
Abstract
DNA lesions, constantly produced by endogenous and exogenous sources, activate the DNA damage response (DDR), which involves detection, signaling and repair of the damage. Autophagy, a lysosome-dependent degradation pathway that is activated by stressful situations such as starvation and oxidative stress, regulates cell fate after DNA damage and also has a pivotal role in the maintenance of nuclear and mitochondrial genomic integrity. Here, we review important evidence regarding the role played by autophagy in preventing genomic instability and tumorigenesis, as well as in micronuclei degradation. Several pathways governing autophagy activation after DNA injury and the influence of autophagy upon the processing of genomic lesions are also discussed herein. In this line, the mechanisms by which several proteins participate in both DDR and autophagy, and the importance of this crosstalk in cancer and neurodegeneration will be presented in an integrated fashion. At last, we present a hypothetical model of the role played by autophagy in dictating cell fate after genotoxic stress.
Similar content being viewed by others


Facts
-
Autophagy can be activated by the DNA damage response (DDR) and influences the processing of genomic lesions; in some cases, autophagy may contribute to cell death after genotoxic stress.
-
By degrading dysfunctional mitochondria and toxic protein aggregates, autophagy contributes to genomic stability, thereby acting as a tumor suppressor mechanism.
-
Genomic stabilizing properties of autophagy can also be achieved through removal of micronuclei and damaged nuclear parts.
Open Questions
-
How does autophagy influences the processing of DNA lesions?
-
Do different types of DNA lesions activate autophagy through specific or shared pathways?
-
What determines whether autophagy will prevent or contribute to cell death after genotoxic stress?
-
Can the interplay between DDR and autophagy be exploited to improve the treatment of cancer or neurodegenerative diseases?
Cells have evolved complex mechanisms to safeguard the genome, which is constantly threatened by environmental and endogenous DNA damage-inducing agents. In the event of genomic assault, the DNA damage response (DDR) takes place, leading to the detection, signaling and repair of lesions. In the case of excessive damage, cells activate apoptosis or senescence, thereby avoiding the proliferation of potentially tumorigenic cells.1, 2, 3, 4
Macroautophagy (hereafter referred to as autophagy) is a lysosome-dependent degradation pathway that promotes cell homeostasis in response to stress such as nutrient deprivation, oxidative stress or DNA damage. This mechanism is centrally controlled by the autophagy-related (atg) family of genes,5 which is modulated by several kinases including mTOR,6 PI3k/Akt,6 AMPK7 and MAPK.8 The protective functions of autophagy are achieved through the recycling of damaged and/or obsolete cellular components, such as dysfunctional mitochondria and toxic protein aggregates, thereby generating metabolic precursors for vital processes such as ATP production and macromolecular synthesis.9, 10, 11 In addition to its role in cell survival, autophagy also contributes to organism homeostasis by clearing apoptotic cells during embryonic development12 and after certain types of DNA damage.13, 14
In this review, we examine the roles proposed for autophagy in preventing genomic instability, as well as the connection of autophagy to DDR and cell fate after DNA damage. We also discuss the roles proposed for autophagy in the development and therapy of cancer and other human diseases.
Autophagy, Mitochondria Metabolism and Tumorigenesis
One of the first evidences linking autophagy to tumorigenesis was described by Schwarze and Seglen in 1985. They observed that the degradation of long-lived proteins during starvation was reduced in hepatocytes from carcinogen-treated rats because of reduced autophagic activity, contributing to cell survival.15 A few years later, a surprising number of reports highlighted the role of autophagy in tumorigenesis. In 1999, Aita et al. reported allelic deletions in the essential autophagy gene beclin 1 (atg6) in a high percentage of breast carcinoma cell lines.16 In the same year, Liang et al. reported that expression of beclin 1 in MCF7 cells, a metastatic human breast cancer cell line with 17q21 loss of heterozygosity, the region where the beclin 1 locus maps, increased contact inhibition, reduced proliferation rates and decreased tumor formation in vivo.17 Conversely, heterozygous disruption of beclin 1 compromised autophagy activation and resulted in increased cellular proliferation18 and spontaneous tumor formation in mice.19
These early observations of the role of beclin 1 in tumorigenesis were extended to other autophagy genes. Expression of the UV irradiation resistance-associated gene (UVRAG) protein, which participates in the autophagosome-formation regulatory complex Bcl-2-Beclin1-PI(3)KC3-UVRAG, increased autophagy, reduced proliferation and suppressed tumorigenicity of HCT116 colorectal carcinoma cells in mice.20 Moreover, lack of Bif-1, which also participates in autophagosome formation during starvation, increased spontaneous tumor formation in mice.21
Despite these evidences, it was not until 2007 that Karantza-Wadsworth et al. and Mathew et al. shed light on the mechanism behind the tumor suppressive function of autophagy. They described that under conditions of metabolic stress, beclin 1+/− cells accumulated mitochondria with structural abnormalities, endoplasmic reticulum chaperones and p62/SQSTM1, which target organelles and proteins to the autophagosome. These cells also underwent a marked increase in reactive oxygen species (ROS) generation, causing DNA damage and increased aneuploidy. Moreover, increased resistance to N-(phophonoacetyl)-L-aspartate (PALA) treatment in autophagy-defective cells suggested higher gene amplification rates, evidence that loss of autophagy increased genomic instability, a driving force behind tumorigenesis.22, 23, 24 Accordingly, a significant association between loss of beclin 1 and amplification of the HER2/NEU oncogene was described in breast carcinoma.25 Quenching ROS with N-acetyl l-cysteine (NAC) delayed the promotion of aneuploidy and improved survival of beclin 1 +/− cells, revealing that ROS contributes to genomic instability in these cells.23, 24 Interestingly, expression of p62 increased ROS and DNA damage in autophagy-defective cells under metabolic stress, thereby revealing that p62 accumulation may potentiate generation of ROS due to dysfunctional mitochondria.26 These evidences suggest that autophagy is an important tumor suppressor mechanism involved in different steps of carcinogenesis (Figure 1).

Overview of the genomic instability caused by autophagy impairment. Autophagy impairment leads to the accumulation of hazardous cellular components, such as dysfunctional mitochondria and toxic protein aggregates, which leads to an increase in ROS production (box i), cell cycle dynamic alterations, DNA damage and, consequently, genomic instability. Autophagy impairment also interferes with DNA repair (box ii) and removal of micronuclei (here referred to as nucleophagy (box iii), contributing to genomic instability. The molecular and cellular mechanisms involved in the role of mitophagy in the context of DNA damage are shown in Figure 2. Pathways that are involved in the crosstalk between DDR and autophagy are summarized in Figure 3 and Table 1, whereas the dual role of DDR-induced autophagy is shown on Figure 4 and Table 2
The mitochondria is central to the model linking autophagy, ROS and DNA (Figure 2). Normal mitochondrial activity inevitably generates ROS as by-products, which may cause damage to cell components, including the DNA. Direct ROS-mediated damage to the mitochondria may result in mitochondrial DNA (mtDNA) damage, alterations in the mitochondrial membrane permeability (MMP) and uncoupling of the respiratory chain, resulting in even more ROS generation in a vicious cycle (Figure 1, box i; Figure 2, #1).27, 28 Mitophagy of injured organelles has a central role in impeding this vicious cycle (Figure 2, #6), a process in which the protein parkin has a central role. Parkin translocates from the cytosol to the injured mitochondria, signaling for mitophagy,29 a process that involves the BCL2/adenovirus E1B 19 kd-interacting protein (BNIP3) in cardiac myocytes (Figure 2, #3).30 Interestingly, mtDNA deletions also trigger autophagy through the increase of oxidized proteins and a reduction of tRNA, leading to reduced levels of ATP and amino acids, triggering AMPK activation and autophagy (Figure 2, #4).31, 32, 33

Mitochondria quality control by mitophagy in the context of DNA damage. Details of the processes are given in the main text. A, autophagosomes; L, lysosomes; AL, autophagolysosomes; MMP, mitochondrial membrane potential
Supporting the importance of autophagy for basal mitochondrial physiology and ROS control, deletion of atg7 in the mouse hematopoietic system resulted in accumulation of mitochondria with high membrane potential, superoxide production, DNA damage and death of hematopoietic stem cells. Atypical myeloid infiltrates were detected in several organs of these animals, showing that loss of autophagy contributes to development of myeloproliferative disorders.34, 35 Further, hepatocytes from atg5 mosaically deleted mice accumulated swollen mitochondria and oxidatively generated DNA damage, in addition to displaying an increase in glutathione-S-transferase in tumor areas as a result of oxidative stress.36 Mice lacking MAP1S, which is involved in autophagosome biogenesis, treated with the hepatocarcinogenesis initiator diethylnitrosamine, displayed similar features.37
ROS can result in genomic instability (Figure 2, #2) through direct damage to the DNA and/or compromising spindle checkpoint maintenance. In contact with DNA, ROS generates base damage, such as 7,8-dihydro-8-oxo-guanine (8-oxo-G), which, if not repaired, increases the chance of mispairing adenine opposite the lesion.38 ROS may also lead to breaks in the phosphodiester chain of DNA, including double-strand breaks (DSBs) that are normally detected by γ-H2AX.39 These extremely toxic and deleterious lesions may cause chromosome alterations or even cell death. The induction of DSBs by oxidative stress is most likely a result of the processing of other types of DNA damage, including the repair of clustered lesions, breakage during the fragile blockage of replication forks by the lesions,40, 41 or the handling of DNA–DNA and DNA–protein crosslinks induced by ROS.42
A second mechanism by which ROS leads to genomic instability is through the degradation of the anaphase blockers securin and cyclin B1, which impede aneuploidy by ensuring correct segregation of chromosomes during mitosis43 and in checkpoint-arrested cells, thereby suspending the spindle checkpoint. In agreement with this, budding yeast cells activate autophagy after the induction of DSBs, accompanied by anaphase arrest. This arrest persists even when phosphorylation of the checkpoint kinase Rad53 is reduced, but is overcome when autophagy is blocked or vacuolar proteolysis is inhibited, suggesting that autophagy is fundamental for DNA damage-induced anaphase arrest, thus avoiding improper chromosome segregation.44 Interestingly, elimination of the mid-body, which is involved in the final stages of cytokinesis, by autophagy was also shown to influence the tumorigenic potential of cancer cells.45 Indeed, autophagy-defective cells exhibited nuclear morphometric alterations, centrosome abnormalities and increased chromosome number under normal culture conditions.24
Altogether, these data show that autophagy has a strong impact on genomic stability, contributing to mitochondria quality control and, as a consequence, modulating ROS levels, ATP production and cell death signaling. These mechanisms are all directly involved in the carcinogenic process and may contribute to the tumor suppressor effect attributed to autophagy.
The Role of Autophagy in DNA Repair
In addition to mitigating DNA damage by controlling ROS production, autophagy can also influence the dynamics of DNA repair by recycling key proteins involved in the processing of lesions.46 Alternatively, autophagy may also provide metabolic precursors for the generation of ATP, which is employed in several steps of DNA repair,47 as well as regulate the supply of dNTPs for DNA synthesis during repair.48
By targeting glycogen, lipids and proteins to lysosomes, autophagy guides the breakdown of these macromolecules, thereby producing metabolic precursors that can sustain oxidative phosphorylation and glycolysis.49, 50 In cancer cells from solid undernourished tumors, response to radiotherapy or DNA-damaging chemotherapy triggers ATP production by autophagy, which may have an essential role in DNA repair (Figure 1, box ii). Supporting this hypothesis, the inhibition of autophagy suppressed ATP generation and increased mitotic catastrophe in glioma cells treated with temozolomide (TMZ). Addition of pyruvate rescued ATP levels and prevented mitotic catastrophe, suggesting that autophagy-sustained ATP generation could be employed by mechanisms that promote genomic integrity, such as DNA repair processes.47 In fact, DNA repair requires ATP at several steps, including DNA unwinding by helicases during nucleotide-excision repair (NER),51 ATP-dependent chromatin remodeling complexes in DSB repair52 and PARP activity, which consumes NAD+ and can cause energy collapse in DNA-damaged cells.53, 54 However, direct evidence to corroborate this hypothesis is still lacking.
Autophagy was also implicated in regulating the dNTP pool levels, which are essential for DNA replication and repair. Upon methyl methane sulfonate (MMS) treatment, yeast trigger autophagy, thereby promoting degradation of ribonucleotide reductase 1 (Rnr1), which associates with other Rnr proteins to regulate the reduction of ribonucleotides to deoxyribonucleotides. This reduction in Rnr1 levels may favor assembly of the most catalytically active form of Rnr, Rnr1-Rnr3, instead of Rnr1-Rnr1 in the final RNR complex, resulting in optimization of RNR activity and dNTP levels, which in turn could be employed as substrates during DNA repair processes, such as mismatch repair (MMR).48 It is also interesting to note that imbalanced levels of dNTPs can increase mutagenesis.55 Thus, it is tempting to speculate that through the degradation of Rnr subunits autophagy may also fight mutagenesis by ensuring a balanced dNTP pool, which is fundamental to avoid stress replication and gene amplification, two characteristics frequently observed in autophagy-deficient cells.23, 24
Besides dNTP recycling and ATP generation, autophagy also participates in the turnover of key proteins involved in the regulation/processing of genomic lesions. Recently, an intricate relationship between histone deacetylases (HDACS) – which are involved in DNA repair and apoptosis,56, 57 – DSB processing and autophagy was shown in budding yeast.46, 58 Treatment with valproic acid (VPA), an HDAC inhibitor, impaired the activation of Rad53 in response to DSBs. In the VPA-treated cells, Mre11, the first factor recruited to DSB sites, remained bound to the DSB site, accompanied by reduced levels of Sae2, which is responsible for removing Mre11 from the DSB region, a step required for the progress of lesion repair. In this context, inhibition of autophagy by the serine protease inhibitor PMSF or deletion of atg1 increased acetylated Sae2 levels, whereas rapamycin, which activates autophagy through mTOR inhibition, decreased it, confirming that autophagy induced by VPA could impair DSB processing through degradation of acetylated Sae2. Moreover, Atg1 inhibition partially rescued sensitivity of an hda1-rpd3 (HDACs) double mutant (which exhibits low levels of Sae2 as well as impaired DSB resection) to camptothecin. These results suggest that, in one hand, autophagy may be involved in destabilizing key factors, such as the acetylated form of Sae2, impairing DSB repair. On the other hand, clearance of Sae2 by autophagy could also help cells in the control of DSB repair pathway by counteracting extensive DSB resection that may be harmful to cells,46 demonstrating the complex role of autophagy in the context of DNA damage and repair.
In the same line of thinking, FIP200 (a focal adhesion kinase that participates in autophagy induction)59 KO MEFS showed persistent nuclear γ-H2AX staining after exposure to ionizing radiation (IR), indicating defective DNA damage repair.60 Although the initial amount of DNA breaks were similar between fip200 KO and WT MEFs immediately after IR, the DNA breaks persisted for a longer period in KO cells. Similar results were obtained in response to other DNA damage-inducing agents (camptothecin and etoposide) and also when autophagy was pharmacollogially inhibited using 3-methyladenine. Interestingly, silencing p62 in these cells improved DNA repair and cell viability in response to IR and camptothecin. Although accumulation of p62 was shown to increase oxidative stress,26 the antioxidant NAC did not improve cell viability in response to camptothecin or etoposide, revealing that the mechanism underlying persistent DNA damage in fip200 KO cells is ROS independent.
These data show that autophagy can influence the resolution of DNA injuries. Although several reports showed that inhibition of autophagy can undermine cells’ resistance to chemo- and radiotherapy, only a few studies provide a more careful look into the effect of this approach over DNA repair dynamics.47 In this sense, spatial and temporal tracking of DNA repair enzymes may provide important clues about the influence of autophagy on the resolution of genomic injuries. In this sense, yeast models can be of great value to create a library of strains in which recruitment of specific DNA repair proteins can be followed. For instance, yeast strains expressing homologous recombination enzymes tagged with fluorescent proteins allowed spatial and temporal localization of these enzymes upon DSB repair activation.61 Thus, by using such approaches, important clues may be revealed that significantly improve our understanding of this exciting yet obscure role of autophagy in DNA repair.
Nucleophagy as a Way to Eliminate Injured DNA
Autophagic removal of whole nuclei is not as common as removal of other organelles because it may cause deleterious loss of genetic information. However, in multinucleated cells of the filamentous fungus Aspergillus oryzae, nucleophagy of entire nuclei contributes to cell maintenance during nutrient deprivation.62 Similarly, removal of nuclei from intestinal epithelial cells of Caernohabiditis elegans also occurs through autophagy.63
Autophagy of nuclear components in eukaryotes, or piecemeal microautophagy of the nucleus (PMN), was initially described in Saccharomyces cerevisae. It is triggered by nutrient deprivation and occurs through the release of nuclear portions into the vacuole,64 followed by digestion by hydrolases.65 Nuclear components targeted for PMN include granular nucleolus enriched in pre-ribosomes and nuclear envelope, nuclear pore complexes or spindle pole bodies.66, 67 PMN involves the core Atg proteins involved in macroautophagy and microautophagy, such as Atg4, 5, 7 and 12, as well as macroautophagy-specific proteins such as Atg17, 29 and 31.64
In mammalian cells, autophagy can degrade nuclear components, thereby contributing to the maintenance of nuclear function and integrity.68 The initial observation of the engulfment of nuclear components points to the presence of perinuclear vacuoles in skeletal and/or cardiac muscle cells from patients or mice with envelopathies,68 disorders caused by mutations in nuclear envelope components.69, 70 Mutated cells presented higher levels of autophagic flow and the vacuoles were positive for Atg5, Atg16L and Atg9. Cells presented giant autophagosomes and autophagolysosomes containing LC3 and DNA, with the presence of histone H1 and γ-H2AX, but not the markers of nuclear envelope, lamin A and B, confirming that the DNA contained in autophagosomes was extranuclear and damaged. In this case, the contribution of autophagy to nuclear stability was clear since its inhibition increased the incidence of nuclear abnormalities, accompanied by a reduction in cell viability.68 Autophagy was also shown to degrade micronuclei generated by treatment with cell cycle blockers. Interestingly, ‘autophagic micronuclei’ co-localized with p62, besides presenting reduced chromatin content and γ-H2AX foci, a DNA damage marker. However, non-autophagic micronuclei appeared p62-negative, suggesting that the presence of DNA damage directly or indirectly signaled for autophagic engulfment (Figure 1, box iii).71
Thus, evolutionarily, the process of nucleophagy may represent a physiological mechanism for the removal of damaged nuclear components and micronuclei, thus contributing to genomic stability. In multinuclear eukaryote cells, it is plausible that autophagy of nuclear components can be triggered under metabolically stressful situations or DNA damage, contributing to genomic stability and cellular homeostasis (Figure 1; box iii).
Pathways Connecting DDR to Autophagy
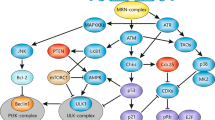
DDR comprises an array of processes triggered by DNA lesions that allows cells to cope with these insults, aiming at safeguarding the integrity of the genome and avoiding propagation of mutated cells.4, 17 The kinases ATM and ATR have key roles sensing DNA breaks and activating downstream components of DDR, such as Chk1, Chk2 and p53.72, 73 p53 is an important protein in DDR, inducing the transcription of key genes involved in cell cycle arrest, DNA repair, apoptosis74, 75 and, more recently described, in autophagy.76
Two central components, p53 and mTOR, link DDR to autophagy (Figure 3 and Table 1). p53 can activate autophagy after DNA damage through transcriptional induction of several genes, including damage-regulated autophagy modulator (dram), UNC-51-like kinase 1/2, (ulk1/2), sestrin1/2, isg20L1 and bnip3, among others.77 p53 targets can regulate autophagy directly, as is the case with the lysosomal proteins DRAM14 and ULK1/2, which interact with Atg13 and FIP200 to induce autophagy,13 or indirectly through Sestrin 1 and 2, which activate AMPK and the TSC1/2 complex, leading to inactivation of mTORC1 and autophagy induction.78 Additionally, ATM was shown to activate AMPK in a p53-independent manner through direct activation of the AMP kinase LKB1.79 Interestingly, cytoplasmic p53 is able to repress autophagy, and deletion or pharmacological inhibition of p53 induces, rather than inhibits, autophagy. Accordingly, induction of autophagy by starvation requires destruction of cytoplasmic p53,80, 81 thereby revealing a complex role for p53 in the regulation of autophagy.

Autophagy modulation in response to DNA damage response (DDR). DDR-activated signaling can result in autophagy modulation. The autophagy box represents the central autophagy regulating genes. DSB, double-strand brake; SSB, single-strand break; MMR, mismatch repair
The other protein placed in the core of DDR to autophagy signaling, mTOR, is an important repressor of autophagy, and inactivation of the mTOR complex 1 (mTORC1) by AMPK-TSC1/2 has an important role in autophagy induction upon starvation. Interestingly, DDR was also shown to participate in autophagy induced by starvation,53 which may increase mitochondria-dependent ROS generation, causing DNA damage, PARP-1 activation and ATP depletion. As a consequence, AMPK is activated, thereby inhibiting mTOR and inducing autophagy.53, 54 It is worth noting that autophagy activation was shown to precede phosphorylation of ATM and p53 and activation of DNA repair proteins in response to capsaicin treatment, revealing an intricate pathway in which autophagy acts upstream, and not just as a consequence, of DDR activation.82
BNIP3 is a Bcl-2 homologous protein and is activated after conditions of stress such as hypoxia. MMR induced by 6-thioguanine activates autophagy through MLH1, p53 activation and transcription of bnip3. Additionally, the TORC1 target p70S6 kinase 1 promotes translation of BNIP3, which induces loss of mitochondrial outer membrane potential and further autophagy activation, possibly through ROS generation, thus triggering mitophagy and preventing apoptosis.83, 84 It is worth noting that in this case mTOR activation, rather than inhibition, activates autophagy.
E2F1 transcriptional activity is activated after DNA damage, most likely due to the removal of C-EBPα repression85, 86 and activates autophagy by directly inducing the transcription of atg1, atg6, atg5 and dram,87 as well as by inducing p73, which is a transcriptional activator of atg5, atg7, ambra, dram and isg20l1.88, 89 Several chemotherapeutic agents induce TA-p73α and TA-p73β expression,90 which are sufficient to activate autophagy through direct transcriptional regulation of the above-mentioned genes. Moreover, p73 induces the expression of several DNA repair genes,91 thus positioning p73 in the interface between DNA repair and autophagy. In strong contrast to p53, p73 is rarely mutated in primary tumors.92 Thus, it is plausible that p73-induced autophagy has an important role in the resistance of p53-compromised cells, making p73 inhibition a good target for chemotherapy sensitization.
As seen in Figure 3, the signaling that links DNA damage to autophagy is complex and redundant, as is the case for signaling pathways fundamental for life. It is unlikely that all these pathways are activated in a given cell by one type of DNA damage. However, the relative contribution of these pathways to the cellular response to different types of damage is not clear and may be an important part to understand the link between DNA damage and cell fate.
The Dual role of Autophagy in the Context of DNA Damage
As previously discussed, autophagy can either contribute to or prevent cell death in response to DNA damage (Figure 4). As summarized in Table 2, the majority of studies showed that inhibition of autophagy in cells treated with DNA damaging agents leads to increased cell death, supporting a protective role for autophagy. We hypothesize that a mechanism based on the severity and/or type of genomic damage could turn on either a pro-survival or pro-death autophagic role. In this scenario, transcription factors such as p53, p73 and E2F1 would have pivotal roles, as they were not only shown to promote DNA repair, cell cycle arrest or apoptosis in response to different degrees of DNA damage, but also to control autophagy.74, 75, 91, 93

Roles for autophagy in regulating cell fate after DNA damage. We propose that after DNA injury, autophagy can influence cell fate, supporting or impairing cell survival. As a cytoprotective mechanism, autophagy may degrade pro-apoptotic proteins and membrane permeabilized mitochondria, enhance ATP and dNTPs generation for DNA repair and also regulate cell cycle arrest. However, autophagy may favor cell death through degradation of anti-apoptotic and DNA repair-related proteins
Thus, we hypothesize that after low doses of DNA damage, autophagy activation by these transcription factors would result in clearance of membrane-permeabilized mitochondria,94 generation of dNTPs and/or ATP for DNA repair activity,47, 48 degradation of pro-apoptotic proteins such as active caspase 895 and elimination of p62, thus preventing p38-hyperactivation.96 Supporting the role of p53 in autophagy and cell survival, p53 mediates the transcription of parkin,97 suggesting that p53 could regulate transcription of mitophagy genes in response to genomic damage, thus counterbalancing mitochondrial apoptotic signaling.
Conversely, autophagy can also promote degradation of anti-apoptotic proteins, thus facilitating cell death. Autophagy-mediated degradation of the inhibitor of apoptosis dBruce during late oogenesis in Drosophila melanogaster98 and degradation of catalase in apoptosis-compromised cells resulting in increased ROS and oxidatively generated damage99 reveal how autophagy can have an impact on cell death. As autophagy was also shown to promote degradation of acetylated Sae2 in VPA-treated yeast cells, thereby influencing, in an intricate manner, the dynamics of DNA DSB repair, it is possible that autophagy activation could contribute to perseverance of DNA damage and further enhancement of apoptotic signaling in mammalian cells by controlling turnover of certain DNA repair-related enzymes.46 In this scenario, autophagy was shown to degrade OGG1, an enzyme that participates in 8-oxoG base-excision repair, in starved myocytes.100
Thus, it is tempting to speculate that, the intensity by which autophagy is activated as well as the targets to be degraded can dictate whether it is going to cooperate with or protect from cell death induced by DNA damage (Figure 4). This dual role of autophagy in cell death can be exemplified by the modulation of autophagy by the MAPK pathway. While transient or moderate activity of MEK/ERK results in mTOR inhibition, weakly beclin 1 increase and protective autophagy, sustained MEK/ERK activation results in inhibition of mTORC1 and mTORC2, stronger beclin 1 activation and toxic autophagy.101 Thus, high levels of DNA damage could induce stronger mTORC1 inhibition, followed by stronger beclin 1 activation, thus resulting in levels of autophagy that contribute to cell death. In fact, overexpression of beclin 1, per se, is able to increase basal as well as induced autophagy in both normal and cancerous tissue and cells.17, 102, 103
Thus, understanding the crosstalk between DDR and autophagy may be essential to understand how autophagy has either a positive or negative role in cell death induction after activation of DDR-induced autophagy. Recent advances in the field of transcription factors and effector proteins are addressing these questions and may aid in the understanding of how cells define their fate in this context.104, 105
Autophagy-DNA damage crosstalk in neurodegeneration, cancer and aging
All of the aforementioned findings raised a natural interest in the pharmacological modulation of autophagy, which could have a significant impact on mitigating genomic damage. This is of particular interest for human syndromes that arise from genetic deficiency in genes related to DNA repair, such as the premature aging disorder Cockayne Syndrome (CS).106 In fact, accumulation of dysfunctional mitochondria, mtDNA mutations and oxidatively generated damage were observed in CS type B (CSB) fibroblasts,107, 108 suggesting that the regulation of autophagy may have an impact on CS pathogenesis. Indeed, the induction of autophagy reduced mitochondrial loading and mitochondrial membrane potential in CSB cells, revealing that pharmacological modulation of this pathway is a promising approach.109 Moreover, autophagy also has a role in stem cells maintenance,110 suggesting that this pathway may also fight accelerated aging by maintaining the health of the stem cell population, avoiding loss of regenerative potential.111
Accumulation of oxidatively generated damage has been implicated in several neurodegenerative diseases such as Parkinson’s and Alzheimer’s diseases.112, 113 Data point to autophagy as an important factor in the context of both the pathogenesis and, consequently, therapy of these pathologies. It has become clear that parkin is necessary for p62 localization to damaged mitochondria and its consequent elimination through beclin-dependent autophagy.29, 114 Deletion of parkin increases ROS generation due to accumulation of dysfunctional organelles, resulting in mtDNA and nDNA damage, which is the basis for parkin deficiency-associated Parkinson’s disease.115, 116 In this sense, induction of autophagy has given promising results in mouse models of Alzheimer’s disease117 and other neurodegenerative diseases.118 Further, the increased genomic instability observed in parkin-deleted cells could also be explained by the observation that parkin translocates from the cytosol to the nucleus where it participates in DDR after DNA damage.115, 116 Therefore, the parkin protein is an important factor in both DDR and autophagic removal of injured mitochondria.
In cancer, heterozygous disruption of beclin 1 compromised autophagy activation and resulted in increased cellular proliferation and increased spontaneous as well as induced tumor formation. Contrary to the normal genetic behavior of classical tumor suppressors, the remaining wild-type allele was neither mutated nor silenced in the formed tumors.18, 19 Accordingly, 40–75% of cases of human sporadic breast, ovarian and prostate cancer had monoallelic deletion of beclin 1.16 Further, genes involved in autophagy are monoallelically inactivated in human cancers or occur in genes whose deletion only partially reduces autophagy. Moreover, frameshift mutations were identified in UVRAG, atg2B, atg5 and atg9B in colorectal and in gastric carcinomas with microsatellite instability (MSI), but not in DNA from normal tissues of the same patients,119, 120, 121 although the effects of these mutations on autophagic flux were not determined.
This genetic evidence points to the scenario in which reduced levels of autophagy favor tumor development, whereas the complete absence of autophagy is anti-tumoral.122 However, it is important to keep in mind that several members of the classical autophagic pathway have autophagy-independent roles. For instance, ATG4C KO mice did not present altered basal or starvation-induced autophagy in several tissues, but an increased methylcholanthren-induced fibrosarcoma formation.123 Similarly, MSI-positive colon cancer cells with monoallelic deletions of UVRAG or UVRAG-KD HEK cells did not show reduced autophagy. Indeed, UVRAG participates in an autophagy-independent manner in preventing centrosome overduplication and chromosome missegregation during anaphase124 as well as in endocytic trafficking of EGFR, whose accumulation may enhance growth factor receptor signaling, thus supporting tumor growth.125 Therefore, an autophagy-centric interpretation must always bear in mind these other functions described for the ‘autophagy’ genes.126
Concluding Remarks
The crosstalk between autophagy and DDR, as well as its role in defining cell fate, is a hot topic that is just beginning to be explored, as can be evidenced by the new and fast-growing body of work related to this theme. Understanding this complex and intricate relationship will have profound impacts on several fields of medical interest, such as cancer, aging and neurodegeneration. Additionally, the majority of studies mentioned in this review focus on cell biology and the roles played by autophagy in response to DNA damage in DDR and survival of the cell. Much more difficult and therefore less clear is the impact of the link between DNA damage and autophagy on the physiology of the whole organism, mainly on aging and cancer, in which elimination of cellular components by high levels of mitophagy or nucleophagy may, in fact, be very beneficial. Notwithstanding, the current evidence linking DNA damage to autophagy indicates that both are involved in the normal physiology as well as in pathological processes and that modulation of the pathways linking DDR to autophagy has to be considered in therapeutic interventions for several diseases.
Abbreviations
- Ambra1:
-
activating molecule in Beclin1-regulated autophagy
- AMPK:
-
AMP-activated protein kinase
- ATG:
-
autophagy-related
- ATM:
-
ataxia telangiectasia mutated
- ATR:
-
ataxia telangiectasia mutated and Rad3 related
- BER:
-
base excision repair
- Bif-1:
-
endophilin-B1
- C-EBPα:
-
CCAAT/enhancer-binding protein alpha
- Bnip3:
-
Bcl-2/adenovirus E1B 19-kDa-interacting protein 3
- DDR:
-
DNA damage response
- DNAPK:
-
DNA-dependent protein kinase
- dNTP:
-
deoxyribonucleoside triphosphate
- DRAM:
-
damage-regulated autophagy modulator
- DSB:
-
double strand break
- E2F1:
-
E2F transcription factor 1
- ERK:
-
extracellular signal-regulated protein kinase
- FIP200:
-
focal adhesion kinase family interacting protein of 200 kDa
- γ-H2AX:
-
phosphorylated histone H2AX
- HDAC:
-
histone deacetylases
- HR:
-
homologous recombination
- ISG20L1:
-
interferon stimulated gene 20- like 1
- LC3:
-
light chain 3
- LKB1:
-
liver kinase B1
- 3-MA:
-
3-methyladenine
- MAPK:
-
mitogen-activated protein kinase
- MLH:
-
MutL-homolog
- MMP:
-
mitochondrial membrane permeabilization
- MMR:
-
mismatch repair
- MSH:
-
MutS-homolog
- mtDNA:
-
mitochondrial DNA
- mTOR:
-
mammalian target of rapamycin
- NAC:
-
N-acetyl l-cysteine
- nDNA:
-
nuclear DNA
- NER:
-
nucleotide excision repair
- NHEJ:
-
non homologous end joining
- OGG1:
-
8-oxoguanine DNA glycosylase 1
- PALA:
-
N-(phophonoacetyl)-L-aspartate
- PARP:
-
poly (ADP-ribose) polymerase
- PCNA:
-
proliferating cell nuclear antigen
- PI3K:
-
phosphatidylinositol 3-kinase
- PINK1:
-
PTEN-induced kinase 1
- PTEN:
-
phosphatase and tensin homolog
- p70S6 kinase:
-
p70 ribosomal protein S6 kinase
- ROS:
-
reactive oxygen species
- SQSTM1:
-
sequestosome 1
- SSB:
-
single strand break
- TSC2:
-
tuberous sclerosis complex 2
- ULK-1:
-
unc-51-like kinase 1
- UVRAG:
-
UV irradiation resistance-associated gene
- VPA:
-
valproic acid
References
Ljungman M . Activation of DNA damage signaling. Mutat Res 2005; 577: 203–216.
Lord CJ, Ashworth A . The DNA damage response and cancer therapy. Nature 2012; 481: 287–294.
Lenz G . Endogenous anticancer mechanisms (EACMs). Front Biosci (Schol Ed) 2012; 4: 1017–1030.
Ljungman M . The DNA damage response-repair or despair? Environ Mol Mutagen 2010; 51: 879–889.
Mizushima N, Yoshimori T, Ohsumi Y . The role of Atg proteins in autophagosome formation. Annu Rev Cell Dev Biol 2011; 27: 107–132.
Shanware NP, Bray K, Abraham RT . The PI3K, metabolic, and autophagy networks: interactive partners in cellular health and disease. Annu Rev Pharmacol Toxicol 2013; 53: 89–106.
Kim J, Kundu M, Viollet B, Guan KL . AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol 2011; 13: 132–141.
Corcelle E, Djerbi N, Mari M, Nebout M, Fiorini C, Fenichel P et al. Control of the autophagy maturation step by the MAPK ERK and p38: lessons from environmental carcinogens. Autophagy 2007; 3: 57–59.
Green DR, Galluzzi L, Kroemer G . Mitochondria and the autophagy-inflammation-cell death axis in organismal aging. Science 2011; 333: 1109–1112.
Kroemer G, Marino G, Levine B . Autophagy and the integrated stress response. Mol Cell 2010; 40: 280–293.
Singh R, Cuervo AM . Autophagy in the cellular energetic balance. Cell Metab 2011; 13: 495–504.
Qu X, Zou Z, Sun Q, Luby-Phelps K, Cheng P, Hogan RN et al. Autophagy gene-dependent clearance of apoptotic cells during embryonic development. Cell 2007; 128: 931–946.
Gao W, Shen Z, Shang L, Wang X . Upregulation of human autophagy-initiation kinase ULK1 by tumor suppressor p53 contributes to DNA-damage-induced cell death. Cell Death Differ 2011; 18: 1598–1607.
Crighton D, Wilkinson S, O'Prey J, Syed N, Smith P, Harrison PR et al. DRAM, a p53-induced modulator of autophagy, is critical for apoptosis. Cell 2006; 126: 121–134.
Schwarze PE, Seglen PO . Reduced autophagic activity, improved protein balance and enhanced in vitro survival of hepatocytes isolated from carcinogen-treated rats. Exp Cell Res 1985; 157: 15–28.
Aita VM, Liang XH, Murty VV, Pincus DL, Yu W, Cayanis E et al. Cloning and genomic organization of beclin 1, a candidate tumor suppressor gene on chromosome 17q21. Genomics 1999; 59: 59–65.
Liang XH, Jackson S, Seaman M, Brown K, Kempkes B, Hibshoosh H et al. Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature 1999; 402: 672–676.
Qu X, Yu J, Bhagat G, Furuya N, Hibshoosh H, Troxel A et al. Promotion of tumorigenesis by heterozygous disruption of the beclin 1 autophagy gene. J Clin Invest 2003; 112: 1809–1820.
Yue Z, Jin S, Yang C, Levine AJ, Heintz N . Beclin 1, an autophagy gene essential for early embryonic development, is a haploinsufficient tumor suppressor. Proc Natl Acad Sci USA 2003; 100: 15077–15082.
Liang C, Feng P, Ku B, Dotan I, Canaani D, Oh BH et al. Autophagic and tumour suppressor activity of a novel Beclin1-binding protein UVRAG. Nat Cell Biol 2006; 8: 688–699.
Takahashi Y, Coppola D, Matsushita N, Cualing HD, Sun M, Sato Y et al. Bif-1 interacts with Beclin 1 through UVRAG and regulates autophagy and tumorigenesis. Nat Cell Biol 2007; 9: 1142–1151.
Hanahan D, Weinberg RA . Hallmarks of cancer: the next generation. Cell 2011; 144: 646–674.
Karantza-Wadsworth V, Patel S, Kravchuk O, Chen G, Mathew R, Jin S et al. Autophagy mitigates metabolic stress and genome damage in mammary tumorigenesis. Genes Dev 2007; 21: 1621–1635.
Mathew R, Kongara S, Beaudoin B, Karp CM, Bray K, Degenhardt K et al. Autophagy suppresses tumor progression by limiting chromosomal instability. Genes Dev 2007; 21: 1367–1381.
Negri T, Tarantino E, Orsenigo M, Reid JF, Gariboldi M, Zambetti M et al. Chromosome band 17q21 in breast cancer: significant association between beclin 1 loss and HER2/NEU amplification. Genes Chromosomes Cancer 2010; 49: 901–909.
Mathew R, Karp CM, Beaudoin B, Vuong N, Chen G, Chen HY et al. Autophagy suppresses tumorigenesis through elimination of p62. Cell 2009; 137: 1062–1075.
Lenaz G . Mitochondria and reactive oxygen species. Which role in physiology and pathology? Adv Exp Med Biol 2012; 942: 93–136.
Lenaz G, Bovina C, D'Aurelio M, Fato R, Formiggini G, Genova ML et al. Role of mitochondria in oxidative stress and aging. Ann N Y Acad Sci 2002; 959: 199–213.
Geisler S, Holmstrom KM, Skujat D, Fiesel FC, Rothfuss OC, Kahle PJ et al. PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat Cell Biol 2010; 12: 119–131.
Lee Y, Lee HY, Hanna RA, Gustafsson AB . Mitochondrial autophagy by Bnip3 involves Drp1-mediated mitochondrial fission and recruitment of Parkin in cardiac myocytes. Am J Physiol Heart Circ Physiol 2011; 301: H1924–H1931.
Prigione A, Cortopassi G, Mitochondrial DNA . deletions and chloramphenicol treatment stimulate the autophagic transcript ATG12. Autophagy 2007; 3: 377–380.
Alemi M, Prigione A, Wong A, Schoenfeld R, DiMauro S, Hirano M et al. Mitochondrial DNA deletions inhibit proteasomal activity and stimulate an autophagic transcript. Free Radic Biol Med 2007; 42: 32–43.
Prigione A, Cortopassi G, Mitochondrial DNA . deletions induce the adenosine monophosphate-activated protein kinase energy stress pathway and result in decreased secretion of some proteins. Aging Cell 2007; 6: 619–630.
Mortensen M, Soilleux EJ, Djordjevic G, Tripp R, Lutteropp M, Sadighi-Akha E et al. The autophagy protein Atg7 is essential for hematopoietic stem cell maintenance. J Exp Med 2011; 208: 455–467.
Mortensen M, Watson AS, Simon AK . Lack of autophagy in the hematopoietic system leads to loss of hematopoietic stem cell function and dysregulated myeloid proliferation. Autophagy 2011; 7: 1069–1070.
Takamura A, Komatsu M, Hara T, Sakamoto A, Kishi C, Waguri S et al. Autophagy-deficient mice develop multiple liver tumors. Genes Dev 2011; 25: 795–800.
Xie R, Wang F, McKeehan WL, Liu L . Autophagy enhanced by microtubule- and mitochondrion-associated MAP1S suppresses genome instability and hepatocarcinogenesis. Cancer Res 2011; 71: 7537–7546.
van Loon B, Markkanen E, Hubscher U . Oxygen as a friend and enemy: How to combat the mutational potential of 8-oxo-guanine. DNA Repair (Amst) 2010; 9: 604–616.
Kinner A, Wu W, Staudt C, Iliakis G . Gamma-H2AX in recognition and signaling of DNA double-strand breaks in the context of chromatin. Nucleic Acids Res 2008; 36: 5678–5694.
Greinert R, Volkmer B, Henning S, Breitbart EW, Greulich KO, Cardoso MC et al. UVA-induced DNA double-strand breaks result from the repair of clustered oxidative DNA damages. Nucleic Acids Res 2012; 40: 10263–10273.
Eccles LJ, O'Neill P, Lomax ME . Delayed repair of radiation induced clustered DNA damage: friend or foe? Mutat Res 2011; 711: 134–141.
Cadet J, Ravanat JL, TavernaPorro M, Menoni H, Angelov D . Oxidatively generated complex DNA damage: tandem and clustered lesions. Cancer Lett 2012; 327: 5–15.
D'Angiolella V, Santarpia C, Grieco D . Oxidative stress overrides the spindle checkpoint. Cell Cycle 2007; 6: 576–579.
Dotiwala F, Eapen VV, Harrison JC, Arbel-Eden A, Ranade V, Yoshida S et al. DNA damage checkpoint triggers autophagy to regulate the initiation of anaphase. Proc Natl Acad Sci USA 2013; 110: E41–E49.
Kuo TC, Chen CT, Baron D, Onder TT, Loewer S, Almeida S et al. Midbody accumulation through evasion of autophagy contributes to cellular reprogramming and tumorigenicity. Nat Cell Biol 2011; 13: 1214–1223.
Robert T, Vanoli F, Chiolo I, Shubassi G, Bernstein KA, Rothstein R et al. HDACs link the DNA damage response, processing of double-strand breaks and autophagy. Nature 2011; 471: 74–79.
Katayama M, Kawaguchi T, Berger MS, Pieper RO . DNA damaging agent-induced autophagy produces a cytoprotective adenosine triphosphate surge in malignant glioma cells. Cell Death Differ 2007; 14: 548–558.
Dyavaiah M, Rooney JP, Chittur SV, Lin Q, Begley TJ . Autophagy-dependent regulation of the DNA damage response protein ribonucleotide reductase 1. Mol Cancer Res 2011; 9: 462–475.
Ferraro E, Pulicati A, Cencioni MT, Cozzolino M, Navoni F, di Martino S et al. Apoptosome-deficient cells lose cytochrome c through proteasomal degradation but survive by autophagy-dependent glycolysis. Mol Biol Cell 2008; 19: 3576–3588.
Rabinowitz JD, White E . Autophagy and metabolism. Science 2010; 330: 1344–1348.
de Laat WL, Jaspers NG, Hoeijmakers JH . Molecular mechanism of nucleotide excision repair. Genes Dev 1999; 13: 768–785.
Bao Y, Shen X . Chromatin remodeling in DNA double-strand break repair. Curr Opin Genet Dev 2007; 17: 126–131.
Rodriguez-Vargas JM, Ruiz-Magana MJ, Ruiz-Ruiz C, Majuelos-Melguizo J, Peralta-Leal A, Rodriguez MI et al. ROS-induced DNA damage and PARP-1 are required for optimal induction of starvation-induced autophagy. Cell Res 2012; 22: 1181–1198.
Munoz-Gamez JA, Rodriguez-Vargas JM, Quiles-Perez R, Aguilar-Quesada R, Martin-Oliva D, de Murcia G et al. PARP-1 is involved in autophagy induced by DNA damage. Autophagy 2009; 5: 61–74.
Kumar D, Abdulovic AL, Viberg J, Nilsson AK, Kunkel TA, Chabes A . Mechanisms of mutagenesis in vivo due to imbalanced dNTP pools. Nucleic Acids Res 2011; 39: 1360–1371.
Henderson C, Mizzau M, Paroni G, Maestro R, Schneider C, Brancolini C . Role of caspases, Bid, and p53 in the apoptotic response triggered by histone deacetylase inhibitors trichostatin-A (TSA) and suberoylanilide hydroxamic acid (SAHA). J Biol Chem 2003; 278: 12579–12589.
Kruszewski M, Szumiel I . Sirtuins (histone deacetylases III) in the cellular response to DNA damage—facts and hypotheses. DNA Repair (Amst) 2005; 4: 1306–1313.
True O, Matthias P . Interplay between histone deacetylases and autophagy—from cancer therapy to neurodegeneration. Immunol Cell Biol 2012; 90: 78–84.
Hara T, Takamura A, Kishi C, Iemura S, Natsume T, Guan JL et al. FIP200, a ULK-interacting protein, is required for autophagosome formation in mammalian cells. J Cell Biol 2008; 181: 497–510.
Bae H, Guan JL . Suppression of autophagy by FIP200 deletion impairs DNA damage repair and increases cell death upon treatments with anticancer agents. Mol Cancer Res 2011; 9: 1232–1241.
Moore DM, Karlin J, Gonzalez-Barrera S, Mardiros A, Lisby M, Doughty A et al. Rad10 exhibits lesion-dependent genetic requirements for recruitment to DNA double-strand breaks in Saccharomyces cerevisiae. Nucleic Acids Res 2009; 37: 6429–6438.
Shoji JY, Kikuma T, Arioka M, Kitamoto K . Macroautophagy-mediated degradation of whole nuclei in the filamentous fungus Aspergillus oryzae. PLoS One 2010; 5: e15650.
McGee MD, Weber D, Day N, Vitelli C, Crippen D, Herndon LA et al. Loss of intestinal nuclei and intestinal integrity in aging C. elegans. Aging Cell 2011; 10: 699–710.
Krick R, Muehe Y, Prick T, Bremer S, Schlotterhose P, Eskelinen EL et al. Piecemeal microautophagy of the nucleus requires the core macroautophagy genes. Mol Biol Cell 2008; 19: 4492–4505.
Kvam E, Gable K, Dunn TM, Goldfarb DS . Targeting of Tsc13p to nucleus-vacuole junctions: a role for very-long-chain fatty acids in the biogenesis of microautophagic vesicles. Mol Biol Cell 2005; 16: 3987–3998.
Kraft C, Reggiori F, Peter M . Selective types of autophagy in yeast. Biochim Biophys Acta 2009; 1793: 1404–1412.
Millen JI, Krick R, Prick T, Thumm M, Goldfarb DS . Measuring piecemeal microautophagy of the nucleus in Saccharomyces cerevisiae. Autophagy 2009; 5: 75–81.
Park YE, Hayashi YK, Bonne G, Arimura T, Noguchi S, Nonaka I et al. Autophagic degradation of nuclear components in mammalian cells. Autophagy 2009; 5: 795–804.
Bonne G, Di Barletta MR, Varnous S, Becane HM, Hammouda EH, Merlini L et al. Mutations in the gene encoding lamin A/C cause autosomal dominant Emery-Dreifuss muscular dystrophy. Nat Genet 1999; 21: 285–288.
Eriksson M, Brown WT, Gordon LB, Glynn MW, Singer J, Scott L et al. Recurrent de novo point mutations in lamin A cause Hutchinson-Gilford progeria syndrome. Nature 2003; 423: 293–298.
Rello-Varona S, Lissa D, Shen S, Niso-Santano M, Senovilla L, Marino G et al. Autophagic removal of micronuclei. Cell Cycle 2012; 11: 170–176.
Smith J, Tho LM, Xu N, Gillespie DA . The ATM-Chk2 and ATR-Chk1 pathways in DNA damage signaling and cancer. Adv Cancer Res 2010; 108: 73–112.
Tichy A, Vavrova J, Pejchal J, Rezacova M . Ataxia-telangiectasia mutated kinase (ATM) as a central regulator of radiation-induced DNA damage response. Acta Medica (Hradec Kralove) 2010; 53: 13–17.
Batista LF, Roos WP, Kaina B, Menck CF . p53 mutant human glioma cells are sensitive to UV-C-induced apoptosis due to impaired cyclobutane pyrimidine dimer removal. Mol Cancer Res 2009; 7: 237–246.
Vousden KH, Prives C . Blinded by the light: the growing complexity of p53. Cell 2009; 137: 413–431.
Tasdemir E, Chiara Maiuri M, Morselli E, Criollo A, D'Amelio M, Djavaheri-Mergny M et al. A dual role of p53 in the control of autophagy. Autophagy 2008; 4: 810–814.
Ryan KM . p53 and autophagy in cancer: guardian of the genome meets guardian of the proteome. Eur J Cancer 2011; 47: 44–50.
Budanov AV, Karin M . p53 target genes sestrin1 and sestrin2 connect genotoxic stress and mTOR signaling. Cell 2008; 134: 451–460.
Alexander A, Cai SL, Kim J, Nanez A, Sahin M, MacLean KH et al. ATM signals to TSC2 in the cytoplasm to regulate mTORC1 in response to ROS. Proc Natl Acad Sci USA 2010; 107: 4153–4158.
Morselli E, Tasdemir E, Maiuri MC, Galluzzi L, Kepp O, Criollo A et al. Mutant p53 protein localized in the cytoplasm inhibits autophagy. Cell Cycle 2008; 7: 3056–3061.
Tasdemir E, Maiuri MC, Galluzzi L, Vitale I, Djavaheri-Mergny M, D'Amelio M et al. Regulation of autophagy by cytoplasmic p53. Nat Cell Biol 2008; 10: 676–687.
Yoon JH, Ahn SG, Lee BH, Jung SH, Oh SH . Role of autophagy in chemoresistance: regulation of the ATM-mediated DNA-damage signaling pathway through activation of DNA-PKcs and PARP-1. Biochem Pharmacol 2012; 83: 747–757.
Zeng X, Kinsella TJ . BNIP3 is essential for mediating 6-thioguanine- and 5-fluorouracil-induced autophagy following DNA mismatch repair processing. Cell Res 20: 665–675.
Zeng X, Kinsella TJ . Mammalian target of rapamycin and S6 kinase 1 positively regulate 6-thioguanine-induced autophagy. Cancer Res 2008; 68: 2384–2390.
Guo R, Chen J, Zhu F, Biswas AK, Berton TR, Mitchell DL et al. E2F1 localizes to sites of UV-induced DNA damage to enhance nucleotide excision repair. J Biol Chem 2001; 285: 19308–19315.
Marabese M, Vikhanskaya F, Rainelli C, Sakai T, Broggini M . DNA damage induces transcriptional activation of p73 by removing C-EBPalpha repression on E2F1. Nucleic Acids Res 2003; 31: 6624–6632.
Polager S, Ofir M, Ginsberg D . E2F1 regulates autophagy and the transcription of autophagy genes. Oncogene 2008; 27: 4860–4864.
Eby KG, Rosenbluth JM, Mays DJ, Marshall CB, Barton CE, Sinha S et al. ISG20L1 is a p53 family target gene that modulates genotoxic stress-induced autophagy. Mol Cancer 2010; 9: 95.
Crighton D, O'Prey J, Bell HS, Ryan KM . p73 regulates DRAM-independent autophagy that does not contribute to programmed cell death. Cell Death Differ 2007; 14: 1071–1079.
Irwin MS, Kondo K, Marin MC, Cheng LS, Hahn WC, Kaelin WG Jr . Chemosensitivity linked to p73 function. Cancer Cell 2003; 3: 403–410.
Zaika E, Wei J, Yin D, Andl C, Moll U, El-Rifai W et al. p73 protein regulates DNA damage repair. FASEB J 2011; 25: 4406–4414.
Ikawa S, Nakagawara A, Ikawa Y . p53 family genes: structural comparison, expression and mutation. Cell Death Differ 1999; 6: 1154–1161.
Guo R, Chen J, Zhu F, Biswas AK, Berton TR, Mitchell DL et al. E2F1 localizes to sites of UV-induced DNA damage to enhance nucleotide excision repair. J Biol Chem 2010; 285: 19308–19315.
Ravikumar B, Sarkar S, Davies JE, Futter M, Garcia-Arencibia M, Green-Thompson ZW et al. Regulation of mammalian autophagy in physiology and pathophysiology. Physiol Rev 2010; 90: 1383–1435.
Hou W, Han J, Lu C, Goldstein LA, Rabinowich H . Autophagic degradation of active caspase-8: a crosstalk mechanism between autophagy and apoptosis. Autophagy 2010; 6: 891–900.
Qiang L, Wu C, Ming M, Viollet B, He YY . Autophagy controls p38 activation to promote cell survival under genotoxic stress. J Biol Chem 2013; 288: 1603–1611.
Zhang C, Lin M, Wu R, Wang X, Yang B, Levine AJ et al. Parkin, a p53 target gene, mediates the role of p53 in glucose metabolism and the Warburg effect. Proc Natl Acad Sci USA 2011; 108: 16259–16264.
Nezis IP, Shravage BV, Sagona AP, Lamark T, Bjorkoy G, Johansen T et al. Autophagic degradation of dBruce controls DNA fragmentation in nurse cells during late Drosophila melanogaster oogenesis. J Cell Biol 2010; 190: 523–531.
Yu L, Wan F, Dutta S, Welsh S, Liu Z, Freundt E et al. Autophagic programmed cell death by selective catalase degradation. Proc Natl Acad Sci USA 2006; 103: 4952–4957.
Siggens L, Figg N, Bennett M, Foo R . Nutrient deprivation regulates DNA damage repair in cardiomyocytes via loss of the base-excision repair enzyme OGG1. FASEB J 2012; 26: 2117–2124.
Wang J, Whiteman MW, Lian H, Wang G, Singh A, Huang D et al. A non-canonical MEK/ERK signaling pathway regulates autophagy via regulating Beclin 1. J Biol Chem 2009; 284: 21412–21424.
Wang ZH, Xu L, Duan ZL, Zeng LQ, Yan NH, Peng ZL . Beclin 1-mediated macroautophagy involves regulation of caspase-9 expression in cervical cancer HeLa cells. Gynecol Oncol 2007; 107: 107–113.
Nascimento-Ferreira I, Santos-Ferreira T, Sousa-Ferreira L, Auregan G, Onofre I, Alves S et al. Overexpression of the autophagic beclin-1 protein clears mutant ataxin-3 and alleviates Machado-Joseph disease. Brain 2011; 134 (Pt 5): 1400–1415.
Rodriguez-Rocha H, Garcia-Garcia A, Panayiotidis MI, Franco R . DNA damage and autophagy. Mutat Res 2011; 711: 158–166.
Hughson LR, Poon VI, Spowart JE, Lum JJ . Implications of therapy-induced selective autophagy on tumor metabolism and survival. Int J Cell Biol 2012; 2012: 872091.
Stevnsner T, Muftuoglu M, Aamann MD, Bohr VA, The role of Cockayne Syndrome group. B (CSB) protein in base excision repair and aging. Mech Ageing Dev 2008; 129: 441–448.
Stevnsner T, Nyaga S, de Souza-Pinto NC, van der Horst GT, Gorgels TG, Hogue BA et al. Mitochondrial repair of 8-oxoguanine is deficient in Cockayne syndrome group B. Oncogene 2002; 21: 8675–8682.
Aamann MD, Sorensen MM, Hvitby C, Berquist BR, Muftuoglu M, Tian J et alCockayne syndrome group. B protein promotes mitochondrial DNA stability by supporting the DNA repair association with the mitochondrial membrane. FASEB J 2010; 24: 2334–2346.
Scheibye-Knudsen M, Ramamoorthy M, Sykora P, Maynard S, Lin PC, Minor RK et alCockayne syndrome group. B protein prevents the accumulation of damaged mitochondria by promoting mitochondrial autophagy. J Exp Med 2012; 209: 855–869.
Vessoni AT, Muotri AR, Okamoto OK . Autophagy in stem cell maintenance and differentiation. Stem Cells Dev 2012; 21: 513–520.
Garcia-Prat L, Sousa-Victor P, Munoz-Canoves P . Functional dysregulation of stem cells during aging: a focus on skeletal muscle stem cells. FEBS J 2013 e-pub ahead of print 2 March 2013; doi:10.1111/febs.12221.
Filipcik P, Cente M, Ferencik M, Hulin I, Novak M . The role of oxidative stress in the pathogenesis of Alzheimer's disease. Bratisl Lek Listy 2006; 107: 384–394.
Nakabeppu Y, Tsuchimoto D, Yamaguchi H, Sakumi K . Oxidative damage in nucleic acids and Parkinson's disease. J Neurosci Res 2007; 85: 919–934.
Khandelwal PJ, Herman AM, Hoe HS, Rebeck GW, Moussa CE . Parkin mediates beclin-dependent autophagic clearance of defective mitochondria and ubiquitinated Abeta in AD models. Hum Mol Genet 2011; 20: 2091–2102.
Kao SY . DNA damage induces nuclear translocation of parkin. J Biomed Sci 2009; 16: 67.
Kao SY . Regulation of DNA repair by parkin. Biochem Biophys Res Commun 2009; 382: 321–325.
Majumder S, Richardson A, Strong R, Oddo S . Inducing autophagy by rapamycin before, but not after, the formation of plaques and tangles ameliorates cognitive deficits. PLoS One 2011; 6: e25416.
Taylor R, Goldman SJ . Mitophagy and disease: new avenues for pharmacological intervention. Curr Pharm Des 17: 2056–2073.
Ionov Y, Nowak N, Perucho M, Markowitz S, Cowell JK . Manipulation of nonsense mediated decay identifies gene mutations in colon cancer cells with microsatellite instability. Oncogene 2004; 23: 639–645.
Kang MR, Kim MS, Oh JE, Kim YR, Song SY, Kim SS et al. Frameshift mutations of autophagy-related genes ATG2B, ATG5, ATG9B and ATG12 in gastric and colorectal cancers with microsatellite instability. J Pathol 2009; 217: 702–706.
Kim MS, Jeong EG, Ahn CH, Kim SS, Lee SH, Yoo NJ . Frameshift mutation of UVRAG, an autophagy-related gene, in gastric carcinomas with microsatellite instability. Hum Pathol 2008; 39: 1059–1063.
Maiuri MC, Tasdemir E, Criollo A, Morselli E, Vicencio JM, Carnuccio R et al. Control of autophagy by oncogenes and tumor suppressor genes. Cell Death Differ 2009; 16: 87–93.
Marino G, Salvador-Montoliu N, Fueyo A, Knecht E, Mizushima N, Lopez-Otin C . Tissue-specific autophagy alterations and increased tumorigenesis in mice deficient in Atg4C/autophagin-3. J Biol Chem 2007; 282: 18573–18583.
Zhao Z, Oh S, Li D, Ni D, Pirooz SD, Lee JH et al. A dual role for UVRAG in maintaining chromosomal stability independent of autophagy. Dev Cell 2012; 22: 1001–1016.
Knaevelsrud H, Ahlquist T, Merok MA, Nesbakken A, Stenmark H, Lothe RA et al. UVRAG mutations associated with microsatellite unstable colon cancer do not affect autophagy. Autophagy 2010; 6: 863–870.
Ni Chonghaile T, Letai A . Who put the "A" in Atg12: autophagy or apoptosis? Mol Cell 2011; 44: 844–845.
Green DR, Kroemer G . Cytoplasmic functions of the tumour suppressor p53. Nature 2009; 458: 1127–1130.
Rosenbluth JM, Pietenpol JA . mTOR regulates autophagy-associated genes downstream of p73. Autophagy 2009; 5: 114–116.
Teitz T, Penner M, Eli D, Stark M, Bakhanashvili M, Naiman T et al. Isolation by polymerase chain reaction of a cDNA whose product partially complements the ultraviolet sensitivity of xeroderma pigmentosum group C cells. Gene 1990; 87: 295–298.
Biswas AK, Johnson DG . Transcriptional and nontranscriptional functions of E2F1 in response to DNA damage. Cancer Res 2012; 72: 13–17.
Stevens C, La Thangue NB . The emerging role of E2F-1 in the DNA damage response and checkpoint control. DNA Repair (Amst) 2004; 3: 1071–1079.
Chen LH, Loong CC, Su TL, Lee YJ, Chu PM, Tsai ML et al. Autophagy inhibition enhances apoptosis triggered by BO-1051, an N-mustard derivative, and involves the ATM signaling pathway. Biochem Pharmacol 2011; 81: 594–605.
Leung M, Rosen D, Fields S, Cesano A, Budman DR . Poly(ADP-ribose) polymerase-1 inhibition: preclinical and clinical development of synthetic lethality. Mol Med 2011; 17: 854–862.
Javle M, Curtin NJ . The role of PARP in DNA repair and its therapeutic exploitation. Br J Cancer 2011; 105: 1114–1122.
Casorelli I, Bossa C, Bignami M . DNA damage and repair in human cancer: molecular mechanisms and contribution to therapy-related leukemias. Int J Environ Res Public Health 2012; 9: 2636–2657.
Zeng X, Yan T, Schupp JE, Seo Y, Kinsella TJ . DNA mismatch repair initiates 6-thioguanine—induced autophagy through p53 activation in human tumor cells. Clin Cancer Res 2007; 13: 1315–1321.
Knizhnik AV, Roos WP, Nikolova T, Quiros S, Tomaszowski KH, Christmann M et al. Survival and death strategies in glioma cells: autophagy, senescence and apoptosis triggered by a single type of temozolomide-induced DNA damage. PLoS One 2013; 8: e55665.
Amaravadi RK, Yu D, Lum JJ, Bui T, Christophorou MA, Evan GI et al. Autophagy inhibition enhances therapy-induced apoptosis in a Myc-induced model of lymphoma. J Clin Invest 2007; 117: 326–336.
Shin SY, Hyun J, Yu JR, Lim Y, Lee YH . 5-Methoxyflavanone induces cell cycle arrest at the G2/M phase, apoptosis and autophagy in HCT116 human colon cancer cells. Toxicol Appl Pharmacol 2011; 254: 288–298.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Edited by E White
Rights and permissions
About this article
Cite this article
Vessoni, A., Filippi-Chiela, E., Menck, C. et al. Autophagy and genomic integrity. Cell Death Differ 20, 1444–1454 (2013). https://doi.org/10.1038/cdd.2013.103
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cdd.2013.103
Keywords
This article is cited by
-
USP24-i-101 targeting of USP24 activates autophagy to inhibit drug resistance acquired during cancer therapy
Cell Death & Differentiation (2024)
-
Autophagy and nuclear morphometry are associated with histopathologic features in esophageal squamous cell carcinoma
Journal of Molecular Medicine (2024)
-
A new vulnerability to BET inhibition due to enhanced autophagy in BRCA2 deficient pancreatic cancer
Cell Death & Disease (2023)
-
Cytotoxic n-Hexane Fraction of the Egyptian Pteris vittata Functions as Anti-breast Cancer Through Coordinated Actions on Apoptotic and Autophagic Pathways
Applied Biochemistry and Biotechnology (2023)
-
CDKL3 Promotes Non-small Cell Lung Cancer by Suppressing Autophagy Via Activation of PI3K/Akt/mTOR Pathway
Molecular Biotechnology (2023)
