33077.
Sherr CJ . Cancer cell cycles. Science 1996; 274:1672–1677.
Sherr CJ, Roberts JM . CDK inhibitors: positive and negative regulators of G1-phase progression. Genes Dev 1999; 13:1501–1512.
Jope RS, Johnson GVW . The glamour and gloom of glycogen synthase kinase-3. Trends Biochem Sci 2004; 29:95–102.
Macheda ML, Rogers S, Best JD . Molecular and cellular regulation of glucose transporter (GLUT) proteins in cancer. J Cell Physiol 2005; 202:654–662.
Gatenby RA, Gawlinski E . The glycolytic phenotype in carcinogenesis and tumor invasion: insights through mathematical models. Cancer Res 2003; 63:3847–3854.
Li Y, Ahmed F, Ali S, et al. Inactivation of nuclear factor kappaB by soy isoflavone genistein contributes to increased apoptosis induced by chemotherapeutic agents in human cancer cells. Cancer Res 2005; 65:6934–6942.
Salvatore C, Camarda G, Maggi CA, et al. NF-kappaB activation contributes to anthracycline resistance pathway in human ovarian carcinoma cell line A2780. Int J Oncol 2005; 27:799–806.
Venkatraman M, Anto RJ, Nair A, et al. Biological and chemical inhibitors of NF-kappaB sensitize SiHa cells to cisplatin-induced apoptosis. Mol Carcinogen 2005; 44:51–59.
Biliran Jr H, Wang Y, Banerjee S, et al. Overexpression of cyclin D1 promotes tumor cell growth and confers resistance to cisplatin-mediated apoptosis in an elastase-myc transgene-expressing pancreatic tumor cell line. Clin Cancer Res 2005; 11: 6075–6086.
Acknowledgements
We thank Dr Y Rao (Northwestern University) for generously providing Axin GID5-6/pCS2MT, Axin GID5-6LP/pCS2MT and GSK-3β S9A/ pCS2 plasmids and YC Jia, CX Huang, Y Hou for critical discussion and ZJ Fan for technical support. This work was supported by Grant from Shanghai Foundation for Key Subject Development of Medical Science (05III016).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Cao, Q., Lu, X. & Feng, YJ. Glycogen synthase kinase-3β positively regulates the proliferation of human ovarian cancer cells. Cell Res 16, 671–677 (2006). https://doi.org/10.1038/sj.cr.7310078
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.cr.7310078
Keywords
This article is cited by
-
Galectin-3 promotes secretion of proteases that decrease epithelium integrity in human colon cancer cells
Cell Death & Disease (2023)
-
Network hub-node prioritization of gene regulation with intra-network association
BMC Bioinformatics (2020)
-
Potential of platinum-resensitization by Wnt signaling modulators as treatment approach for epithelial ovarian cancer
Journal of Cancer Research and Clinical Oncology (2020)
-
HOXA4, down-regulated in lung cancer, inhibits the growth, motility and invasion of lung cancer cells
Cell Death & Disease (2018)
-
Stress-induced phosphoprotein 1 acts as a scaffold protein for glycogen synthase kinase-3 beta-mediated phosphorylation of lysine-specific demethylase 1
Oncogenesis (2018)
