
Abstract
Purpose
Pulmonary arteriovenous malformations (pAVMs) are major contributors to morbidity and mortality in hereditary hemorrhagic telangiectasia (HHT). Mutations in ENG and ACVRL1 underlie the vast majority of clinically diagnosed cases. The aims of this study were to characterize and compare the clinical and morphologic features of pAVMs between these two genotype groups.
Methods
Sixty-six patients with HHT and affected family members were included. Genotype, phenotypic data, and imaging were obtained from medical records. Morphologic features of pAVMs were analyzed using computed tomography angiography. HHT symptoms, pAVM imaging characteristics, frequency of procedural intervention, and HHT severity scores were compared between ENG and ACVRL1 genotype groups.
Results
ENG mutation carriers were more likely than ACVRL1 mutation carriers to have pAVMs (P < 0.001) or multiple lesions (P = 0.03), and to undergo procedural intervention (P = 0.02). Additionally, pAVMs in ENG carriers were more likely to exhibit bilateral lung involvement and growth over time, although this did not reach statistical significance. The HHT severity score was significantly higher in ENG than in ACVRL1 (P = 0.02).
Conclusion
The propensity and multiplicity of ENG-associated pAVMs may contribute to the higher disease severity in this genotype, as reflected by the HHT severity score and the frequency of interventional procedures.
Similar content being viewed by others


Introduction
Hereditary hemorrhagic telangiectasia (HHT), previously known as Osler-Weber-Rendu syndrome, is an autosomal dominant vascular malformation disorder characterized by arteriovenous malformations (AVMs) occurring in the mucosal membranes and visceral organs, including the brain, spine, lungs, liver, and gastrointestinal tract. The diagnosis of HHT is based on the Curaçao criteria: (i) spontaneous and recurrent epistaxis, (ii) multiple mucocutaneous telangiectasia, (iii) visceral AVMs in the gastrointestinal tract, lungs, liver, brain, or spine, and (iv) a first-degree relative with HHT. Satisfaction of at least three of these criteria is considered to indicate “definite HHT” and of two of the criteria to indicate “possible HHT.”1, 2 Pulmonary AVMs (pAVMs) are common in HHT, and are found in up to 24–50% of affected individuals.3, 4 PAVMs in HHT range in size, number, and location, as well as in the number of feeding arteries,5 and account for many of the complications seen in HHT, including polycythemia, hemothorax, cerebral abscess, stroke, hemoptysis, and myocardial infarction.6, 7, 8, 9, 10, 11, 12
Several genes in the TGF-β pathway (ENG, ACVRL1, SMAD4, and GDF2/BMP9) have been associated with an HHT-like phenotype. Of these, ENG and ACVRL1 mutations account for up to 96% of individuals meeting strict Curaçao clinical diagnostic criteria,13 and each accounts for a similar percentage of total HHT cases.3 Increasing evidence suggests that ENG-associated and ACVRL1-associated HHT may be more phenotypically distinct than previously thought. For instance, previous studies have suggested a higher rate of visceral AVMs in ENG versus ACVRL1 mutation carriers, although ACVRL1 mutation carriers are more likely to exhibit hepatic AVMs and have higher rates of GI bleeding.14, 15 Among ENG mutation carriers, PAVMs were noted at higher frequencies,14, 16 tended to be larger,17 and were associated with higher complication rates15, 16 than those for ACVRL1 mutation carriers. Despite these emerging genotype–phenotype correlations, current management guidelines recommend the same screening for ENG and ACVRL1 mutation carriers.1
Several characteristics of pAVM morphology have been studied as possible links to clinical outcomes. For example, prior studies have demonstrated that the multiplicity,18 feeding artery diameter,19 diffuse form,12 and bilateral lung involvement of pAVMs were associated with increased complications and poorer outcomes,6 although these studies did not distinguish between mutation types. The present study aims to further characterize and compare pAVM imaging features and morphology based on genotype, to correlate imaging findings with clinical severity, and to provide additional evidence supporting the phenotypic distinction between ENG and ACVRL1 mutation carriers.
Materials and methods
Study subjects
Sixty-six patients with HHT were characterized genetically from January 2008 to June 2016 at the Johns Hopkins HHT Center of Excellence. Retrospective medical-record review was performed. This study was approved by the Institutional Review Board of the Johns Hopkins University, as part of a larger imaging protocol for HHT patients.
Clinical evaluation
Phenotypic data were collected from medical intake and screening data at the initial assessment as well as at follow-up appointments. All new patients in the HHT Center without prior screening data underwent brain magnetic resonance imaging and magnetic resonance angiography, and saline bubble contrast echocardiography (the recommended screening for pAVM).1 If the bubble study was positive or equivocal, patients further underwent chest computed tomography (CT) or CT angiography (CTA). Records of patients who had recently been screened at an outside institution before entering the HHT Center were collected and reviewed. Retrospective chart review on the frequency of endovascular or surgical treatment was performed and used to make comparisons between the patients with ENG and those with ACVRL1 genotypes. All individuals with definite pathogenic mutations in ENG and ACVRL1 underwent comprehensive evaluation according to the International HHT Guidelines,1 including assessment with the Epistaxis Severity Score,20 mucocutaneous telangiectasia, pulmonary, hepatic, and gastrointestinal symptoms, as well as screening for the presence of pulmonary, cerebral, and hepatic AVMs.
Molecular genetic analysis
Genetic testing was offered only to individuals meeting definite Curaçao criteria, with the exception of children and young adults, who may not yet have demonstrated symptoms owing to the age-related penetrance in HHT (n = 10). Individuals without prior genetic testing were offered ENG and ACVRL1 sequencing and deletion/duplication testing, as well as the 5’ untranslated region of ENG (after 2011).21 If this was negative, SMAD4 and BMP9/GDF2 gene sequencing were offered (the latter gene being included since it was associated with an HHT phenotype in 2013).22 A few cases were presumed to be obligate mutation carriers if they met Curaçao criteria and had a known familial mutation. All molecular genetic testing was performed on whole blood samples by ARUP Laboratories (Salt Lake City, UT). This included Sanger sequencing of entire coding regions and intron-exon boundaries of each gene, as well as deletion/duplication analysis using exonic oligonucleotide-based comparative genomic hybridization microarray. Some participants had undergone genetic testing prior to establishing care with the HHT Center.
Severity score
The overall disease burden was assessed independently by two of the authors (W.M. and D.L.) using the recently developed HHT Severity Score.23 This measure takes into account chronic nasal or gastrointestinal bleeding, presence of visceral AVMs, and whether the visceral AVMs are diffuse or cause symptomatic liver involvement. The score ranges from 0 (mild) to 7 (severe), and correlates with adverse outcomes associated with HHT as a validation measurement. Currently, the HHT Severity Score is the only published tool for measurement of quantitative disease burden that is specific to HHT.
Pulmonary arteriovenous malformation morphology data
Of individuals who had pAVMs, 15 of 28 in the ENG genotype group and 6 of 34 in the ACVRL1 genotype group had imaging data, including chest CTA with three-dimensional rendering, and digital subtraction angiography, for further evaluation of suspected pAVMs after screening by saline bubble echocardiogram. Based on pAVM features correlating with clinical outcomes in the literature, seven parameters were selected for characterization. These included the following:
1. Presence of diffuse pAVMs (defined as involvement of every segmental artery in at least one lobe)1, 5
2. Presence of multiple (defined two or more) pAVMs
3. Simple or complex (complex defined as having two or more feeding arteries, and two or more draining veins)2, 24
4. Presence of a feeding artery greater than or equal to 2 mm (this measure is used as the threshold for offering embolization at the Johns Hopkins HHT Center)
5. Presence of lesions greater than 10 mm in the longest dimension across the entire AVM
6. Presence of bilateral lung involvement
7. Indication of growth over time, including enlargement of previously identified and treated pAVMs
Statistical methods
Statistical analysis was completed using Statistica 7 (StatSoft, Tulsa, OK). Data are expressed as mean values, with standard error where appropriate. Percent affected data were calculated from complete data. Differences in phenotypic and imaging characteristics between groups were assessed by Fisher’s exact test. The Mann-Whitney test was used to compare differences in HHT Severity Scores between groups. A P value <0.05 was considered significant.
Results
Molecular genetic characterization
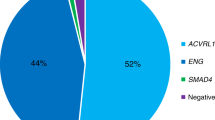
Sixty patients completed genetic testing. ENG mutations were identified in 42.4% of individuals who had undergone genetic testing; ACVRL1 mutations in 51.5% (Table 1). Together, 93.9% of all genotyped individuals had a mutation in either ENG or ACVRL1. Equal numbers of male and female patients had positive genetic testing for each genotype group. Mean ages were 47.8 years for ENG mutation carriers and 50.0 years for ACVRL1 mutation carriers. There were no significant differences in sex or age; this is consistent with existing literature. Deletion, followed by missense and then splice defect, were the most common types of mutation in ENG. “Large deletion” indicates a deletion of one or more exons, whereas “deletion” indicates deletion of a few base pairs. Missense was the most common mutation type in ACVRL1.
One patient was identified as having a variant of unknown significance in BMP9; one patient had a SMAD4 mutation that had previously been identified by another center. Two patients who met definite Curaçao criteria had no definitive pathogenic mutations identified in ENG or ACVRL1 by sequencing and deletion/duplication testing. One had negative ENG and ACVRL1 testing but died before additional testing could be offered; the other had a variant of unknown significance identified in ENG. Owing to the limited number of patients found to have SMAD4 or BMP9 mutations, analysis in this study focused on ENG and ACVRL1.
HHT features
ENG mutation carriers were significantly more likely to have pAVMs than ACVRL1 mutation carriers (72% v. 18.2%; P < 0.001) (Table 2). No statistically significant differences were found in percentages of patients with epistaxis (96.4% in ENG, 91.2% in ACVRL1 carriers), mucocutaneous telangiectasia (85.0% in ENG, 84.0% in ACVRL1 carriers), and cerebral AVMs (17.4% in ENG, 12.1% in ACVRL1 carriers).
Characterizations of pAVMs
Patients with an underlying ENG genotype were more likely to demonstrate multiple pAVMs (P = 0.03) (Table 3). There were two cases of diffuse pAVMs, both in patients with the ENG genotype. In addition, there was a higher proportion of ENG-associated pAVMs demonstrating bilateral lung involvement (11/15 in ENG v. 1/5 in ACVRL1, P = 0.1) as well as growth over time on subsequent imaging (6/12 in ENG v. 0/5 in ACVRL1, P = 0.1) than of ACVRL1-associated pAVMs, although these did not reach statistical significance. For the growth-over-time parameter, the range of interval imaging follow-up ranged from 1 to 8 years, with a median of 5.5 years. No significant differences were found in the complexity of pAVMs, diameters of arterial feeders, or size of the pAVM. Figure 1 shows representative chest CTAs of two select cases: an ENG mutation carrier with multiple and bilateral pAVMs, in contrast to an ACVRL1 mutation carrier with a single AVM.

Selective chest CTA images of ENG and ACVRL1. (a) 14-year-old male ENG mutation carrier presenting with epistaxis, dyspnea on exertion, and hypoxia, with SpO2 as low as 70%. CTA (coronal maximal intensity projection) revealed multiple AVMs bilaterally (white arrows). The left lower lobe AVM (curved outline arrow) is complex and measures up to 4.4 cm; one of the two feeding arteries measures 11 mm in diameter, the other 5.6 mm. (b) 63-year-old female ACVRL1 mutation carrier presented asymptomatically. Routine screening revealed a single 1.3 × 1 cm pAVM (outline arrow) in the right lower lobe fed by an anterior basal segment pulmonary artery measuring 4.5 mm in diameter and a smaller branch arterial feeder measuring 2.9 mm. AVM, arteriovenous malformation; CTA, chest computed tomography angiography; pAVM, pulmonary arteriovenous malformation.
Likelihood of procedural interventions
The vast majority of the procedures in this population were embolizations; a few had lobectomy before 1990. ENG mutation carriers were more likely than ACVRL1 mutation carriers to have had procedural interventions for pAVMs (either embolization or lobectomy; it is of note that lobectomy is no longer performed routinely) (P = 0.01) (Table 3). No significant difference was found in the need for multiple procedural interventions.
Disease burden comparison
Patients with ENG mutations had a significantly higher average HHT Severity Score than did ACVRL1 mutation carriers (2.25 v. 1.57; P = 0.02).
Discussion
HHT is a clearly defined and common hereditary vascular syndrome that affects up to 1 in 5000 individuals.25 Four known genes (ENG, ACVRL1, SMAD4, and BMP9/GDF2) have been associated with the syndrome,24 and current screening recommendations for it are uniform, regardless of genotype.1 However, a small but growing body of literature suggests that the rate and severity of visceral AVMs differ between ENG and ACVRL1 mutation carriers.
The genotype and phenotype correlations in this study support and expand on the existing literature. Targeted genetic testing in this cohort yielded a high pickup rate of ENG and ACVRL1 mutations, with 94% of all patients who had completed genetic testing possessing a mutation in one of these two genes. This mutation pickup rate is consistent with findings in current literature, and is similar to the findings by McDonald et al.13, 26 of a 96% pickup rate when the Curaçao criteria are strictly applied. In HHT, no specific mutation hot spots have been identified, with known mutations spanning all exons of both genes and mutations often unique to extended families.13 Additionally, no specific phenotypes are correlated with individual mutations or mutation types, and despite nearly 100% penetrance, wide variable expressivity exists within each HHT family.26 This study’s results are consistent with the literature in that there were no shared mutations among patients that were not due to founder effect. The most common type of mutations observed in each genotype (i.e., splice-defect mutations in ENG) are also consistent with established databases.27
Prior studies have demonstrated that pAVMs in ENG mutation carriers were noted at higher frequencies than in ACVRL1 mutation carriers.14, 16 The current study’s findings are consistent with this, but the disparity in frequency is considerably larger (72.0% in ENG versus 18.2% in ACVRL1). Previous larger studies, with more than 100 subjects, have reported pAVM frequencies of 75% versus 44%,16 67.5% versus 48%,28 and 59% versus 28.9%.17 Letteboer et al. reported 48.7% versus 5.3%, although pAVMs were diagnosed clinically.14 The French–Italian HHT Network reported pAVMs in 54% of ENG mutation carriers versus 12.8% of ACVRL1 mutation carriers in asymptomatic patients, and 34.4% versus 5.2% in symptomatic patients.16 Significant differences were nevertheless found in studies similar in size to the present study.29, 30 Variations in interstudy prevalence may be attributed to geographically differing screening protocols and demographics, including age and sex, which have been reported to influence phenotype.31, 32
The current study demonstrated that ENG-associated pAVMs were more likely to exhibit multiple and bilateral pAVMs, although the latter did not reach statistical significance. These specific morphologic differences between the genotype groups are novel findings, and suggest that ENG mutations may be associated with a more severe phenotype. Individuals with multiple pAVMs have been reported to run an increased risk for stroke and abscess than those with a single AVM.18 Bayrak-Toydemir et al.17 noted that pAVMs were larger in ENG mutation carriers. The current study did not detect a significant difference in size, but its findings suggest an increased likelihood of growth in ENG-associated pAVMs upon follow-up imaging.
Individuals with ENG mutations in this population were found to have a higher HHT Severity Score than those with ACVRL1 mutations. This is in alignment with prior findings in which almost all respiratory and neurological complications were found in ENG mutation carriers,15 and individuals with ENG mutations were more likely to be symptomatic.16 It is acknowledged, however, that the Severity Score may be underestimated in asymptomatic hepatic AVMs, which are known to be present at a higher frequency in ACVRL1 mutation carriers than in ENG carriers, but are not routinely diagnosed by imaging or other screening methods. Conceivably the additionally characterized features of size, growth, multiplicity, and bilaterality of pAVMs may also contribute to the higher phenotypic severity in ENG mutation carriers. While the frequency of diffuse forms was not significantly different between ENG and ACVRL1 carriers in the current study, the only two cases of diffuse form pAVMs in this population were associated with ENG. Faughnan et al. and Lacombe et al. demonstrated higher rates of stroke and brain abscess in diffuse forms;5, 12 the two cases in the present study did not have acute cerebral events, as both presented at young ages, and pAVMs were diagnosed on screening and then promptly treated. In concordance with the increased severity in the ENG mutation carriers, this study also demonstrated a higher rate of procedural interventions to embolize or surgically resect pAVMs in the ENG genotype group, which may again reflect the higher clinical severity of ENG-associated pAVMs and the consequent necessity for treatment.
In summary, ENG mutation carriers demonstrated a higher frequency of occurrence of pAVMs. as well as multiple pAVMs, compared to those with ACVRL1 mutation carriers, possibly one of the factors contributing to a more severe phenotype as demonstrated by a significantly higher HHT Severity Score and a higher frequency of requirement for interventional procedures to treat pAVMs. To our knowledge, these data also represent the first study to identify a potential difference in pAVM morphology, as well as to suggest a difference in frequency of surgical intervention based on genotype.
Limitations of this study include the small sample size for identified mutations, especially for participants who were mutation-positive and had prior imaging data for analysis. Additional limitations include the low frequency of certain imaging features in this study to detect significant differences, including the low number of cerebral AVMs, hepatic AVMs, diffuse and complex forms of pAVMs, and pAVMs that demonstrated growth. Nevertheless, presence or absence of all visceral AVMs, including symptomatic liver involvement, were taken into account when calculating the HHT Severity Score comparing ENG and ACVRL1.23 Taken together, although the limited data set represents some ascertainment bias, the novel differences are consistent with and expand on prior literature describing size and morphology.
The current study and others before it have clearly demonstrated phenotypic differences in pAVMs between ENG and ACVRL1 mutation carriers, which may be relevant to ongoing management for HHT. The present study contributes novel differences, such as the likelihood of procedural intervention for pAVMs and overall disease severity, and may be helpful in guiding clinical decision making for genotyped HHT patients. The dissimilarity in pAVM morphology raises questions about separations in pathogenesis between these two major genotype groups in HHT and warrants further exploration. Future studies into pAVM incidence, structure, growth, and associated clinical outcomes could determine whether there is a genotype group with a higher rate of morbidity.
References
Faughnan ME, Palda VA, Garcia-Tsao G et al. International guidelines for the diagnosis and management of hereditary haemorrhagic telangiectasia. Genet Med 2011;48:73–87.
Shovlin CL, Guttmacher AE, Buscarini E et al. Diagnostic criteria for hereditary hemorrhagic telangiectasia (Rendu-Osler-Weber syndrome). Am J Med Genet A 2000;91:66–67.
Bayrak-Toydemir P, Mao R, Lewin S, McDonald J . Hereditary hemorrhagic telangiectasia: an overview of diagnosis and management in the molecular era for clinicians. Genet Med 2004;6:175–191.
Kjeldsen AD, Oxhøj H, Andersen PE, Green A, Vase P . Prevalence of pulmonary arteriovenous malformations (PAVMs) and occurrence of neurological symptoms in patients with hereditary haemorrhagic telangiectasia (HHT). J Intern Med 2000;248:255–262.
Labombe P, Lacout A, Marcy PY et al. Diagnosis and treatment of pulmonary arteriovenous malformations in hereditary hemorrhagic telangietasia: an overview. Diagn Interv Imaging 2013;94:835–48.
Pierucci P, Murphy J, Henderson KJ, Chyun DA, White RI . New definition and natural history of patients with diffuse pulmonary arteriovenous malformations: twenty-seven-year experience. Chest 2008;133:653–661.
Faughnan ME, Lui YW, Wirth JA et al. Diffuse pulmonary arteriovenous malformations: characteristics and prognosis. Chest 2000;117:31–8.
Todo K, Moriwaki H, Higashi M, Kimura K, Naritomi H . A small pulmonary arteriovenous malformation as a cause of recurrent brain embolism. Am J Neuroradiol 2004;25:428–30.
Cottin V, Dupuis-Girod S, Lesca G, Cordier JF . Pulmonary vascular manifestations of hereditary hemorraghic telangiectasia (Rendu-Osler disease). Respiration 2007;74:361–78.
Trerotola SO, Pyeritz RE, Bernhardt BA . Outpatient single-session pulmonary arteriovenous malformation embolization. J Vasc Interv Radiol 2009;20:1287–91.
Clark K, Pyeritz RE, Trerotola SO . Angina pectoris or myocardial infarctions, pulmonary arteriovenous malformations, hereditary hemorrhagic telangiectasia, and paradoxical emboli. Am J Cardiol 2013;112:731–4.
Donaldson JW, McKeever TM, Hall IP . Complications and mortality in hereditary hemorrhagic telangiectasia: A population-based study. Neurology 2015;84:1886–1893.
McDonald J, Wooderchak-Donahue W, Webb CV, Whitehead K, Stevenson DA, Bayrak-Toydemir P . Hereditary hemorrhagic telangiectasia: genetics and molecular diagnostics in a new era. Front Genet 2015;6:1–8.
Letteboer TGW, Mager JJ, Snijder RJ et al. Genotype-phenotype relationship in hereditary hemorrhagic telangiectasia. J Med Genet 2006;43:371–377.
Sabbà C, Pasculli G, Lenato GM et al. Hereditary hemorrhagic telangiectasia: clinical features in ENG and ALK1 mutation carriers. J Thromb Haemost 2007;5:1149–57.
Lesca G, Olivieri C, Burnichon N et al. Genotype-phenotype correlations in hereditary hemorrhagic telangiectasia: data from the French-Italian HHT network. Genet Med 2007;9:14–22.
Bayrak-Toydemir P, McDonald J, Markewitz B et al. Genotype-phenotype correlation in hereditary hemorrhagic telangiectasia: mutations and manifestations. Am J Med Genet A 2006;140A:463–470.
Moussouttas M, Fayad P, Rosenblatt M et al. Pulmonary arteriovenous malformations: cerebral ischemia and neurologic manifestations. Neurology 2000;55:959–64.
Rosenblatt M, Pollak JS, Fayad PB et al. Pulmonary arteriovenous malformations: what size should be treated to prevent embolic stroke. Radiology 1992;185:134.
Hoag JB, Terry P, Mitchell S, Reh D, Merlo CA . An epistaxis severity score for hereditary hemorrhagic telangiectasia. Laryngoscope 2010;120:838–43.
Damjanovich K, Langa C, Blanco FJ et al. 5’UTR mutations of ENG cause hereditary hemorrhagic telangiectasia. Orphanet J Rare Dis 2011;6:85.
Wooderchak-Donahue WL, McDonald J, O’Fallon B et al. BMP9 mutations cause a vascular-anomaly syndrome with phenotypic overlap with hereditary hemorrhagic telangiectasia. Am J Hum Genet 2013;93:531–537.
Brain Vascular Malformation Consortium HHT Investigator Group Latino GA Brain Vascular Malformation Consortium HHT Investigator Group Kim H Brain Vascular Malformation Consortium HHT Investigator Group Nelson J Brain Vascular Malformation Consortium HHT Investigator Group Pawlikowska L Brain Vascular Malformation Consortium HHT Investigator Group Young W Brain Vascular Malformation Consortium HHT Investigator Group Faughnan ME Brain Vascular Malformation Consortium HHT Investigator Group. Severity score for hereditary hemorrhagic telangiectasia. Orphanet J Rare Dis 2014;9:188–194.
Carette MF, Nedelcu C, Tassart M, Grange JD, Wislez M, Khalil A . Imaging of hereditary hemorrhagic telangiectasia. Cardiovasc Intervent Radiol 2009;32:745–57.
Govani FS, Shovlin CL . Hereditary hemorrhagic telangiectasia: a clinical and scientific review. Eur J Hum Genet 2009;17:860–871.
McDonald J, Pyeritz RE. Hereditary hemorrhagic telangiectasia. In: Pagon RA, Adam MP, Ardinger HH et al (eds). GeneReviews. University of Washington: Seattle, WA, 2017.
Hereditary Hemorrhagic Telangiectasia Mutation Database. Salt Lake City, UT: University of Utah Department of Pathology, 2006. http://www.arup.utah.edu/database/HHT. Accessed 9 February 2017.
Bossler AD, Richards J, George C, Godmilow L, Ganguly A . Novel mutations in ENG and ACVRL1 identified in a series of 200 individuals undergoing clinical genetic testing for hereditary hemorrhagic telangiectasia (HHT): correlation of genotype with phenotype. Hum Mut 2006;27:667–675.
Berg J, Porteous M, Reinhardt D et al. Hereditary haemorrhagic telangiectasia: a questionnaire based study to delineate the different phenotypes caused by endoglin and ALK1 mutations. J Med Genet 2003;40:585–590.
Kieldsen AD, Moller TR, Brusgaard K et al. Clinical symptoms according to genotype amongst patients with hereditary haemorrhagic telangiectasia. J Intern Med 2005;258:349–355.
White RI, Lynch-Nyhan A, Terry P et al. Pulmonary arteriovenous malformations: techniques and long-term outcome of embolotherapy. Radiology 1988;169:633–669.
Trembath RC, Thornson JR, Machado RD et al. Clinical and molecular genetic features of pulmonary hypertension in patients with hereditary hemorrhagic telangiectasia. N Engl J Med 2001;345:325–334.
Acknowledgements
This work was made possible by the collaboration of the staff and faculty at the Johns Hopkins HHT Center of Excellence. Clinical genetic testing was performed by, and mutation characteristics were derived from databases hosted and curated by, the ARUP Institute for Clinical and Experimental Pathology, Salt Lake City, Utah. We thank our patients and their families for participating.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article

Cite this article
Mu, W., Cordner, Z., Yuqi Wang, K. et al. Characterization of pulmonary arteriovenous malformations in ACVRL1 versus ENG mutation carriers in hereditary hemorrhagic telangiectasia. Genet Med 20, 639–644 (2018). https://doi.org/10.1038/gim.2017.160
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/gim.2017.160
