
Abstract
We report a Belgian patient with early-onset cerebellar ataxia, progressive spasticity, learning difficulties and moderate perceptive hearing loss. Array-Comparative Genomic Hybridisation (aCGH) detected a 1.54 Mb deletion on chromosome 13q12.12. This microdeletion occurred de novo and encompasses the SACS gene. Mutations in SACS are known to cause a recessive condition, similar to the patient's phenotype, called autosomal recessive spastic ataxia of Charlevoix–Saguenay (ARSACS). Sequencing of the remaining SACS allele revealed a hemizygous mutation c.10517T>C in exon 9, resulting in an amino-acid substitution (p.F3506S). This is the first patient with ARSACS that carries a de novo chromosomal deletion comprising SACS. We demonstrate the presence of homologous segmental duplications at the breakpoint-containing regions. This suggests non-allelic homologous recombination as the mechanism generating this deletion and explains the previous description of copy number variations of this region. This finding confirms the contribution of aCGH to gene identification in autosomal recessive disorders.
Similar content being viewed by others

Introduction
Autosomal recessive spastic ataxia of Charlevoix–Saguenay (ARSACS) is a distinctive form of hereditary early onset spastic ataxia that is characterised by spasticity, dysarthria, nystagmus, distal muscle wasting, and finger and foot deformities. Hypermyelination of retinal nerve fibres and atrophy of the superior part of the vermis are the main neuropathological findings. Initially, ARSACS was described among French Canadians in the Charlevoix–Saguenay region of Quebec. After the identification of the SACS gene on proximal chromosome 13q as the causative gene,1 mutations in this gene were detected in patients from other populations, notably Tunisia,2 Turkey,3 Italy,4, 5 Spain,6 Japan7, 8, 9, 10 and Belgium.11 Up to now, approximately 30 different loss-of-function mutations have been reported in the SACS gene.12, 13 We here report a Belgian patient with ARSACS who is a compound heterozygote for an inherited point mutation in the SACS gene on the paternal allele and a de novo microdeletion of maximally 1.47 Mb, comprising the SACS gene, on the maternal chromosome.
Case report
This 28-year-old Belgian male was born as the first child of healthy, non-consanguineous parents, after caesarean section due to breech presentation. Birth weight and length were normal, 3450 g (50th centile) and 51 cm (50th centile), respectively. He started walking at the age of 19 months but never acquired a normal gait, initially showing pelvic instability and progressively declining owing to weakness and contractures of the lower limb muscles. Neurological examination at age 16 revealed muscular hypotrophy of the four limbs, especially of the distal muscles, and spastic hypertonia and paresis of the lower limbs. Muscle strength of the upper limbs was slightly diminished, especially of the proximal muscles. Although patellar reflexes were very intense, ankle jerks were absent and plantar reflexes were bilaterally indifferent. Osteotendon reflexes of the upper limbs were weak. The patient was walking independently with effort, limping with endorotation of the feet. Sensation was not affected. He showed mild dysarthria and was enlisted in special education for mild learning difficulties, with a full scale IQ of 88 on the Wechsler Adult Intelligence Scale (WAIS). Ophthalmologic evaluation was normal. Electromyography showed signs of an axonal sensorimotor polyneuropathy. He had a moderate perceptive bilateral hearing loss. Although brain auditory evoked potentials were normal on the right side, on the left side, waves II and III were absent and there was a prolonged I–V interpeak latency. No scalp response was detected by somatosensory evoked potentials of the nervus tibialis. The peripheral response to somatosensory stimulation of the nervus medianus showed a bilateral minor increase in latency, whereas central conduction was severely prolonged.
At age 26, neurological re-examination revealed progressive spasticity and ataxic gait. At that time, he was walking with crutches. Brain MRI showed atrophy of the vermis superior and the superior cerebellar peduncles; otherwise, brain parenchyma was normal and there were no white matter abnormalities (Figure 1).

Brain MRI at the age of 26 years showing atrophy of the vermis superior (black arrows) and the superior cerebellar peduncles. No anomalies of the cerebral hemispheres were detected.
Materials and methods
Conventional karyotyping with G-banding was performed according to standard techniques. Array-Comparative Genomic Hybridisation (aCGH) was carried out as described.14 We used a microarray containing 3625 BAC/PACs chosen genome wide at 1 Mb intervals and a full tiling array for chromosome 13, with a resolution of 100 kb, containing 1010 BAC/PACs obtained from BACPAC Resources Center (Children's Hospital, Oakland, CA, USA). Fluorescent in situ hybridisation (FISH) using probe RP11-760M1 and real-time quantitative PCR (qPCR) using primers A, B, C, D, E, F and G (Figure 3; Table 1) were performed as described.15 The sequences of the breakpoint-containing regions were investigated using Miropeats (thresholds 400 bp).16 Homology of low copy repeats was investigated using AlignX with standard settings (Vector NTI Advance 10.0.1, Invitrogen). The polymorphic repeat D13S232 was analysed by PCR using specific primers (Table 1) on 50 ng of genomic DNA using Expand Long Template (Roche) and buffer 3 according to the manufacturer's instructions. Mutation analysis of the SACS gene was performed as described.17 This study was approved by the Medical Ethical Committees of University Hospital Leuven and Jichi Medical University.

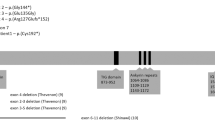
Overview of the deleted region on 13q12.12 comprising six genes, SGCG, SACS, TNFRSF19, MIPEP, SPATA13 and C1QTNF9 (black arrows). PARP4 is not deleted (white arrow). The location of polymorphic marker D13S232 and qPCR primers A, B, C, D, E, F and G is indicated by red vertical lines (primers inside the deleted region are displayed in red). The deleted BAC/PACs (RP11-760M1 and 32k clones RP11-357N10 till RP11-530B16) are displayed in black and the flanking non-deleted BAC/PACs as white boxes. The minimal 1.37 Mb deleted region is shown below as a black box. Grey boxes indicate the breakpoint-containing regions. Red boxes at the bottom line indicate homologous sequences in the breakpoint-containing regions. (See online version for colour figure).
Results
Conventional karyotyping showed a normal result. Higher-resolution molecular karyotyping by aCGH, using a BAC/PAC microarray with 1 Mb resolution, detected a deletion of a single DNA clone RP11-760M1, located on chromosome 13q12.12, partially overlapping the SACS gene. This microdeletion was confirmed by FISH using probe RP11-760M1 (Figures 2b and 3). Involvement of the SACS gene was demonstrated by qPCR using primer pair A (Figure 3; Table 1). The deleted region was further delineated by means of a chromosome 13 specific tiling array. The deletion spans maximally 1.54 Mb and affects 6 genes (Figures 2a and 3), with breakpoints mapping to a region 22335637-22383193 (proximal) to 23801734-23879339 (distal).

(a) Microarray containing BAC/PAC clones covering chromosome 13 with a tiling resolution of 100 kb, showing a maximal deletion of 1.54 Mb on 13q12.12 and spanning clones from RP11-357N10 to RP11-530B16. (b) The microdeletion was confirmed by FISH using probe RP11-760M1. (c) Analysis of the polymorphic marker D13S232 using patient and parental DNA revealed that the 13q12.12 microdeletion occurred de novo and was located on the maternal chromosome. (See online version for colour figure).
Analysis of the polymorphic marker D13S232 on patient and parental DNA revealed that the 13q12.12 microdeletion occurred de novo and was located on the maternally inherited chromosome 13 (Figure 2c).
The sequences of the breakpoint-containing regions were investigated using Miropeats and AlignX. This revealed the presence of two highly homologous sequences. A first 14 kb sequence starting from 22 384 kb was found to be 94% homologous to a 14 kb sequence starting from 23 796 kb. A second 43 kb sequence starting from 22 400 kb was found to be 91% homologous to a 44 kb sequence starting from 23 822 kb. To fine map the breakpoints, qPCR primers were designed flanking these four regions (primers B, C, D, E, F and G (Figure 3; Table 1)). This demonstrated that the breakpoint-containing regions are located between the primers F and G (proximal) and C and D (distal) and thus coincide with the second 44/43 kb homologous sequences: 22399440–22442680 (proximal) and 23813131–23866606 (distal).
Sequencing of the SACS gene revealed a hemizygous mutation in exon 9 (c.10517T>C) (GeneBank accession number, AL157766) in the SACS gene resulting in an amino-acid change, F3506S (Figure 4). The father was carrier of this mutation, which was not detected in 100 normal Belgian control chromosomes.

(a) Partial results of nucleotide sequence analysis of exon 9 of the SACS gene. A T-to-C change in the patient and his father with a heterozygous state, and the normal sequence in his mother are shown. (b) The PCR products after BtsC1 digestion; lane M, 100 bp size markers; lane 1, the father with a heterozygous T-to-C change; lane 2, the patient with a heterozygous T-to-C change; lane 3, the mother with normal sequence. The wild-type BtsC1-digested PCR products gave four bands (260, 125, 122 and 106 bp). The BtsC1-digested PCR products with a heterozygous T-to-C change gave six bands (260, 216, 125, 122, 106 and 44 bp). As the patient had a microdeletion of chromosome 13q in one allele, his BtsC1-digested PCR products gave five bands (216, 125, 122, 106 and 44 bp). The BtsC1-digested band of 44 bp cannot be observed.
Discussion
In this patient with a complicated form of spastic paraparesis, a de novo microdeletion of maximally 1.54 Mb, encompassing six genes, was detected using aCGH, a high-resolution molecular cytogenetic technique. The SACS gene was located in this region, and the patient's phenotype was typical of ARSACS. In addition, brain imaging revealed the characteristic cerebellar anomalies. This diagnosis was formally confirmed by the demonstration of a paternally inherited point mutation on the other allele. The F3506S variant, caused by c.10517T>C, is predicted by the PolyPhen tool to be probably damaging to the function of the Sacsin protein (PSIC score difference 2.225), as it is situated in a conserved ARM repeat.18
Up to now, all reported mutations in ARSACS patients were point mutations or deletions of a few nucleotides in the SACS gene, resulting in a loss of function. We here report the first case of ARSACS where one of the mutations is a gross chromosomal deletion comprising the SACS gene. Besides SACS, the deleted region contains five other genes, namely SGCG, TNFRSF19, MIPEP, SPATA13 and C1QTNF9. Interestingly, the only feature by which the present patient differs from other cases with ARSACS is the presence of moderate perceptive hearing loss. None of the five genes has a known role in hearing (loss), and therefore it remains uncertain whether the hearing loss is a rare manifestation of ARSACS or is caused by haploinsufficiency of one of the flanking genes (Figure 3).
The aberrant region most likely coincides with 2 formerly described copy number variants, one deletion and one duplication. One 1.4 Mb deletion was described by Pinto et al19 in a cohort of 506 unrelated healthy Caucasian individuals. Two duplications comprising SACS entirely were found by Simon-Sanchez et al20 in DNA extracted from EBV immortalised cell lines, derived from lymphocytes of 276 elderly neurologically normal subjects. In one subject, the duplication was confirmed in DNA extracted directly from the blood.20 The presence of the segmental duplication suggests non-allelic homologous recombination as a recurrent mechanism generating the imbalances found in our patient and the aforementioned subjects. Therefore, ARSACS can be added to the growing list of genomic disorders.
The present observation illustrates that in addition to its usefulness in the detection of candidate genes for autosomal dominant disorders, aCGH may contribute to gene identification for autosomal recessive disorders, as discussed by Coman and Gardner.21
In conclusion, we report on the first case of ARSACS carrying an atypical mutation, that is a de novo chromosomal deletion comprising SACS. This finding underscores the need to include dosage analysis of the SACS gene in mutation analysis in patients with spastic ataxia.
Accession codes
References
Engert JC, Berube P, Mercier J et al: ARSACS, a spastic ataxia common in northeastern Quebec, is caused by mutations in a new gene encoding an 11.5-kb ORF. Nat Genet 2000; 24: 120–125.
El Euch-Fayache G, Lalani I, Amouri R et al: Phenotypic features and genetic findings in sacsin-related autosomal recessive ataxia in Tunisia. Arch Neurol 2003; 60: 982–988.
Richter AM, Ozgul RK, Poisson VC, Topaloglu H : Private SACS mutations in autosomal recessive spastic ataxia of Charlevoix–Saguenay (ARSACS) families from Turkey. Neurogenetics 2004; 5: 165–170.
Grieco GS, Malandrini A, Comanducci G et al: Novel SACS mutations in autosomal recessive spastic ataxia of Charlevoix–Saguenay type. Neurology 2004; 62: 103–106.
Criscuolo C, Banfi S, Orio M et al: A novel mutation in SACS gene in a family from southern Italy. Neurology 2004; 62: 100–102.
Criscuolo C, Sacca F, De Michele G et al: Novel mutation of SACS gene in a Spanish family with autosomal recessive spastic ataxia. Mov Disord 2005; 20: 1358–1361.
Ogawa T, Takiyama Y, Sakoe K et al: Identification of a SACS gene missense mutation in ARSACS. Neurology 2004; 62: 107–109.
Hara K, Onodera O, Endo M et al: Sacsin-related autosomal recessive ataxia without prominent retinal myelinated fibers in Japan. Mov Disord 2005; 20: 380–382.
Yamamoto Y, Hiraoka K, Araki M et al: Novel compound heterozygous mutations in sacsin-related ataxia. J Neurol Sci 2005; 239: 101–104.
Yamamoto Y, Nakamori M, Konaka K et al: Sacsin-related ataxia caused by the novel nonsense mutation Arg4325X. J Neurol 2006; 253: 1372–1373.
Ouyang Y, Segers K, Bouquiaux O et al: Novel SACS mutation in a Belgian family with sacsin-related ataxia. J Neurol Sci 2008; 264: 73–76.
Takiyama Y : Sacsinopathies: sacsin-related ataxia. Cerebellum 2007; 28: 1–7.
Takado Y, Hara K, Shimohata T, Tokiguchi S, Onodera O, Nishizawa M : New mutation in the non-gigantic exon of SACS in Japanese siblings. Mov Disord 2007; 22: 748–749.
Thienpont B, Mertens L, de Ravel T et al: Submicroscopic chromosomal imbalances detected by array-CGH are a frequent cause of congenital heart defects in selected patients. Eur Heart J 2007; 28: 2778–2784.
Menten B, Maas N, Thienpont B et al: Emerging patterns of cryptic chromosomal imbalance in patients with idiopathic mental retardation and multiple congenital anomalies: a new series of 140 patients and review of published reports. J Med Genet 2006; 43: 625–633.
Parsons JD : Miropeats: graphical DNA sequence comparisons. Comput Appl Biosci 1995; 11: 615–619.
Ouyang Y, Takiyama Y, Sakoe K et al: Sacsin-related ataxia (ARSACS): expanding the genotype upstream from the gigantic exon. Neurology 2006; 66: 1103–1104.
Ramensky V, Bork P, Sunyaev S : Human non-synonymous SNPs: server and survey. Nucleic Acids Res 2002; 30: 3894–3900.
Pinto D, Marshall C, Feuk L, Scherer SW : Copy-number variation in control population cohorts. Hum Mol Genet 2007; 16 Spec No. 2: R168–R173.
Simon-Sanchez J, Scholz S, Fung HC et al: Genome-wide SNP assay reveals structural genomic variation, extended homozygosity and cell-line induced alterations in normal individuals. Hum Mol Genet 2007; 16: 1–14.
Coman DJ, Gardner RM : Deletions that reveal recessive genes. Eur J Hum Genet 2007; 15: 1103–1104.
Acknowledgements
We thank Drs Haruo Shimazaki and Junko Honda, Division of Neurology, Department of Internal Medicine, Jichi Medical University, Japan, and Karin Segers, Department of Human Genetics, Hôpital du Sart Tilman, Liege, Belgium, for their cooperation and the reviewers for their constructive comments. This work was partially supported by a grant from the Research Committee for Ataxic Diseases (YT) of the Ministry of Health, Labor and Welfare, Japan. This work was enabled by a grant of the Research Foundation-Flanders (FWO).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Breckpot, J., Takiyama, Y., Thienpont, B. et al. A novel genomic disorder: a deletion of the SACS gene leading to Spastic Ataxia of Charlevoix–Saguenay. Eur J Hum Genet 16, 1050–1054 (2008). https://doi.org/10.1038/ejhg.2008.58
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2008.58
Keywords
This article is cited by
-
Documenting manifestations and impacts of autosomal recessive spastic ataxia of Charlevoix–Saguenay to develop patient-reported outcome
Orphanet Journal of Rare Diseases (2022)
-
Clinical and Molecular Findings of Autosomal Recessive Spastic Ataxia of Charlevoix Saguenay: an Iranian Case Series Expanding the Genetic and Neuroimaging Spectra
The Cerebellum (2022)
-
Genetic spectrum and clinical features in a cohort of Chinese patients with autosomal recessive cerebellar ataxias
Translational Neurodegeneration (2021)
-
An inherited large-scale rearrangement in SACS associated with spastic ataxia and hearing loss
neurogenetics (2009)
