
Abstract
The short-chain, non-methane hydrocarbons propane and butane can contribute significantly to the carbon and sulfur cycles in marine environments affected by oil or natural gas seepage. In the present study, we enriched and identified novel propane and butane-degrading sulfate reducers from marine oil and gas cold seeps in the Gulf of Mexico and Hydrate Ridge. The enrichment cultures obtained were able to degrade simultaneously propane and butane, but not other gaseous alkanes. They were cold-adapted, showing highest sulfate-reduction rates between 16 and 20 °C. Analysis of 16S rRNA gene libraries, followed by whole-cell hybridizations with sequence-specific oligonucleotide probes showed that each enrichment culture was dominated by a unique phylotype affiliated with the Desulfosarcina-Desulfococcus cluster within the Deltaproteobacteria. These phylotypes formed a distinct phylogenetic cluster of propane and butane degraders, including sequences from environments associated with hydrocarbon seeps. Incubations with 13C-labeled substrates, hybridizations with sequence-specific probes and nanoSIMS analyses showed that cells of the dominant phylotypes were the first to become enriched in 13C, demonstrating that they were directly involved in hydrocarbon degradation. Furthermore, using the nanoSIMS data, carbon assimilation rates were calculated for the dominant cells in each enrichment culture.
Similar content being viewed by others

Introduction
The short-chain alkanes propane and butane occur as constituents of natural gas and are also found, in smaller amounts, dissolved in crude oil (Tissot and Welte, 1984). Propane and butane are gaseous under normal standard temperature and pressure conditions, and liquid at low temperatures (at 0.1 MPa, the boiling point of propane is −42 °C and that of butane −0.5 °C) or at high pressure (at 20 °C, ⩾0.75 MPa for propane, and ⩾0.22 MPa for butane) (Schwarzenbach et al., 2003). In places where plate tectonics causes fracturing in the crust, gas and oil from deep-seated reservoirs are able to migrate upwards through the sediment as free gas or as dissolved components of geothermal fluids (Hovland et al., 1993; Sassen et al., 2001). Propane and butane are moderately soluble in water (saturation at 20 °C, 1.76 mM propane, 1.46 mM butane), with solubility increasing at lower temperatures and higher pressures (Dean, 1992; Chapoy et al., 2004), conditions often prevailing at deep-sea environments. Seepage in such low temperature and high pressure environments may lead to entrapment of significant amounts of short-chain alkanes in structure II gas hydrates (Sloan, 2003). These formations, such as those found in the Gulf of Mexico, may contain up to 36% propane and up to 4% butane (Orcutt et al., 2004; Sassen et al., 2004).
The degradation of propane and butane under oxic conditions has been studied in detail and numerous strains of propane- or butane-degrading bacteria and fungi have been described (Shennan 2006 and references therein). Recent studies have shown that propane is degraded in situ along with methane and ethane in oxic, hydrocarbon-contaminated marine sediments (Kinnaman et al., 2007; Redmond et al., 2010) and in the water column after an oil spill (Valentine et al., 2010). Geochemical studies of anoxic deep-sea sediments provided the first indications that propane and butane are also degraded under anoxic conditions, suggesting a significant influence on the carbon and sulfur cycles in hydrocarbon-impacted environments. These data include the 13C-enrichment of propane and butane in sediment interstitial waters atop gas hydrates (Sassen et al., 2004) or at deep sea mud volcanoes (Mastalerz et al., 2009), sulfate-reduction rates much higher than could be accounted for by anaerobic oxidation of methane in hydrothermal sediments or at cold seeps (Joye et al., 2004; Kallmeyer and Boetius, 2004; Orcutt et al. 2004, 2010; Kleindienst et al., 2012), or the apparent consumption of short-chain alkanes at mud volcanoes (Niemann et al., 2006). In addition, oxidation of propane under anoxic conditions has been determined in sediments collected from marine gas seeps (Quistad and Valentine, 2011). Anaerobic degradation of short-chain alkanes was demonstrated recently with a pure culture of a marine sulfate-reducing bacterium strain BuS5, isolated from Guaymas Basin hydrothermal vent sediments (Kniemeyer et al., 2007), and with several enrichment cultures obtained from sediments around marine and terrestrial hydrocarbon seeps (Kniemeyer et al., 2007; Savage et al., 2010). Strain BuS5 can degrade both propane and butane and is phylogenetically affiliated with the Desulfosarcina-Desulfococcus cluster of the Deltaproteobacteria. Analysis of metabolites showed that propane and butane are activated similar to higher alkanes by addition to fumarate (Kropp et al., 2000; Rabus et al., 2001) primarily at the secondary carbon atom yielding iso-propyl- and (1-methylpropyl)-succinate, respectively (Kniemeyer et al., 2007; Savage et al., 2010).
Despite the potentially important role in carbon and sulfur cycling at marine hydrocarbon seeps, the diversity of anaerobic, propane- and butane-degrading bacteria is largely unknown. To address this gap in knowledge, one goal of the present study was to enrich for, identify and characterize novel propane- and butane-degrading bacteria from marine gas and oil cold seeps. The enrichment cultures were characterized with respect to their hydrocarbon substrate spectrum and optimum temperature for sulfate reduction. The propane- and butane-degrading bacteria were identified directly in the enrichment cultures using 16S rRNA gene-targeted oligonucleotide probes and incubations with 13C-labeled substrates followed by nanoSIMS analysis. Moreover, hydrocarbon assimilation rates were calculated for single cells directly in the enrichment cultures.
Materials and methods
Source of organisms, media and cultivation techniques
Anoxic, marine sediment was collected around hydrocarbon seeps at Hydrate Ridge and in the Gulf of Mexico. The sediment samples from Hydrate Ridge were collected during the RV SONNE cruise, leg 148-1, in July 2000 at 777 m water depth from Station 28 and 29 Beggiatoa field (44° 34′ N, 125° 09′ W; Treude et al., 2003). The sediment samples from the Gulf of Mexico were collected using the RV Seward Johnson I and the Johnson Sea Link submersible in July 2001 from the brine-influenced site GC233 (650 m water depth, 27° 44′ N, 92° 17′ W; Joye et al., 2004). The cultures were set up in 100 ml flat bottles sealed with butyl rubber stoppers, containing 50 ml of bicarbonate-buffered artificial seawater medium (Widdel and Bak, 1992; Widdel, 2010), under a N2:CO2 headspace (9:1, v/v). Propane and butane (Air Liquide, Düsseldorf, Germany) were added to 50% v/v of the headspace (final pressure 1.5 atm). The bottles were inoculated with 5 ml homogenized sediment, and incubated at 12 and 28 °C nearly horizontally in order to increase the hydrocarbon diffusion surface (Rabus and Widdel, 1995). Control incubations without the addition of hydrocarbons were set up under the same conditions. Sediment-containing cultures were incubated without shaking, while sediment-free cultures were incubated with slow (40–50 r.p.m.) horizontal shaking. For subcultivation, 10% v/v of an active culture was used as inoculum. Sediment-free enrichment cultures were amended with 3 ml l−1 trace element solution (Widdel and Bak, 1992). Strain BuS5 and the butane-degrading enrichment culture But12-GMe were cultivated as previously described (Kniemeyer et al., 2007). The optimum temperature for sulfate reduction was determined with duplicate 10 ml cultures incubated in a temperature gradient block, across a temperature range from 0–40 °C. For each culture, sulfide concentrations were determined in triplicate at 24 h intervals. The sulfate-reduction rate at each temperature was calculated from the slope of the sulfide production curve, and normalized to the highest rate determined.
To test the ability of the enrichment cultures to degrade other hydrocarbons, cultures were set up in 120 ml serum bottles containing 79 ml artificial seawater medium. The bottles were provided with methane, ethane, propane, butane, isobutane (methylpropane; 6 ml in the headspace each), or pentane (1% v/v in 2,2,4,4,6,8,8-heptamethylnonane, HMN, as an inert carrier phase). To test for possible co-oxidation of other alkanes in the presence of propane or butane, additional substrate utilization tests were done with mixtures of (a) methane, ethane, propane and butane (6 ml each), (b) isobutane and butane or propane (6 ml each), and (c) pentane (1% v/v in HMN) and butane or propane (6 ml). Abiotic control bottles supplied with hydrocarbons but no inoculum, and inoculated bottles lacking hydrocarbon additions were used as controls. The bottles were pre-incubated for 48 h with slow shaking (40 r.p.m.) to allow equilibration of the hydrocarbon with the culture media, and subsequently inoculated with 1 ml of a 10 × concentrated cell suspension. To prepare concentrated cell suspensions, 150 ml of grown cultures were collected and centrifuged (20 min at 14 300 g; Beckman Coulter Avanti J-26 XP, Krefeld, Germany) under anoxic conditions. The cell pellets were homogeneously resuspended in fresh, anoxic culture medium, under a N2:CO2 headspace (9:1); cultures forming aggregates were partly homogenized by repeatedly passing the aggregates through a hypodermic needle (gauge 0.6 mm) attached to an N2-flushed syringe.
For halogen in situ hybridization-secondary ion mass spectrometry (HISH-SIMS) analysis, 15 ml cultures in 22-ml cultivation tubes were supplied with propane and 13C1-propane (yielding 11.7% 13C) or butane and 13C1-butane (11.7% 13C for But12-HyR enrichment culture and 25% for But12-GMe enrichment culture). Before the incubation the aggregate forming cultures were partly homogenized under anoxic conditions using a glass mortar and pestle (0.1 ml, Wheaton, Millville, NJ, USA). 13C1-propane and 13C1-butane (99 atom %) were purchased from ISOTEC (Miamisburg, OH, USA). Samples (0.5 ml) for mass spectrometry analysis of the bulk 13C-incorporation were obtained every 24 h and added onto precombusted glass fiber filters. In addition, 0.5 ml samples were fixed for whole-cell hybridization.
Analytical techniques
Sulfide was quantified photometrically as colloidal CuS (Cord-Ruwisch, 1985). Alkane concentrations in the headspace were determined by gas chromatography using a Shimadzu GC-14B gas chromatograph (Shimadzu, Duisburg, Germany) equipped with a 30-m Supel-Q PLOT column (0.53 mm inner diameter, film thickness 30 μm; Supelco, Bellefonte, PA, USA) and a FID. The carrier gas was N2 at a flow rate of 3 ml min−1. The detector was operated at 280 °C and the injector at 150 °C. For quantification of single hydrocarbons, the oven was operated at 140 °C. For analysis of mixtures of alkanes, the oven was maintained for 1 min at 35 °C, and then heated with a rate of 20 °C min−1 to 140 °C where it was maintained for 1 min. Headspace samples (0.1 ml) were withdrawn with N2-flushed gastight glass syringes. Measurements of hydrocarbon and sulfide concentrations in all incubations were done in triplicate. The glass fiber filters with cells collected from the labeling experiments with 13C-alkanes were analyzed for bulk abundance of 13C and 12C with an automated elemental analyzer (Thermo Flash EA, 1112 Series, Thermo Fischer, Dreieich, Germany) and a Finnigan Delta Plus Advantage mass spectrometer (Thermo Fischer).
Determination of cellular carbon content
The carbon content per cell volume was determined for strain BuS5 as a model organism using the carbon content and the cell number per ml culture, and an average cellular volume. Defined volumes (5, 10 and 15 ml) of a grown (13 mM H2S, OD660 nm=0.2) culture of strain BuS5 were transferred in triplicate onto precombusted glass fiber filters, dried and analyzed for total carbon content using a Carlo Erba NA-1500 CNS analyzer (Thermo Fischer), and sulfonilamide as a standard. For the same culture the total cell number per ml was determined by counting cells stained with DAPI (4′,6-diamidino-2-phenylindole). Average cellular volumes of strain BuS5 and of the dominant bacteria in the enrichment cultures were calculated by measuring the length and width of at least 30 cells and considering the volume formula of a rod with hemispherical ends. Cells of strain BuS5 were measured from phase contrast photomicrographs. The cells in the enrichment cultures were measured using epifluorescence microscope images after hybridization with sequence-specific oligonucleotide probes and DAPI staining.
Clone libraries, sequencing and phylogenetic analysis
Genomic DNA extracted from the enrichment cultures (Zhou et al., 1996) was used to amplify almost full length 16S rRNA genes using bacteria-specific primers 8f (Hicks et al., 1992) and 1496r (Kane et al., 1993). The PCR products were purified using the QIAquick Gel Extraction kit (Qiagen, Hilden, Germany), cloned into the pCR4 vector (TOPO-TA cloning kit Invitrogen, Groningen, The Netherlands) and transformed into E. coli Top 10 competent cells (Invitrogen). Positive clones were sequenced using the ABI Prism BigDye Terminator v 3.0 cycle sequencing kit and an ABI Prism 3100 Genetic Analyzer (Applied Biosystems, Darmstadt, Germany). Sequences were assembled with the DNA Baser software (www.dnabaser.com). The 16S rRNA gene sequences were aligned to those of the Silva database (Pruesse et al., 2007). Phylogenetic trees were constructed using the ARB software (Ludwig et al., 2004) by neighbor joining and maximum likelihood with application of different sets of filters, and in addition by maximum likelihood using the RAxML algorithm (Stamatakis et al., 2008; http://phylobench.vital-it.ch/raxml-bb). The sequence data obtained in this study have been deposited in the DDBJ, EMBL and GenBank databases under accession numbers FR823363–FR823378.
Fluorescence in situ hybridization
For fluorescence in situ hybridization 0.5 ml samples were fixed with 0.5 ml 2% paraformaldehyde in 1 × phosphate-buffered saline (PBS) for 1 h at room temperature, washed with 1 × PBS and stored in 1 × PBS:ethanol (1:1) at −20 °C. Aliquots of fixed cells were filtered onto 0.2 μm pore GTTP polycarbonate filters (Millipore, Eschborn, Germany). Fixed samples of aggregate forming cultures were in addition homogenized using a glass mortar and pestle (0.1 ml, Wheaton) before filtration. Alternatively, aggregates were double embedded in agar and paraffin blocks (Musat et al., 2007; Supplementary Information) and thin sections were cut using a Leica RM 2165 microtome (Leica, Nussloch, Germany). Cells on filters or cross sections were hybridized with Cy3-labeled 16S rRNA targeted oligonucleotide probes, stained with DAPI and microscopically counted as previously described (Snaidr et al., 1997). The hybridized aggregate sections were analyzed with a Zeiss Axioskop 2mot plus fluorescence microscope (Zeiss, Oberkochen, Germany), and the number of hybridized cells was estimated using the PHLIP software (Mueller et al., 2006). The oligonucleotide probes used in this study, Prop12-GMe-133 (TATCCCGATTTCCAGGGT, 50% formamide (FA), this study), But12-HyR-193 (GAGGCCACCTTTAATCTG, 40% FA, this study), But12-1275 (Kniemeyer et al., 2007), DSS658 (Manz et al., 1998), EUB338 and NON338 (Amann et al., 1990) were purchased from Biomers GmbH (Ulm, Germany). The sequence-specific probes designed in this study (in ARB, after Hugenholtz et al., 2001), were evaluated for specificity in hybridization assays with increasing FA concentrations (0–60%, with 10% increment). Signal intensities were compared with those of Desulfovibrio aespoensis (two mismatches vs Prop12-GMe-133; a strain with one mismatch was not available) and strain BuS5 (one mismatch vs But12-HyR-193). The probes Prop12-GMe-133 and But12-HyR-193 had at least four, respectively, three mismatches with non-target clones in the respective enrichment cultures.
Halogen in situ hybridization, nanoSIMS analyses and calculation of assimilation rates
Incubations with 13C-labeled substrates followed by hybridization with sequence-specific, HRP-labeled oligonucleotide probes and deposition of F-containing tyramides was used to quantify C-assimilation by the phylotypes of the dominant cells (Behrens et al., 2008; Musat et al., 2008). Aliquots of fixed samples from the incubations with 13C-labeled alkanes were added onto Au/Pd-coated GTTP filters. Hybridization and tyramide signal amplification were done according to Musat et al. 2008 (we further refer to the method as HISH-SIMS). Hybridized samples were analyzed with a NanoSIMS50L (Cameca, Gennevilliers, France). To ensure sputtering equilibrium (Audinot et al., 2011), the sample was pre-sputtered with Cs+ ions for 3–5 min before analysis. The primary ion beam of ∼2 pA was focused to a nominal spot size of ca. 100 nm and stepped over the sample with a 512 × 512 pixel raster and a dwell time of 1 ms per pixel. A mass resolving power above 6000 was used to resolve isobaric interferences. Secondary ion images of 12C−, 13C−, 19F− and 12C14N− were simultaneously recorded using four electron multipliers and a raster size of 20 × 20 μm2 or 10 × 10 μm2. For each region analyzed, 15–150 scans were acquired. The depth of the removed material was estimated to 120–600 nm, without considering the material removed during pre-sputtering (Lechene et al., 2006). The data were analyzed with the Look@NanoSIMS software (Polerecky et al., 2012). For each field analyzed, individual scans were accumulated and drift corrected. Regions of interest were manually drawn around the inner margins of individual cells (to avoid including filter-derived material in the ratio calculations) using the 12C14N− and 19F− ion images as a mask. The regions of interests were used to calculate 19F/12C and 13C/12C ratios. On average, 70 cells were analyzed for every time point. The 19F/12C ratios were used to identify cells of the dominant phylotype; cells with 19F/12C ratios similar with those of the filter background were considered contaminant cells, while cells with 19F/12C ratios 1 order of magnitude higher than the filter background were considered as dominant phylotype cells (Supplementary Table S1). The 13C-abundance in individual cells was calculated from the 13C/12C ratios as follows: 13C/(13C+12C)=(13C/12C)/(1+13C/12C). The rate of carbon assimilation for individual cells was calculated using the 13C-enrichment, the percent label of each substrate, the cellular carbon content, and the incubation time (Δt) as previously described (Musat et al., 2008). For the calculation, we assumed that the C-assimilated was homogeneously distributed within the cells, and that cells of the dominant phylotypes had the same C-content as determined for strain BuS5.
Results and discussion
Enrichment and determination of the optimum temperature
We report the enrichment of propane- and butane-degrading sulfate-reducing bacteria from sediments associated with cold marine hydrocarbon seeps in the Gulf of Mexico and with sediment associated with structure II gas hydrates at Hydrate Ridge. Incubations were started at 12 °C, similar to in situ temperature (<10 °C), and also at 28 °C to cover a higher range of temperature-adapted microorganisms. After 6 months at 12 °C, incubations with propane and Gulf of Mexico sediments (Prop12-GMe) and incubations with butane and Hydrate Ridge sediments (But12-HyR) produced up to 18 mM sulfide vs controls lacking addition of substrate, which did not exceeded 3 mM (not shown). Within the same incubation time, the sulfide production in bottles incubated with propane or butane at 28 °C did not exceeded that of substrate-free controls. Sediment-free enrichment cultures obtained by repeated transfers (10% v/v inoculum) in fresh medium formed ∼15 mM sulfide within 5–6 weeks. The successful enrichment at 12 but not at 28 °C suggested that these short-chain alkane-degrading bacteria are rather cold-adapted, reflecting the in situ temperature conditions. To test this hypothesis, sulfate-reduction rates over a wide range of temperatures (0–40 °C) were determined for the enrichment cultures Prop12-GMe and But12-HyR, and for a previously described enrichment culture obtained in our laboratory with butane from Gulf of Mexico sediments (But12-GMe; Kniemeyer et al., 2007). For comparison, temperature-dependent sulfate-reduction rates were determined for the mesophilic strain BuS5, isolated from hydrothermal vent-associated sediments (Kniemeyer et al., 2007). All enrichment cultures displayed significant substrate-dependent sulfate-reduction rates between 4 and 25 °C, with maximum rates between 16 and 20 °C (Figure 1). At 28 °C, the sulfate-reduction rates of the enrichment cultures Prop12-GMe and But12-HyR dropped below 5% of the maximum, while the enrichment culture But12-GMe retained about 20% of the maximum rate. No sulfate-reduction rates could be measured at temperatures ⩾30 °C. In contrast, strain BuS5 showed butane-dependent sulfate-reduction rates between 10 and 35 °C, with highest rates at 27–30 °C (Figure 1). The temperature profiles show that the propane- and butane-degrading microorganisms in these enrichment cultures are rather cold-adapted (psychrotrophs) and not true psychrophiles (Isaksen and Jørgensen, 1996). The determined optimum temperature is significantly higher than the reported in situ temperature at cold seeps and gas hydrates (4–8 °C; Suess et al., 1999; Orcutt et al., 2004). Similar findings have been reported in previous studies (Isaksen and Jørgensen, 1996; Finke and Jørgensen, 2008; Hubert et al., 2009). It was commonly observed that microorganisms living in arctic environments displayed optimum growth or respiration rates at temperatures significantly higher than those encountered in situ, whereas microorganisms from hot environments exhibit optimum temperatures closer to the actual in situ temperatures (Stetter et al., 1990; Jørgensen et al., 1992). The reason for these observations likely involve lowered enzymatic affinities at low temperatures, an impaired ability of enzymes to undergo conformational changes, or impaired fluidity of cellular membranes affecting the transport of substrates and products (Wiegel, 1990).

Temperature-dependent sulfate reduction rates (SRR) of the propane- and butane-degrading enrichment cultures from marine hydrocarbon seeps. Incubations were done for up to 11 days. The SRR are expressed as percent of the highest rate determined. The maximum sulfate reduction rates (mM H2S day-1) were 0.62 (Prop12-GMe), 0.82 (But12-GMe), 0.9 (But12-HyR) and 1.12 (strain BuS5).
Growth tests with other hydrocarbons
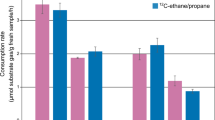
Quantitative growth experiments showed that the enrichment cultures degraded propane or butane completely to CO2, with stoichiometric reduction of sulfate to sulfide, as described before (Kniemeyer et al., 2007; Savage et al., 2010). The enrichment cultures were tested for their ability to degrade methane, the dominant hydrocarbon at gas seeps and other alkanes found in relatively high concentrations in such environments, for example, ethane, isobutane and pentane. Incubations with individual hydrocarbons or with mixtures of methane, ethane, propane and butane showed that the enrichment cultures Prop12-GMe and But12-HyR degraded propane and butane simultaneously, whereas methane and ethane were not degraded (Figure 2, Supplementary Figure S1). In similar incubations, the culture But12-GMe degraded only butane. None of the enrichment cultures degraded isobutane or pentane when supplied as single substrates or together with propane or butane (Figure 2, Supplementary Figure S2). In sterile control incubations, the concentration of the hydrocarbons tested decreased slightly (Supplementary Figure S3); a similar decrease, not associated with an increase of sulfide concentrations, was observed for methane, ethane, isobutane and pentane in incubations with the enrichment cultures (Figure 2, Supplementary Figures S1, S2). This was interpreted as abiotic (physical) loss during the relatively long incubation time. During the incubations with mixtures of alkanes the abundance of the dominant phylotype was stable, as shown by hybridizations with fluorescently-labeled, sequence-specific oligonucleotide probes (see Identification of dominant phylotypes; Supplementary Table S2). The results show that these microorganisms were apparently specialized, capable of degrading only propane and butane, similar to strain BuS5. This further supports in situ observations reporting that propane and butane appeared to be biologically degraded in oil and gas reservoirs (James and Burns, 1984; Boreham et al., 2001; Head et al., 2003; Larter et al., 2005) and at marine hydrocarbon-rich sites (Orcutt et al., 2010; Quistad and Valentine, 2011), whereas ethane, isobutane and pentane appeared rather resistant to degradation.

Consumption of propane ( ) and butane (
) and butane ( ) coupled to the production of sulfide (
) coupled to the production of sulfide ( ) by the enrichment culture Prop12-GMe. Methane (
) by the enrichment culture Prop12-GMe. Methane ( ), ethane (
), ethane ( ), pentane (
), pentane ( ) and isobutane (
) and isobutane ( ) were not degraded if supplied as single substrates or in mixtures with propane or butane. No sulfide was produced in controls without substrate (
) were not degraded if supplied as single substrates or in mixtures with propane or butane. No sulfide was produced in controls without substrate ( ). Cultures were set up in 120 ml serum bottles supplied with 79 ml artificial seawater medium and 1 ml inoculum from a dense cell suspension (10 × concentrated).
). Cultures were set up in 120 ml serum bottles supplied with 79 ml artificial seawater medium and 1 ml inoculum from a dense cell suspension (10 × concentrated).
Identification of dominant phylotypes
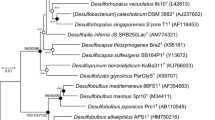
The sediment-free enrichments Prop12-GMe and But12-HyR formed macroscopic, dark-red aggregates with a diameter of up to 1 mm. Microscopic analysis showed that aggregates of both enrichment cultures consisted of densely packed, mainly oval, slightly curved cells, of 3 μm length–1 μm diameter (on average; Prop12-GMe) or 2.5 μm length–0.8 μm diameter (But12-HyR) (Figure 3a and b). Further description of the apparently dominant morphotypes was attempted using 16S rRNA-based methods. Construction and analysis of 16S rRNA gene libraries showed a relatively high diversity of sulfate-reducing bacteria in both enrichment cultures (Supplementary Table S3, Supplementary Figure S4). Sequences were closely related to those of Desulfobacula, Desulfococcus and Desulfosarcina species, including strain BuS5 and the dominant phylotype in the butane-degrading enrichment culture, But12-GMe (Kniemeyer et al., 2007). Sequences obtained by direct sequencing of PCR products from increasingly diluted (10−2) DNA template were identical to Prop12-GMe clone 235 and to But12-HyR clone 303 (Figure 4) suggesting that these phylotypes were dominant in the Prop12-GMe and But12-HyR enrichment culture, respectively. This hypothesis was verified by a top-to-bottom fluorescence in situ hybridization approach (Amann et al., 1995). Hybridizations of aggregate sections with the group-specific probe DSS658 showed that phylotypes affiliated with the Desulfosarcina-Desulfococcus group of the Deltaproteobacteria represented ∼80% of the Prop12-GMe (Figure 3c) and ∼95% of the But12-HyR enrichment culture (Figure 3d, Supplementary Figure S5). Furthermore, hybridizations of homogenized aggregate samples with sequence-specific oligonucleotide probes showed that Prop12-GMe clone 235 accounted for up to 74% of the total cell number of the Prop12-GMe enrichment culture (Figure 3e), and But12-HyR clone 303 for up to 69% of the total cell number of the But12-HyR enrichment culture (Figure 3f, Supplementary Figure S5). Prop12-GMe clone 235 and But12-HyR clone 303 shared 95.5% sequence identity and were closely related to strain BuS5 (93.1% and 94.5%, respectively) and to the dominant phylotype in the enrichment culture But12-GMe (94.3% and 95.3%, respectively; Kniemeyer et al., 2007) (Figure 4). Other close relatives include phylotypes retrieved from marine mud volcanoes (Heijs et al., 2007; Lösekann et al., 2007), hydrocarbon seep-associated sediments (Orphan et al., 2001; Lloyd et al., 2006; Kleindienst et al., 2012) or marine sediments associated with gas hydrates (Knittel et al., 2003; Orcutt et al., 2010) (Figure 4). The high abundance of a distinct phylotype in each enrichment culture suggests that the corresponding microorganisms were most likely responsible for the degradation of propane or butane. This hypothesis was further investigated by incubating the enrichment cultures with 13C-labeled propane or butane followed by HISH-SIMS analyses.

Microscopic images of the enrichment cultures Prop12-GMe (a, c, e), and But12-HyR (b, d, f). Phase contrast images show densely packed, mostly oval-shaped cells in both enrichment cultures (a, b). These cells affiliated with the Desulfosarcina-Desulfococcus cluster of the Deltaproteobacteria, as demonstrated by hybridization of aggregate cross sections with the probe DSS658 (c, d). Hybridizations of partly homogenized aggregates with sequence-specific oligonucleotide probes further indicated that each of the enrichment cultures was dominated by a unique phylotype (e, f). The hybridization images (c – f) show overlays of probe (orange) and DAPI (blue) signals. Scale bars = 5 μm.

Phylogenetic affiliation of the dominant phylotypes in the enrichment cultures Prop12-GMe and But12-HyR (marked in boldface). Sequences affiliated with the Desulfosarcina-Desulfococcus cluster of the Deltaproteobacteria and those targeted by the DSS658 oligonucleotide probe are marked. The phylogenetic tree was calculated by Maximum Likelihood using only nearly full length sequences (>1400 bp). The numbers next to the nodes indicate bootstrap values (100 replications); only values larger than 50% are shown.
Incubations with 13 C-labeled hydrocarbons and HISH-SIMS analyses
The enrichment cultures incubated with labeled propane or butane became increasingly enriched in 13C, as revealed by mass spectrometry analyses of the bulk biomass (Figure 5). Based on the bulk mass spectrometry analyses, samples were further selected for single-cell analyses by nanoSIMS. Using the 12C14N− ion images (Figures 6a, d and g), regions of interests were drawn around individual cells and used to determine 13C/12C ratios for the dominant and accompanying cells. Cells of the dominant phylotype were identified by the presence of 19F− (Figures 6b, e and h; Supplementary Table S1). In all enrichment cultures, the cells of the dominant phylotype were the first to assimilate 13C from the labeled substrate, while the accompanying cells showed no or only minor label incorporation (Figure 5; Figures 6c, f and i). With longer incubation time, increasing 13C-labeling was observed in the accompanying cells (Figure 5). These results demonstrate that the main phylotypes were responsible for the degradation of propane or butane, while the accompanying bacteria were most probably growing at the expense of excreted degradation intermediates or cellular constituents from lysing dominant cells.

Abundance of 13C as determined by bulk analysis of biomass (□) and by nanoSIMS in single cells of the enrichment cultures Prop12–GMe, But12–GMe, and But12–HyR incubated with 13C-propane (Prop12-GMe) or 13C-butane (But12-GMe and But12-HyR). In order to distinguish cells of the dominant phylotype (•) from accompanying cells ( ), the values were plotted with an off-scale on the time axis of ±0.2. The dominant cells were identified based on the presence of 19F signals. Shaded area indicates the range of 13C-abundance values obtained for cells at 0 incubation days (natural abundance of 13C).
), the values were plotted with an off-scale on the time axis of ±0.2. The dominant cells were identified based on the presence of 19F signals. Shaded area indicates the range of 13C-abundance values obtained for cells at 0 incubation days (natural abundance of 13C).

NanoSIMS images of the enrichment cultures Prop12-GMe (a – c), But12-GMe (d – f), and But12-HyR (g – i) after incubation with 13C-labeled propane or butane for 3, 5 and 3 days, respectively. Individual cells were identified in the total biomass (12C14N−) images (a, d, g), and highlighted by drawings around cell edges, for easier tracking in the ratio images. Cells of the dominant phylotypes were identified based on detection of 19F (b, e, h) introduced into the cells by deposition of F-containing tyramides after hybridization with HRP-labelled oligonucleotide probes. The ratio images of 13C/12C show incorporation of label by individual cells (c, f, i). Scale bars = 1 μm.
For each enrichment culture, the abundance of 13C in the cells of the dominant phylotype ranged from natural abundance (no incorporation of substrate) to relatively high values (Figure 5). This range could be explained as physiological heterogeneity within populations, also observed in other populations from natural environments (Musat et al., 2008). In addition, propane and butane as hydrocarbons have low water solubility, which could lead to diffusion and consequently substrate-availability limitations, especially for cells inside aggregates, which would tend to settle away from the hydrocarbon-water interface. For the aggregate forming enrichment cultures (Prop12-GMe and But12-HyR), the incubations were done with partly homogenized aggregates to facilitate single-cell analysis by nanoSIMS. The cells inside of these aggregates could have been strongly limited in both sulfate and substrate, and due to the relatively short incubation may have remained in a lag phase of growth at the time of analysis.
Using the 13C/12C ratios, substrate-specific carbon assimilation rates were calculated for the dominant cells in each enrichment culture (Supplementary Table S4). For the calculation, the carbon content per cell volume of the dominant phylotypes was considered the same as that determined for the phylogenetically and physiologically related strain BuS5 (13.6 fmol C per cell, 4.6 fmol C μm−3). The average biovolumes of the dominant phylotype cells were 2.2 μm3 (Prop12-GMe), 2.8 μm3 (But12-GMe) and 1.1 μm3 (But12-HyR). Average assimilation rates were calculated for cells showing enrichment in 13C vs cells at t0. The average values obtained ranged from 9±8 amol C per cell per day (But12-GMe) to 50±35 amol C per cell per day (Prop12-GMe), and were similar across incubation times of each enrichment culture (Supplementary Table S4; Supplementary Figure S6). To our knowledge, this is the first study reporting C-assimilation rates for single cells of propane- and butane-degrading microorganisms. A similar approach could be used to identify and quantify the activity of microorganisms degrading gaseous alkanes directly in the environment.
Conclusions
The propane and butane degraders identified in the present study further expand our knowledge on the diversity of short-chain alkane-degrading, sulfate-reducing bacteria. Phylogenetic analyses showed that these bacteria formed an apparent cluster of gaseous alkane degraders. Such ‘metabolic’ clustering of anaerobic hydrocarbon degraders, observed also for sulfate- or nitrate-reducing bacteria degrading aromatic hydrocarbons or higher alkanes (Musat et al., 2009; Widdel et al., 2010), can be explained by evolutionary divergence from a common ancestor.
The cold-adapted gaseous alkane degraders described here are part of the Desulfosarcina-Desulfococcus group within the Deltaproteobacteria, which are found in relatively high abundance at gas and oil hydrocarbon seeps, very often reaching over 10% of the total free cells (for example, Orcutt et al., 2010; Kleindienst et al., 2012). Sulfate-reduction rates determined at such sites often exceeded the rates attributed to anaerobic oxidation of methane by several fold (Bowles et al., 2011), and have been proposed to be due to oxidation of hydrocarbons other than methane, including short-chain alkanes (Joye et al., 2004; Kallmeyer and Boetius 2004; Orcutt et al., 2005, 2010; Niemann et al., 2006; Kleindienst et al., 2012). Propane- and butane-degrading sulfate reducers may contribute significantly to the excess sulfate-reduction rates, and could have an important role for the in situ carbon and sulfur cycling.
References
Amann RI, Krumholz L, Stahl DA . (1990). Fluorescent-oligonucleotide probing of whole cells for determinative, phylogenetic, and environmental-studies in microbiology. J Bacteriol 172: 762–770.
Amann RI, Ludwig W, Schleifer KH . (1995). Phylogenetic identification and in situ detection of individual microbial cells without cultivation. Microbiol Rev 59: 143–169.
Audinot JN, Cabin-Flaman A, Philipp P, Legent G, Wirtz T, Migeon HN . (2011). NanoSIMS50 imaging of thin samples coupled with neutral cesium deposition. Surf Interface Anal 43: 302–305.
Behrens S, Losekann T, Pett-Ridge J, Weber PK, Ng WO, Stevenson BS et al (2008). Linking microbial phylogeny to metabolic activity at the single-cell level by using enhanced element labeling-catalyzed reporter deposition fluorescence in situ hybridization (EL-FISH) and nanoSIMS. Appl Environ Microbiol 74: 3143–3150.
Boreham BCJ, Hope JM, Hartung-Kagi B . (2001). Understanding source, distribution and preservation of Australian natural gas: a geochemical perspective. APPEA J 41: 523–547.
Bowles MW, Samarkin VA, Bowles KM, Joye SB . (2011). Weak coupling between sulfate reduction and the anaerobic oxidation of methane in methane-rich seafloor sediments during ex situ incubation. Geochim Cosmochim Acta 75: 500–519.
Chapoy A, Mokraoui S, Valtz A, Richon D, Mohammadi AH, Tohidi B . (2004). Solubility measurement and modeling for the system propane−water from 277.62 to 368.16 K. Fluid Phase Equilibr 226: 213–220.
Cord-Ruwisch R . (1985). A quick method for the determination of dissolved and precipitated sulfides in cultures of sulfate-reducing bacteria. J Microbiol Methods 4: 33–36.
Dean JA . (1992) Lange’s Handbook of Chemistry. McGraw-Hill: New York, NY, USA.
Finke N, Jørgensen BB . (2008). Response of fermentation and sulfate reduction to experimental temperature changes in temperate and Arctic marine sediments. ISME J 2: 815–829.
Head IM, Jones DM, Larter SR . (2003). Biological activity in the deep subsurface and the origin of heavy oil. Nature 426: 344–352.
Heijs SK, Haese RR, van der Wielen PWJJ, Forney LJ, van Elsas JD . (2007). Use of 16S rRNA gene based clone libraries to assess microbial communities potentially involved in anaerobic methane oxidation in a Mediterranean cold seep. Microbiol Ecol 53: 384–398.
Hicks RE, Amann RI, Stahl DA . (1992). Dual staining of natural bacterioplankton with 4′,6-diamidino-2-phenylindole and fluorescent oligonucleotide probes targeting kingdom-level 16S ribosomal-RNA sequences. Appl Environ Microbiol 58: 2158–2163.
Hovland M, Judd AG, Burke RA . (1993). The global flux of methane from shallow submarine sediments. Chemosphere 26: 559–578.
Hubert C, Loy A, Nickel M, Arnosti C, Baranyi C, Bruchert V et al (2009). A constant flux of diverse thermophilic bacteria into the cold Arctic seabed. Science 325: 1541–1544.
Hugenholtz P, Tyson GW, Blackall LL . (2001). Design and evaluation of 16S rRNA-targeted oligonucleotide probes for fluorescence in situ hybridization. In: De Muro A, Rapley M, Totowa R, (eds) Methods in Molecular Biology–Gene Probes: Principles and Protocols 179. Humana Press Inc.: Totowa, NJ, pp 29–41.
Isaksen MF, Jørgensen BB . (1996). Adaptation of psychrophilic and psychrotrophic sulfate-reducing bacteria to permanently cold marine environments. Appl Environ Microbiol 62: 408–414.
James AT, Burns BJ . (1984). Microbial alterations of subsurface natural gas accumulations. Am Assoc Pet Geol Bull 68: 957–960.
Joye SB, Boetius A, Orcutt BN, Montoya JP, Schulz HN, Erickson MJ et al (2004). The anaerobic oxidation of methane and sulfate reduction in sediments from Gulf of Mexico cold seeps. Chem Geol 205: 219–238.
Jørgensen BB, Isaksen MF, Jannasch HW . (1992). Bacterial sulfate reduction above 100 °C in deep-sea hydrothermal vent sediments. Science 258: 1756–1757.
Kallmeyer J, Boetius A . (2004). Effects of temperature and pressure on sulfate reduction and anaerobic oxidation of methane in hydrothermal sediments of Guaymas Basin. Appl Environ Microbiol 70: 1231–1233.
Kane MD, Poulsen LK, Stahl DA . (1993). Monitoring the enrichment and isolation of sulfate-reducing bacteria by using oligonucleotide hybridization probes designed from environmentally derived 16S ribosomal-RNA sequences. Appl Environ Microbiol 59: 682–686.
Kinnaman FS, Valentine DL, Tyler SC . (2007). Carbon and hydrogen isotope fractionation associated with the aerobic microbial oxidation of methane, ethane, propane and butane. Geochim Cosmochim Acta 71: 271–283.
Kleindienst S, Ramette A, Amann R, Knittel K . (2012). Distribution and in situ abundance of sulfate-reducing bacteria in diverse marine hydrocarbon seep sediments. Environ Microbiol 14: 2689–2710.
Kniemeyer O, Musat F, Sievert SM, Knittel K, Wilkes H, Blumenberg M et al (2007). Anaerobic oxidation of short-chain hydrocarbons by marine sulphate-reducing bacteria. Nature 449: 898–901.
Knittel K, Boetius A, Lemke A, Eilers H, Lochte K, Pfannkuche O et al (2003). Activity, distribution, and diversity of sulfate reducers and other bacteria in sediments above gas hydrate (Cascadia margin, Oregon). Geomicrobiol J 20: 269–294.
Kropp KG, Davidova IA, Suflita JM . (2000). Anaerobic oxidation of n-dodecane by an addition reaction in a sulfate-reducing bacterial enrichment culture. Appl Environ Microbiol 66: 5393–5398.
Larter SR, Head IM, Huang H, Bennet B, Jones M, Aplin AC et al (2005). Biodegradation, gas destruction and methane generation in deep subsurface petroleum reservoirs: an overview. Geological Society, London, Petroleum Geology Conference series 6: 633–639.
Lechene C, Hillion F, McMahon G, Benson D, Kleinfeld AM, Kampf JP et al (2006). High-resolution quantitative imaging of mammalian and bacterial cells using stable isotope mass spectrometry. J Biol 5: 20.
Lloyd KG, Lapham L, Teske A . (2006). Anaerobic methane-oxidizing community of ANME-1b archaea in hypersaline Gulf of Mexico sediments. Appl Environ Microbiol 72: 7218–7230.
Lösekann T, Knittel K, Nadalig T, Fuchs B, Niemann H, Boetius A et al (2007). Diversity and abundance of aerobic and anaerobic methane oxidizers at the Haakon Mosby mud volcano, Barents Sea. Appl Environ Microbiol 73: 3348–3362.
Ludwig W, Strunk O, Westram R, Richter L, Meier H, Yadhukumar et al (2004). ARB: a software environment for sequence data. Nucleic Acids Res 32: 1363–1371.
Manz W, Eisenbrecher M, Neu TR, Szewzyk U . (1998). Abundance and spatial organization of Gram-negative sulfate-reducing bacteria in activated sludge investigated by in situ probing with specific 16S rRNA targeted oligonucleotides. FEMS Microbiol Ecol 25: 43–61.
Mastalerz V, de Lange GJ, Dahlmann A . (2009). Differential aerobic and anaerobic oxidation of hydrocarbon gases discharged at mud volcanoes in the Nile deep-sea fan. Geochim Cosmochim Acta 73: 3849–3863.
Mueller L, de Brouwer J, Almeida J, Stal L, Xavier J . (2006). Analysis of a marine phototrophic biofilm by confocal laser scanning microscopy using the new image quantification software PHLIP. BMC Ecol 6: 1.
Musat F, Galushko A, Jacob J, Widdel F, Kube M, Reinhardt R et al (2009). Anaerobic degradation of naphthalene and 2-methylnaphthalene by strains of marine sulfate-reducing bacteria. Environ Microbiol 11: 209–219.
Musat N, Giere O, Gieseke A, Thiermann F, Amann R, Dubilier N . (2007). Molecular and morphological characterization of the association between bacterial endosymbionts and the marine nematode Astomonema sp from the Bahamas. Environ Microbiol 9: 1345–1353.
Musat N, Halm H, Winterholler B, Hoppe P, Peduzzi S, Hillion F et al (2008). A single-cell view on the ecophysiology of anaerobic phototrophic bacteria. Proc Natl Acad Sci USA 105: 17861–17866.
Niemann H, Duarte J, Hensen C, Omoregie E, Magalhaes VH, Elvert M et al (2006). Microbial methane turnover at mud volcanoes of the Gulf of Cadiz. Geochim Cosmochim Acta 70: 5336–5355.
Orcutt BN, Boetius A, Lugo SK, MacDonald IR, Samarkin VA, Joye SB . (2004). Life at the edge of methane ice: microbial cycling of carbon and sulfur in Gulf of Mexico gas hydrates. Chem Geol 20: 239–251.
Orcutt B, Boetius A, Elvert M, Samarkin V, Joye SB . (2005). Molecular biogeochemistry of sulfate reduction, methanogenesis and the anaerobic oxidation of methane at Gulf of Mexico cold seeps. Geochim Cosmochim Acta 6: 4267–4281.
Orcutt BN, Joye SB, Kleindienst S, Knittel K, Ramette A, Reitz A et al (2010). Impact of natural oil and higher hydrocarbons on microbial diversity, distribution, and activity in Gulf of Mexico cold-seep sediments. Deep-Sea Res Pt II 57: 2008–2021.
Orphan VJ, Hinrichs KU, Ussler W, Paull CK, Taylor LT, Sylva SP et al (2001). Comparative analysis of methane-oxidizing archaea and sulfate-reducing bacteria in anoxic marine sediments. Appl Environ Microbiol 67: 1922–1934.
Polerecky L, Adam B, Milucka J, Musat N, Vagner T, Kuypers MMM . (2012). Look@NanoSIMS—a tool for the analysis of nanoSIMS data in environmental microbiology. Environ Microbiol 14: 1009–1023.
Pruesse E, Quast C, Knittel K, Fuchs BM, Ludwig WG, Peplies J et al (2007). SILVA: a comprehensive online resource for quality checked and aligned ribosomal RNA sequence data compatible with ARB. Nucleic Acids Res 35: 7188–7196.
Quistad SD, Valentine DL . (2011). Anaerobic propane oxidation in marine hydrocarbon seep sediments. Geochim Cosmochim Acta 75: 2159–2169.
Rabus R, Widdel F . (1995). Anaerobic degradation of ethylbenzene and other aromatic hydrocarbons by new denitrifying bacteria. Arch Microbiol 16: 96–103.
Rabus R, Wilkes H, Behrends A, Armstroff A, Fischer T, Pierik AJ et al (2001). Anaerobic initial reaction of n-alkanes in a denitrifying bacterium: Evidence for (1-methylpentyl)succinate as initial product and for involvement of an organic radical in n-hexane metabolism. J Bacteriol 183: 1707–1715.
Redmond MC, Valentine DL, Sessions AL . (2010). Identification of novel methane-, ethane-, and propane-oxidizing bacteria at marine hydrocarbon seeps by stable isotope probing. Appl Environ Microbiol 76: 6412–6422.
Sassen R, Losh SL, Cathles L, Roberts HH, Whelan JK, Milkov AV et al (2001). Massive vein-filling gas hydrate: relation to ongoing gas migration from the deep subsurface in the Gulf of Mexico. Mar Petrol Geol 18: 551–560.
Sassen R, Roberts HH, Carney R, Milkov AV, DeFreitas DA, Lanoil B et al (2004). Free hydrocarbon gas, gas hydrate, and authigenic minerals in chemosynthetic communities of the northern Gulf of Mexico continental slope: relation to microbial processes. Chem Geol 205: 195–217.
Savage KN, Krumholz LR, Gieg LM, Parisi VA, Suflita JM, Allen J et al (2010). Biodegradation of low-molecular-weight alkanes under mesophilic, sulfate-reducing conditions: metabolic intermediates and community patterns. FEMS Microbiol Ecol 72: 485–495.
Schwarzenbach RP, Gschwend PM, Imboden DM . (2003) Environmental Organic Chemistry. John Wiley & Sons, Inc: New Jersey.
Shennan JL . (2006). Utilisation of C2–C4 gaseous hydrocarbons and isoprene by microorganisms. J Chem Technol Biot 81: 237–256.
Sloan ED . (2003). Fundamental principles and applications of natural gas hydrates. Nature 426: 353–359.
Snaidr J, Amann R, Huber I, Ludwig W, Schleifer KH . (1997). Phylogenetic analysis and in situ identification of bacteria in activated sludge. Appl Environ Microbiol 63: 2884–2896.
Stamatakis A, Hoover P, Rougemont J . (2008). A rapid bootstrap algorithm for the RAxML web servers. Syst Biol 57: 758–771.
Stetter KO, Fiala G, Huber G, Huber R, Segerer A . (1990). Hyperthermophilic microorganisms. FEMS Microbiol Rev 75: 117–124.
Suess E, Torres ME, Bohrmann G, Collier RW, Greinert J, Linke P et al (1999). Gas hydrate destabilization: enhanced dewatering, benthic material turnover and large methane plumes at the Cascadia convergent margin. Earth Planet Sci Lett 170: 1–15.
Tissot BP, Welte DH . (1984) Petroleum formation and occurrence. Springer: Berlin, Germany.
Treude T, Boetius A, Knittel K, Wallmann K, Jørgensen BB . (2003). Anaerobic oxidation of methane above gas hydrates at Hydrate Ridge, NE Pacific Ocean. Mar Ecol-Prog Ser 264: 1–14.
Valentine DL, Kessler JD, Redmond MC, Mendes SD, Heintz MB, Farwell C et al (2010). Propane respiration jump-starts microbial response to a deep oil spill. Science 330: 208–211.
Widdel F, Bak F . (1992). Gram-negative mesophilic sulfate-reducing bacteria. In: Balows A, Trüper HG, Dworkin M, Harder W, Schleifer K-H, (eds) The prokaryotes. Springer: New York, NY, USA, pp 2278–3352.
Widdel F . (2010). Cultivation of anaerobic microorganisms with hydrocarbons as growth substrates. In: Timmis KN, (ed) Handbook of Hydrocarbon and Lipid Microbiology. Springer: Berlin, Heidelberg, Germany, pp 3787–3798.
Widdel F, Knittel K, Galushko A . (2010). Anaerobic hydrocarbon-degrading microorganisms: an overview. In: Timmis KN, (ed) Handbook of Hydrocarbon and Lipid Microbiology. Springer: Berlin Heidelberg, Germany, pp 1998–2021.
Wiegel J . (1990). Temperature spans for growth: Hypothesis and discussion. FEMS Microbiol Lett 75: 155–169.
Zhou JZ, Bruns MA, Tiedje JM . (1996). DNA recovery from soils of diverse composition. Appl Environ Microbiol 62: 316–322.
Acknowledgements
We thank the Crews of the RV Seward Johnson I and the RV SONNE-148/1 for their support with work at sea. We also thank Antje Boetius for providing the Hydrate Ridge samples, and Samantha Joye for providing the Gulf of Mexico samples; Ramona Appel and Daniela Franzke for technical support; Tomas Vagner and Rachel Foster for help with nanoSIMS analyses; Gaute Lavik and Gabriele Klockgether for bulk 13C-measurements; Andrea Schipper for bulk carbon measurements and Dennis Fink for help with the PHLIP software. This project was funded by the Deutsche Forschungsgemeinschaft (Priority Program SPP1319, grant MU 2950/1-1 to F Musat) and by the Max Planck Society.
Author information
Authors and Affiliations
Corresponding author
Additional information
Supplementary Information accompanies this paper on The ISME Journal website
Supplementary information
Rights and permissions
About this article
Cite this article
Jaekel, U., Musat, N., Adam, B. et al. Anaerobic degradation of propane and butane by sulfate-reducing bacteria enriched from marine hydrocarbon cold seeps. ISME J 7, 885–895 (2013). https://doi.org/10.1038/ismej.2012.159
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ismej.2012.159
Keywords
This article is cited by
-
Genomic insights into cryptic cycles of microbial hydrocarbon production and degradation in contiguous freshwater and marine microbiomes
Microbiome (2023)
-
Anaerobic oxidation of propane coupled to nitrate reduction by a lineage within the class Symbiobacteriia
Nature Communications (2022)
-
Verrucomicrobial methanotrophs grow on diverse C3 compounds and use a homolog of particulate methane monooxygenase to oxidize acetone
The ISME Journal (2021)
-
Deep sea sediments associated with cold seeps are a subsurface reservoir of viral diversity
The ISME Journal (2021)
-
Thermogenic hydrocarbon biodegradation by diverse depth-stratified microbial populations at a Scotian Basin cold seep
Nature Communications (2020)
