Grajevis Bakatunkanda’s mother knew the signs: when her son lost interest in dinner, that meant the pain was on its way. It would strike, like clockwork, nearly every week. Soon the shy, skinny boy would be at the hospital near their home in the Democratic Republic of the Congo, where doctors would provide morphine for the pain and invariably diagnose him with malaria.
It turns out the doctors were wrong. The culprits were not parasites, but Bakatunkanda’s own red blood cells. Normally soft and springy, some of the boy’s cells were becoming deformed and stiff, like splinters of wood. They would lodge in his capillaries, choking the blood flow to vital organs and sending waves of crushing pain into his back and chest.
It wasn’t until the family immigrated to Cape Town, South Africa, in 2003, that they learned Bakatunkanda had sickle-cell anaemia, one of the world’s most prevalent genetic disorders, and one that has been studied for more than a century. But the diagnosis did little to ease the boy’s pain: the cocktail of drugs that he was prescribed — each of them in use for more than half a century and none developed specifically for sickle-cell disease — failed to break the cycle.
Now, Bakatunkanda is 22, and modern solutions are on the horizon in the form of gene therapies. After decades of work and some painful setbacks, techniques that involve altering a person’s genome have begun to win approval for a handful of rare disorders. Scientists are now working to extend the latest advances — including some that use newer gene-editing technologies — to sickle-cell disease, a condition that affects some 20 million people worldwide (see ‘How to stop sickling’). There are more than half a dozen active clinical trials, and more are planned. “The studies are just literally coming back to back now,” says Lakshmanan Krishnamurti, a paediatric haematologist at Emory University in Atlanta, Georgia. “It’s a very exciting time.”

But sickle-cell disease could challenge the gene-therapy field both ethically and technologically. Gene therapies that have been approved for other conditions have come with price tags in excess of US$1 million. But sickle-cell disease is concentrated in regions of the world such as sub-Saharan Africa, India and the Caribbean, where few have the resources to foot such a hefty bill. The experimental treatments for sickle cell are also complex, requiring long hospital stays and the expertise of large academic medical centres. Even for people who can access such resources, the risks might not always be worth it.
As data drift in from early trials, scientists are working to improve their approaches, and funders have already begun to tackle the equity question. On 23 October, the US National Institutes of Health (NIH) and the Bill & Melinda Gates Foundation announced that they would invest at least $200 million over the next four years to bring gene-based treatments for sickle-cell disease and HIV to low-resource settings.
Bakatunkanda, who founded a support group for people with sickle-cell disease, is confident that gene therapy, if shown to be effective, will one day reach his country, despite its high cost and daunting complexity. “Definitely it will,” he says. “Because South Africa is rising.”
He and others must keep a tight rein on their expectations. “My patients with Internet access, now they are coming to me: ‘Can we go for gene therapy?’” says Dipty Jain, a paediatrician at the Government Medical College in Nagpur, India. “I advise them, ‘This is not yet for you’.”
A medical revolution
The elongated, oddly shaped blood cells typical of sickle-cell disease were first noted in 1910 in a young dental student from Grenada, West Indies, named Walter Clement Noel1. Forty years later, the underpinnings of the disease began to come into view, when biochemist Linus Pauling and his colleagues reported that changes in the structure of haemoglobin, the oxygen-carrying protein found in red blood cells, were altering the shape of the cells2.
The publication marked the first time the effects of a genetic disorder had been traced to their molecular roots. Pauling dubbed the condition “a molecular disease”. Some years later, researchers identified changes in the β-globin protein as responsible3. A mutation in both copies of the gene encoding for this protein results in disease; a single mutated copy correlates with few symptoms and protects the bearer from blood-dwelling parasites such as those causing malaria. This, in part, is why the disease exists in relatively high rates where malaria is endemic. “This is really the basis from where everything that we know today on human medical genetics has been developed,” says Ambroise Wonkam, a geneticist at the University of Cape Town.
Seventy years after Pauling’s discovery, sickle-cell disease is still underdiagnosed in many African countries, says haematologist Olu Akinyanju, the founder and first chairperson of the Sickle Cell Foundation Nigeria in Lagos. Yet early diagnosis can save lives. More than 300,000 people are born with the disease each year, and without prophylactic antibiotics and vaccines to help ward off other infections, most will die before the age of five. Those who survive face a lifetime of risk for pain crises, stroke and infection.
Sickle cell disease’s close association with low-income countries has meant that it has historically received little attention from pharmaceutical companies and governments in richer regions. Many African nations have such pressing public-health needs that it has been difficult to push sickle-cell disease to the top of their priority lists, says Akinyanju, who has campaigned for decades to get African governments to establish treatment plans.

Bakatunkanda takes a cocktail of older drugs to fight his disease. Treatment with gene-therapy sounds attractive, but he knows there are risks.Credit: Aurélie Marrier d'Unienville for Nature
Over the past ten years, however, Akinyanju and others have noticed a shift. As advocates and clinicians push for newborn screening and early intervention, people with sickle-cell disease have begun living longer. The condition is not as stigmatized as it once was. Akinyanju proudly ticks off friends with sickle-cell disease who have lived into their 60s and beyond, becoming doctors, judges and world travellers.
The World Health Organization and the American Society of Haematology have also worked to bring the disease to the attention of researchers and pharmaceutical companies. And Bakatunkanda and other immigrants have raised awareness in wealthier nations, says Wonkam. There are signs that this attention is paying off. On 25 November, the US Food and Drug Administration approved a drug for sickle-cell that aims to reduce clumping between haemoglobin molecules.
But although gene therapy might seem a rational approach for one of the world’s best-known genetic diseases, the field has faced its setbacks. Early attempts were marred by the high-profile death of Jesse Gelsinger in 1999, who was participating in one of the first gene-therapy clinical trials. A procedure used during the trial to replace immune-system genes in blood stem cells caused leukaemia in several of the participants.
Against that backdrop, some felt that it was premature to apply gene therapy to sickle-cell disease, says haematologist David Williams at Boston Children’s Hospital in Massachusetts. “Sickle cell is not an immediately lethal disease,” he says. “In some ways, it wouldn’t be ethical to treat those patients with a highly risky experimental approach.”
Furthermore, the tools were not yet up to the task, says Donald Kohn, a specialist in paediatric bone-marrow transplants at the University of California, Los Angeles. If researchers were to shuttle in a normal haemoglobin gene, it would need to be able to crank out large amounts of protein to sufficiently mute the effects of the sickled version. Early gene-therapy technologies were not able to express genes in human cells at such high levels, says Kohn.
But despite the setbacks, some gene-therapy researchers pushed on, developing safer and more potent ways to shuttle genes into cells. They broke through in 2016, when the European Commission approved a gene therapy for treating ADA-SCID, a rare immune disorder that often kills children before their first birthday. Then in 2017, the US Food and Drug Administration approved a gene therapy to treat a rare form of blindness.
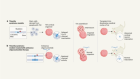
By this time, some researchers had turned their attention back to sickle cell, armed with improved tools and with the backing of the biotechnology industry. Current trials are taking a variety of approaches. Kohn is trying to insert a copy of the β-globin gene that has been modified to resist sickling. So is Bluebird Bio, a company in Cambridge, Massachusetts. The firm looks set to be the first to win approval to market such a treatment in the United States, according to Yaron Werber, a biotechnology analyst at Cowen, a financial-services company in New York City.
Others are introducing modified copies of the genes that encode fetal haemoglobin, a form of the protein that is produced in the developing fetus but usually shuts off soon after birth. Fetal haemoglobin is an attractive option because it works about as well as the adult version, and it prevents defective haemoglobins from clumping together.
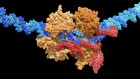
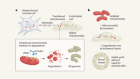
A third approach seeks to block a mechanism that switches off production of fetal haemoglobin after birth. The usual off-switch is a protein called BCL11A, and suppressing it in mice with sickle-cell disease can keep fetal-haemoglobin levels high well into adulthood and prevent symptoms of the disease4. In Boston, Williams has licensed technology to Bluebird Bio that uses a technique called RNA interference to dial down expression of the gene encoding BCL11A in blood stem cells. Sangamo Therapeutics in Richmond, California, in partnership with Sanofi in Paris, is using gene-editing tools called zinc-finger nucleases to create mutations that disable the gene. And Vertex Pharmaceuticals in Boston has teamed up with CRISPR Therapeutics in Cambridge, Massachusetts, to do much the same using the CRISPR–Cas9 gene-editing technique. In all three approaches, blood-producing stem cells are removed from the body, genetically altered — often with the help of a virus — and then reintroduced into the bone marrow. Before the cells are replaced, participants are typically treated with a chemotherapy called busulfan to destroy the remaining diseased stem cells and help the reintroduced, genetically altered cells to take over.
That kind of regimen is risky: participants can develop acute and severe anaemia. The treatment wipes out their white blood cells, and wreaks havoc on the lining of the gut, potentially leaving them dependent on intravenous nutrition. Many will need to stay in the hospital for more than a month. The chemotherapy also causes infertility, and can cause cancer later in life.
This means that gene therapy would probably be used only in those with the most serious forms of sickle-cell disease. Yet many of those people will also have heart, kidney or liver damage that would make the chemotherapy too dangerous.

Ageing brings some benefits to people with sickle-cell disease. Bakatunkanda now deals with fewer pain crises and can even lift weights and hike.Credit: Aurélie Marrier d'Unienville for Nature
Sickle-cell disease complicates the therapy in other ways, too. In many cases, when doctors harvest bone marrow, patients first receive a drug that makes it easier to collect blood stem cells. But that is too dangerous to use in people with sickle-cell disease because it raises the risk of pain crises. And because diseased red blood cells die faster than healthy ones, the stem cells in a person with sickle-cell disease must work harder to produce new blood cells. This can leave them in poor condition for harvest and growth in laboratory cultures. As a result, participants often need blood transfusions just before harvest to ease the stress on their stem cells. Despite these challenges, early signs of success have been making headlines. One of the men in Williams’s RNA-interference trial has been symptom-free for one year. And the first patient in the CRISPR trial has now left the hospital after completing the gruelling therapy. On 19 November, Vertex and CRISPR Therapeutics announced that the person has not experienced any pain crises and has maintained a high level of fetal haemoglobin for four months. Both trials have generated excitement on social media — too much, in some cases. “I have difficulty right now with folks being excited about the discharge of a patient from the hospital, as if that were tantamount to a cure,” says Alexis Thompson, a haematologist at Northwestern University in Chicago, Illinois. “That’s a pretty low bar.”
Still, there is cause for cautious optimism. So far, none of these trials has been stopped for safety concerns. And Bluebird Bio has treated 13 people, some of whom have been monitored for a year after treatment with no severe pain crises, the company reported in June. The gene therapy used was approved in the European Union in June to treat some people with a related genetic blood disorder called β-thalassaemia.
But a major concern for many people is cost. The treatment for β-thalassaemia runs to roughly $1.8 million — not including the hospital stay and other associated costs.
This is still potentially cheaper than standard treatments over the course of a lifetime, says Mani Foroohar, an analyst at the investment bank SVB Leerink in Boston, Massachusetts. Also, Bluebird Bio has established an unusual fee structure: payments are made over the course of five years, and can be halted if the treatment stops working. Still, Foroohar says, it’s not clear whether the same model will be possible in other regions.
The price tag is certainly well beyond the means of many of Jain’s patients in central India, who come to her hospital because they can’t otherwise afford the roughly $3 per month that it costs for standard treatments. Even in the United States, access to the gene therapies is likely to be a challenge. This is particularly true for Black Americans, who tend to have more limited access to health care than White Americans. Although the trials are still in their early days, Krishnamurti urges interested people to begin advocating immediately for access to the therapies. “It’s an enormous ethical issue,” says Krishnamurti, who counsels people with sickle-cell disease each week from his hospital in Atlanta. “In my community conversations, I say, ‘You had better be at the table, otherwise these decisions will be made without your input.’”
At the Cincinnati Children’s Hospital in Ohio, haematologist Punam Malik is hoping to take the first steps towards making gene therapy cheaper and simpler. Malik trained as a doctor in India, where she saw many people with sickle-cell disease and related conditions. When she immigrated to the United States about 30 years ago, she vowed to make sure that her research would benefit people in resource-poor countries.
Now, Malik is leading a trial that introduces stem cells that produce fetal haemoglobin. It uses low doses of a drug called melphalan to remove diseased cells from the bone marrow, which should make the treatment less toxic than the usual busulfan. Her hope is that the technique will reduce the need for a long hospital stay, making the treatment cheaper, safer and more practical.
But the approach has been criticized by others, who worry that the low-dose approach might leave behind some uncorrected cells, and make the therapy less effective. “You want to do as well as you can,” says Stuart Orkin, who studies blood disorders at Boston Children’s Hospital.
Malik counters that once a high dose has been established as effective, it is hard to scale it back. She points to the example of cancer chemotherapy: in some cancers, researchers are reducing the dose of some drugs and finding that they work just as well as, if not better than, the higher doses tried initially. But it has taken oncologists decades to take that step, she notes. “I might fall flat on my face, and I might have to dial up. But it will be very difficult for the others to dial down,” she says.
Her trial has also run up against the practical realities of exporting gene therapies to regions with fewer resources. Her team received FDA approval to carry out the trial only at Cincinnati Children’s Hospital. But after Malik gave a talk at a conference in Jamaica, someone with sickle-cell disease approached her asking for help and describing multiple hospital visits for pain crises.
So, Malik developed a collaboration in Jamaica. “I felt we had to,” she says. It took the team about two years to get the necessary approvals and funding. And then the clinical team in Jamaica ran up against another problem: lack of reliable blood for transfusion.
The team reported in April that its first patient has experienced only two pain crises in the 18 months since treatment ended and has maintained high levels of haemoglobin. The team has since treated a second person, and two more are lined up to take part, Malik says.
The trouble is not only the expense and practicalities, but also the availability of clinicians and facilities who can handle stem-cell transplants. Rural regions already struggle to supply people with with hydroxyurea, a relatively cheap medicine that reduces the rate of pain crises. It’s hard to imagine these regions having enough personnel to monitor recipients of gene therapy over the long term, says anthropologist Duana Fullwiley at Stanford University in California.
Some argue that it is too early to think about such issues. “If we refine the technology, it will be affordable in the long run,” says Wonkam. “The price right now for me is not the problem. The focus needs to be on the efficiency.”
But others think that the time to start thinking about global access is now. To do otherwise “would be almost unethical”, says NIH director Francis Collins.
Collins thinks that the key to fulfilling the NIH’s project with the Gates foundation will be in finding ways to deliver the corrected genes or gene-editing tools to bone-marrow stem cells that don’t involve having to remove the cells first, making therapies cheaper and easier to deliver. It is an ambitious goal — and one that is occasionally met with scepticism, Collins says. “Sometimes there was a vague sense of, ‘Boy, you’re just outside the boundaries of reality there, Collins’,” he says.
There are already suggestions that the viruses typically used to shuttle genes into cells in a dish can be modified to insert genes into blood-producing stem cells while they’re still in the body, notes Kohn. “It’s a great lofty goal,” he says. “I think the science is advancing pretty rapidly.”
For Bakatunkanda, his salvation turned out to be ageing, not medicine. Some people with sickle-cell disease fare worse as children than as adults, he says, and he thinks he is one of them. He still has crises, but not nearly as often. In recent months, he has taken on activities such as hiking and bodybuilding that he once thought were off-limits. “I just know how far I can push myself,” he says.
But he would prefer a life without the constant threat of pain crises and strokes. He is aware of the promise of gene therapies, but knows that it is not yet clear whether they will provide a cure. “I would prefer that,” he says. “But at the moment it’s not a guarantee.”
 First CRISPR editing trial results assuage safety concerns
First CRISPR editing trial results assuage safety concerns
 CRISPR deployed to combat sickle-cell anaemia
CRISPR deployed to combat sickle-cell anaemia
 First gene therapy for β-thalassemia approved
First gene therapy for β-thalassemia approved
 Scientists use gene-edited stem cells to treat HIV — with mixed success
Scientists use gene-edited stem cells to treat HIV — with mixed success