
Abstract
The goal of this study was to determine if fecal metabolite and microbiota profiles can serve as biomarkers of human intestinal diseases, and to uncover possible gut microbe–metabolite associations. We employed proton nuclear magnetic resonance to measure fecal metabolites of healthy children and those diagnosed with diarrhea-predominant irritable bowel syndrome (IBS-D). Metabolite levels were associated with fecal microbial abundances. Using several ordination techniques, healthy and irritable bowel syndrome (IBS) samples could be distinguished based on the metabolite profiles of fecal samples, and such partitioning was congruent with the microbiota-based sample separation. Measurements of individual metabolites indicated that the intestinal environment in IBS-D was characterized by increased proteolysis, incomplete anaerobic fermentation and possible change in methane production. By correlating metabolite levels with abundances of microbial genera, a number of statistically significant metabolite–genus associations were detected in stools of healthy children. No such associations were evident for IBS children. This finding complemented the previously observed reduction in the number of microbe–microbe associations in the distal gut of the same cohort of IBS-D children.
Similar content being viewed by others

Introduction
Gastrointestinal (GI) tract of humans is a complex environment characterized by a high diversity of inhabiting microorganisms, complex metabolic and spatial interactions among community members, and intricate interplay between microbes and the host. Changes in the human gut microbiota have now been associated with a number of GI diseases, including Crohn’s disease and ulcerative colitis, colorectal cancer and obesity (Sekirov et al., 2010). Many of these microbial effects are exerted through the metabolic interactions between microbes and host tissues. Complex carbohydrates from the diet are degraded by gut microbes in anaerobic fermentation to formate, lactate, pyruvate, succinate (intermediates) and short-chain fatty acids (SCFAs, end products; Karasov and Carey, 2009). SCFAs have a major role in the microbiota–host interactions, for example, by inhibiting inflammation and malignant growth (Guilloteau et al., 2010). Microbial degradation of proteins produces branched-chain fatty acids, amino acids, amines, as well as harmful phenolic compounds, ammonia and hydrogen disulfide (Bernalier-Donadille, 2010).
Thus, simultaneous interrogation of microbes and metabolites of the intestinal environment can provide valuable insight into possible gut microbial interactions and allow better understanding of how microbiota alterations in diseases such as irritable bowel syndrome (IBS) lead to detrimental effects on patient’s health. IBS is one of the most common disorders of the human GI system affecting 10–20% of the population with prevalence among adolescents (Quigley et al., 2012). In this study, we obtained quantitative metabolite signatures and carried out correlative microbe–metabolite analysis of fecal samples from healthy children and those diagnosed with diarrhea-predominant IBS (IBS-D).
Results
Metabolite profiling of fecal extracts
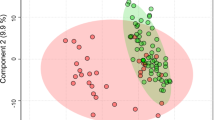
We extracted water-soluble metabolites from 44 fecal samples obtained from healthy and IBS-D children (designated kHLT and kIBS, respectively, 22 samples per group) and subsequently subjected these fecal extracts to proton nuclear magnetic resonance-based metabolomics analysis (see Supplementary Figure S1). All spectra were divided into ∼300 bins (see detailed description of all methods in Supplementary Materials), and these binned data were provided as input to several ordination analyses to reduce data set dimensionality and visualize sample dissimilarity (Figure 1). Using a non-supervised principal components analysis, a partial, but statistically significant, separation of samples from kIBS and kHLT groups was achieved (P=0.01, Figure 1a). We previously revealed a similar partial separation of the same set of fecal samples based on the microbiota abundances obtained with phylogenetic Microbiota Array (Shankar et al., 2013). To evaluate if both principal components analyses produced congruent sample distributions, Procrustes analysis, which seeks to superimpose, align and scale multiple graphical structures (Rohlf and Slice, 1990), was performed and revealed a strong congruency of sample separations based on metabolite and microbial signatures, respectively (P<0.001, Figure 1b). As expected, because supervised orthogonal projections to latent structures discriminant analysis (OPLS-DA) takes into consideration the known group assignment (for example, kIBS versus kHLT) for each sample, OPLS-DA analysis of the binned spectral data revealed a considerably better sample separation between groups (Figure 1c, model significance P<0.00002).

Separation of healthy and IBS fecal samples based on fecal metabolite profiling. Sample separation was assessed by the following ordination techniques: principal components analysis (PCA, a and b), correspondence analysis (CA, d and g), canonical discriminant analysis (CDA, e and h) and orthogonal projection to latent structures discriminant analysis (OPLS-DA, c, f and i). Sample separations in a–c are based on binned spectral data; sample separations in d–i are based on the levels of quantified metabolites. Panels g–i display CA, CDA and OPLS-DA sample distributions with the area of each sample spot proportional to the combined level of tyrosine and formate metabolites in that sample. P-values indicate the statistical significance of group separation. Where shown, group clouds represent areas of three standard errors around the centroid for each group. Panel b shows the results of Procrustes comparison of PCA separation of healthy and IBS samples based on binned metabolomic and microbial phylotype data sets. The distance between each sample position on two PCA plots is indicated by connecting line. Shorter lines represent more similar sample positioning on both plots. P-value indicates the statistical significance of sample separation congruency between two PCAs. kIBS, children diagnosed with IBS-D; kHLT, healthy chidlren.
Focusing our attention on SCFAs, amino acids and compounds involved in energy metabolism, we quantified relative levels of 19 discrete fecal metabolites (see Supplementary Table 1 in Supplementary Materials). No significant difference in average total metabolite levels was evident between kIBS and kHLT groups. Statistically significant differences were observed for individual metabolites including formate, glucose, lactate, pyruvate and several amino acids (tyrosine, lysine and leucine). With an exception of pyruvate, kIBS samples contained higher levels of these metabolites. Total bile acids, one of the proposed causative agents of IBS development (Duboc et al., 2012), were also higher in IBS, but the differences were not wide enough to overcome sample-to-sample variability in a significance test. Applying ordination techniques to the data set of measured metabolites revealed a statistically significant separation of samples between two groups (Figure 1d–i). OPLS-DA model as well as Random Forest analysis indicated that formate and tyrosine were the top two discriminating compounds.
Putative associations between intestinal metabolites and microbes
We computed the Spearman rank-based correlation matrix between metabolites measured here and microbial genus abundances assessed in the same set of samples in our previous report (Rigsbee et al., 2012). We previously reported that microbe–microbe interactions were severely limited in IBS samples (Shankar et al., 2013), therefore, metabolite–microbe associations were calculated separately for the kIBS and kHLT sample sets. Although many strong associations were discovered in the kHLT data set (Figure 2a), associations were weak in the kIBS sample cohort (Figure 2b). Simultaneous microbe- and metabolite-based bi-clustering of the correlation matrix revealed several local clusters of similar relationships in the kHLT data set. For example, all amino acids were clustered together, which can be attributed to their concurrent release through microbial proteolysis. Several groupings of microbial genera were also evident. In contrast, little noticeable clustering was observed in the kIBS correlation matrix.

Correlations of genus and metabolite abundances among healthy and IBS samples. (a and b) Euclidean distance-based bi-clustering of Spearman rank correlation matrices between microbial genus abundances (46 genera with average abundance among all samples above 0.1%) and fecal metabolites in kHLT (a) and kIBS (b) samples. Each square represents a correlation value between specific genus and specific metabolite based on the color gradient as shown in the legend. Statistically significant correlations are highlighted with a yellow border. (c) A bipartite graph of the statistically significant metabolite–genus associations identified in the kHLT sample set. Positive and negative relationships are designated by red and blue colors, respectively; the line width and transparency are proportional to the absolute value of the Spearman rank correlation. Metabolite level is indicated by a node background color gradient as shown in the legend. The area of each genus node is proportional to the square root of the average genus abundance among all healthy samples. Genus assignment to a particular phylum is represented by the color of the node (see legend).
By employing a false discovery rate of 10% to control for multiple hypothesis testing, 21 significant metabolite–microbe relationships were identified in healthy teenagers (Figure 2c); none were found in the kIBS sample set. Among the statistically significant relationships revealed in the kHLT data set, several have been previously uncovered. These included strong positive association between glucose level and Ruminococcus abundance (glucose is released by ruminococci during fermentation), negative relationship between glucose and Coprococcus (some coprococci use glucose under anaerobic conditions), and positive associations of Acidaminobacter, Coprococcus and Prevotella with acetate, valerate and fumarate, respectively, likely explained by the observed production of these metabolites by the members of these genera (Holdeman and Moore, 1974, Bergey and Holt, 1994, Takahashi and Yamada, 2000, Duncan et al., 2002, Iakiviak et al., 2011). Abundances of genera Bacteroides and Bifidobacterium, which contain many members with large repertoires of carbohydrate-degrading enzymes, were not associated significantly with any of the measured fermentation-derived metabolites, likely due to the ability of these organisms to switch from one carbohydrate to another based on luminal availability and thus produce a variety of fermentation products (Martens et al., 2011). The revealed associations of other genera with specific metabolites shown in Figure 2c have not yet been reported and represent putative novel interactions.
Discussion
The present study was undertaken to obtain quantitative metabolite signatures of fecal samples from healthy and IBS children, and to evaluate possible microbe–metabolite associations in the human gut. Overall, fecal samples from healthy and IBS adolescents could be distinguished based on the metabolite measurements of fecal extracts. Levels of formate, most of the identified amino acids, glucose and lactate differed between IBS and healthy samples. The presence of the higher levels of amino acids might be an indication of increased luminal proteolysis by IBS gut microbiota utilizing either diet-derived proteins or host mucins (Steck et al., 2011). Elevated levels of fermentation intermediates in IBS stools might point to a disbalance of the gut environment and intestinal microbiota in this disease, leading to reduced metabolite cross-feeding among microbes and a resulting incomplete fermentation process. Because formate is utilized in the gut by methanogenic microbes, increased fecal formate is consistent with a lower methanogenic activity in the IBS-D lumen (Chatterjee et al., 2007). And as intestinal methane adversely affects gut transit time (Ghoshal et al., 2011), lower methane production in IBS-D would facilitate faster gut transit resulting in a more frequent bowel movement.
A case for the loss of microbial interactions in IBS was also supported by our analysis of microbe–metabolite interactions. Although many strong associations were observed in healthy samples, such interactions were significantly weaker in IBS, with none reaching the level of statistical significance (see Figure 2). A concordant loss of microbe–microbe relative abundance associations in IBS was previously revealed (Shankar et al., 2013). We offer several possible explanations of these findings. If IBS is associated with a disturbance of the gut environment, the resulting shifts in relative abundances of microbial taxa (Rigsbee et al., 2012) may lead to a loss of the ‘steady-state’ microbial interactions within the healthy gut. The disruption of microbial metabolite exchanges would result in altered luminal and fecal metabolite levels we observed (Watanabe et al., 2012). Alternatively, the lack of strong metabolite–microbe interactions in IBS cohort might be an effect of the broad etiology of IBS. Different pathophysiological conditions that lead to IBS symptoms may result in unique shifts of microbiota community structure and/or functional capacity in different individuals. This, in turn, will either produce differentially altered luminal metabolite profiles, or a similar metabolic environment will be created by microbial communities of dissimilar compositions. Both cases will lead to an overall loss of observable cohort-wide interactions. Finally, although it is tempting to infer that the observed metabolite and microbial differences between healthy and IBS-D fecal samples are indicative of IBS pathophysiology, this syndrome is associated with many confounding factors such as change of eating habits or consumed diets, and alteration in stool consistency and frequency of defecation. Even though none of the children in IBS-D cohort were on any restricted diet or reported altered eating patterns at the time of sample collection, and there was only a small difference in the average water content of the collected stool samples between healthy and IBS-D groups, both diet changes and diarrhea have been shown to cause alterations in human gut microbiota (David et al., 2014, Gorkiewicz et al., 2013). Thus, some of the observed IBS-associated effects on the gut environment might be manifestations of the concurrent changes in eating and bowel evacuation patterns in individuals with IBS-D.
References
Bergey DH, Holt JG . (1994) Bergey's Manual of Determinative Bacteriology 9th edn. Williams & Wilkins: Baltimore.
Bernalier-Donadille A . (2010). Fermentative metabolism by the human gut microbiota. Gastroenterol Clin Biol 34: S16–S22.
Chatterjee S, Park S, Low K, Kong Y, Pimentel M . (2007). The degree of breath methane production in IBS correlates with the severity of constipation. Am J Gastroenterol 102: 837–841.
David LA, Maurice CF, Carmody RN, Gootenberg DB, Button JE, Wolfe BE et al. (2014). Diet rapidly and reproducibly alters the human gut microbiome. Nature 505: 559–563.
Duboc H, Rainteau D, Rajca S, Humbert L, Farabos D, Maubert M et al. (2012). Increase in fecal primary bile acids and dysbiosis in patients with diarrhea-predominant irritable bowel syndrome. Neurogastroenterol Motil 24: 513–520.
Duncan SH, Barcenilla A, Stewart CS, Pryde SE, Flint HJ . (2002). Acetate utilization and butyryl coenzyme A (CoA):acetate-CoA transferase in butyrate-producing bacteria from the human large intestine. Appl Environ Microbiol 68: 5186–5190.
Ghoshal UC, Srivastava D, Verma A, Misra A . (2011). Slow transit constipation associated with excess methane production and its improvement following rifaximin therapy: a case report. J Neurogastroenterol Motil 17: 185–188.
Gorkiewicz G, Thallinger GG, Trajanoski S, Lackner S, Stocker G, Hinterleitner T et al. (2013). Alterations in the colonic microbiota in response to osmotic diarrhea. PLoS One 8: e55817.
Guilloteau P, Martin L, Eeckhaut V, Ducatelle R, Zabielski R, Van Immerseel F . (2010). From the gut to the peripheral tissues: the multiple effects of butyrate. Nutr Res Rev 23: 366–384.
Holdeman LV, Moore WEC . (1974). New genus, Coprococcus, twelve new species, and emended descriptions of four previously described species of bacteria from human feces. Int J Syst Evol Microbiol 24: 260–277.
Iakiviak M, Mackie RI, Cann IK . (2011). Functional analyses of multiple lichenin-degrading enzymes from the rumen bacterium Ruminococcus albus 8. Appl Environ Microbiol 77: 7541–7550.
Karasov WH, Carey HV . (2009). Metabolic teamwork between gut microbes and hosts. Microbe 4: 323–328.
Martens EC, Lowe EC, Chiang H, Pudlo NA, Wu M, McNulty NP et al. (2011). Recognition and degradation of plant cell wall polysaccharides by two human gut symbionts. PLoS Biol 9: e1001221.
Quigley EM, Abdel-Hamid H, Barbara G, Bhatia SJ, Boeckxstaens G, De Giorgio R et al. (2012). A global perspective on irritable bowel syndrome: a consensus statement of the World Gastroenterology Organisation Summit Task Force on irritable bowel syndrome. J Clin Gastroenterol 46: 356–366.
Rigsbee L, Agans R, Shankar V, Kenche H, Khamis HJ, Michail S et al. (2012). Quantitative profiling of gut microbiota of children with diarrhea-predominant irritable bowel syndrome. Am J Gastroenterol 107: 1740–1751.
Rohlf FJ, Slice D . (1990). Extensions of the procrustes method for the optimal superimposition of landmarks. Syst Zool 39: 40–59.
Sekirov I, Russell SL, Antunes LC, Finlay BB . (2010). Gut microbiota in health and disease. Physiol Rev 90: 859–904.
Shankar V, Agans R, Holmes B, Raymer M, Paliy O . (2013). Do gut microbial communities differ in pediatric IBS and health? Gut Microbes 4: 347–352.
Steck N, Mueller K, Schemann M, Haller D . (2011). Bacterial proteases in IBD and IBS. Gut 61: 1610–1618.
Takahashi N, Yamada T . (2000). Pathways for amino acid metabolism by Prevotella intermedia and Prevotella nigrescens. Oral Microbiol Immunol 15: 96–102.
Watanabe Y, Nagai F, Morotomi M . (2012). Characterization of Phascolarctobacterium succinatutens sp. nov., an asaccharolytic, succinate-utilizing bacterium isolated from human feces. Appl Environ Microbiol 78: 511–518.
Acknowledgements
We are thankful to Andrew Neuforth for participation in nuclear magnetic resonance data processing. This work was supported by the National Institutes of Health grants AT003423 and HD065575 to OP.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on The ISME Journal website
Rights and permissions
About this article

Cite this article
Shankar, V., Homer, D., Rigsbee, L. et al. The networks of human gut microbe–metabolite associations are different between health and irritable bowel syndrome. ISME J 9, 1899–1903 (2015). https://doi.org/10.1038/ismej.2014.258
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ismej.2014.258
This article is cited by
-
Shedding light on biological sex differences and microbiota–gut–brain axis: a comprehensive review of its roles in neuropsychiatric disorders
Biology of Sex Differences (2022)
-
Metabolic adaption to extracellular pyruvate triggers biofilm formation in Clostridioides difficile
The ISME Journal (2021)
-
Microbial and metabolomic profiles in correlation with depression and anxiety co-morbidities in diarrhoea-predominant IBS patients
BMC Microbiology (2020)
-
Analysis of brain networks and fecal metabolites reveals brain–gut alterations in premenopausal females with irritable bowel syndrome
Translational Psychiatry (2020)
-
Predictive metabolomic profiling of microbial communities using amplicon or metagenomic sequences
Nature Communications (2019)

{kind=link}