
Abstract
Hermansky–Pudlak syndrome (HPS) is an autosomal recessive disorder characterized by oculocutaneous albinism, platelet dysfunction and ceroid deposition. We report suspected ocular albinism in two Japanese sisters, caused by mutations in the HPS6 (Hermansky–Pudlak syndrome 6) gene. Trio-based whole-exome sequencing (WES) identified novel compound heterozygous mutations in HPS6 (c.1898delC: mother origin and c.2038C>T: father origin) in the two sisters. To date, 10 associated mutations have been detected in HPS6. Although we detected no general manifestations, including platelet dysfunction, in the sisters, even in long-term follow-up, we established a diagnosis of HPS type 6 based on the HPS6 mutations and absence of dense bodies in the platelets, indicating that WES can identify cases of HPS type 6. To the best of our knowledge, this is the first report of HPS6 mutations in Japanese patients.
Similar content being viewed by others

Main
Hermansky–Pudlak syndrome (HPS) is an autosomal recessive disorder characterized by oculocutaneous albinism (OCA), platelet dysfunction and ceroid deposition.1 Most HPS patients are Puerto Rican,2 with an estimated incidence of 1 in 1800 in Puerto Rico3 and an incidence of 1 in 500 000–1 000 000 worldwide. A diagnosis of HPS is made based on the decreased visual acuity, nystagmus, hypopigmentation and the absence of platelet-dense bodies. Genetic testing can confirm the diagnosis. Nine genes (HPS1, AP3B1, HPS3–6, DTNBP1, BLOC1S3 and BLOC1S6) have been identified as causative genes in HPS.3, 4, 5, 6, 7, 8, 9 HPS types 1 and 3 (HPS-1 and HPS-3) are common subtypes of HPS, and HPS-1 is the relatively more common subtype in the Japanese population.10 In this study, we examined two sisters presenting with symptoms resembling ocular albinism (OA) or non-syndromic OCA and not HPS. They were previously analyzed for all exons and surrounding areas of five genes (TYR, OCA2, TYRP1, SLC45A2 and GPR143) that are causative for OA and non-syndromic OCA by Sanger sequencing, with no mutations detected.11 Recently, exome sequencing has been used for the genetic diagnosis of ocular disorders at the molecular level.12, 13 Adoption of a whole-exome sequencing (WES) approach is a particularly powerful strategy that has contributed to the identification of many new disease-causing genes.14 We carried out WES and identified novel deleterious mutations in HPS6 that are associated with syndromic OCA.
The proband was a girl aged 3 years and 11 months; she was of Japanese descent and had poor vision. Visual acuity in the right and left eyes was 20/100 and 20/70, respectively. In addition, we examined her younger sister—a 6-month-old girl—who also had impaired vision (see Supplementary Information and Figure 1 for more clinical details). We reviewed hospital records, including clinical data from biochemical and genetic analyses, ultrasound imaging, whole-mount electron microscopy imaging (JEM-1220; JEOL, Tokyo, Japan), and general medical and ophthalmologic examinations such as slit-lamp examination, indirect ophthalmoscopy, fundus photography and optic coherence tomography (Cirrus HD-OCT; Carl Zeiss, Wetzlar, Germany).

Fundus photography and optic coherence tomography (OCT) imaging. (a) Fundus photograph of the elder sister’s (proband) right eye. (b) Fundus photograph of the younger sister’s right eye. (c) OCT imaging of the elder sister’s right eye. (d) OCT imaging of the younger sister’s right eye. Fundus hypopigmentation and lack of macular ring reflexes were observed by fundus photography, whereas OCT imaging revealed foveal hypoplasia with an absence of plexiform layer extrusion, foveal pit, outer segment lengthening and outer nuclear layer widening. (e) Electron microscopy imaging of platelet from a normal control with dense bodies (arrow). (f) Electron microscopy imaging of platelet from patient II-1 (elder sister/proband). (g) Electron microscopy imaging of platelet from Patient II-2 (younger sister). Grids were viewed on a JEM-1220 transmission electron microscope at an accelerating voltage of 100 kV. The absence of dense bodies is an important manifestation in Hermansky–Pudlak syndrome. Scale bar, 1 μm.
Genomic DNA was extracted from peripheral lymphocytes of the two sisters and their parents using standard procedures. Trio samples (one affected individual and both parents) were analyzed by WES as previously described.15 Given that there were two affected female children from the same healthy parents, autosomal recessive inheritance was presumed; therefore, homozygous and compound heterozygous variants were hypothesized as being causative mutations after filtering out variants with a minor allele frequency ⩾0.005 using ExAC (http://exac.broadinstitute.org/), Exome Variant Server (http://evs.gs.washington.edu/EVS/), Human Genetic Variation Database (http://www.genome.med.kyoto-u.ac.jp/SnpDB/), and in-house exome data (n=575) and synonymous variants. The identified potential pathogenic mutations were confirmed to be co-segregated with the disease within this family by Sanger sequencing. Platelets were separated by centrifugation from blood samples. Grids were viewed on an electron microscope.16
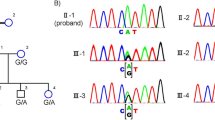
WES data of the proband (II-1 in Figure 2) and parents (I-1 and I-2) were analyzed based on the autosomal recessive model (Supplementary Tables 1 and 2). We identified two homozygous missense variants (c.1181C>A in TRIM49 [NM_020358.2] and c.3955A>C in LRRC37A [NM_014834.4]) and one compound heterozygous truncating variant (c.[1898delC];[2038C>T] in HSP6 [NM_024747.5]) (Supplementary Tables 1–3). The missense variant of TRIM49 was predicted to be non-pathogenic by SIFT and MutationTaster; 12 individuals with this homozygous variant are registered in the 1000 Genome project (http://browser.1000genomes.org; Supplementary Table 3). The missense LRRC37A variant was also predicted to be benign by three software programs (Supplementary Table 3). Thus, these two homozygous variants were excluded from the candidates, and the compound heterozygous variants of HPS6 remained: c.1898delC (p.P633Lfs*76) and c.2038C>T (p.Q680*), which were inherited from the mother and father, respectively (Figure 2; Table 1). HPS6 mutations are known to cause HPS (MIM #614075). According to the Human Gene Mutation Database (https://portal.biobase-international.com/cgi-bin/portal/login.cgi), 10 associated mutations have been registered to date; the two potential pathogenic mutations identified in this family have not been previously described. The two mutations were confirmed using Sanger sequencing (Supplementary Figure 1).

Pedigree of a family with suspected ocular albinism. Genotypes of affected siblings (II-1 and II-2) and their parents (I-1 and I-2) are shown. Nucleotide numbering reflects complementary DNA numbering, with +1 corresponding to A of the ATG translation initiation codon (designated as codon 1) in the reference sequence NM_024747.5, according to the nomenclature recommended by the Human Genome Variation Society (www.hgvs.org/mutnomen). Square and circles indicate male and females, respectively. Filled symbols indicate individuals affected by Hermansky–Pudlak syndrome. This retrospective study was approved by the Institutional Review Board of the Hamamatsu University School of Medicine and Yokohama City University Graduate School of Medicine. The study protocol adhered to the principles of the Declaration of Helsinki. Written informed consent was obtained from the patients’ parents before any study procedure or examination was performed.
Optic coherence tomography imaging revealed an absence of plexiform layer extrusion, foveal pit, outer segment lengthening and outer nuclear layer widening (Figure 1). Electron microscopy analyses of platelets revealed absence of dense bodies (Figure 1).
The 10 HPS6 mutations, including a variety of mutation types (frameshift, nonsense and missense), that have been detected to date show phenotypic variations. It is difficult to establish a clear genotype–phenotype correlation because of the limited number of HPS-6 patients. In this study, the proband and her sister with novel HPS6 mutations had no early bleeding manifestations in the 66-month follow-up period, despite the absence of dense bodies in their platelets. The fact that they were non-syndromic and their skin color was fair suggested that they had OA. However, we diagnosed them with HPS6. These clinical findings can thus broaden the phenotypic definition of HPS.
HPS subtype classification from genetic and diagnostic approaches can be used to predict patient prognosis. HPS is a genetically heterogeneous disorder characterized by intracellular vesicle biogenesis. Most causative genes for HPS, except for AP3B1, encode subunits of three protein complexes, termed biogenesis of lysosome-related organelles complexes (BLOC1–3). These proteins are involved in the biogenesis of lysosome-related organelles such as melanosomes in melanocytes and dense granules in platelets. BLOCs are required for the production of specialized organelles of the endosomal-lysosomal system.17 The complexes are ubiquitous structures of mostly unknown function that exist in plasma membrane-anchored and soluble cytoplasmic forms.18 BLOC-2 is composed of at least the HPS3, HPS-5 and HPS6 proteins, and BLOC-3 is composed of at least the HPS1 and HPS-4 proteins. Pathological effects are dependent on the disrupted BLOC subtype. Patients with genetic alterations affecting BLOC-2 have a mild HPS phenotype (that is, HPS-3, HPS-5 or HPS-6) with minor clinical manifestations;19 life-threatening symptoms such as pulmonary fibrosis mainly occur in HPS-1 and HPS-4, which are associated with BLOC-3. HPS-6 is not related to pulmonary fibrosis;6, 20 the reason for lung involvement is unclear, but likely reflects the role of BLOC-3 in the biogenesis and function of pneumocytic lamellar bodies.21 The fact that the two sisters were diagnosed with HPS-6 proved useful in terms of genetic counseling in this case.
In conclusion, we report novel HPS6 mutations as the first report of HPS6 mutations in the Japanese population. The clinical features in the two sisters suggest OA. Although the patients in this study showed no bleeding problem, we could establish a diagnosis of HPS-6 by WES.
References
Wei, A. H. & Li, W. Hermansky–Pudlak syndrome: pigmentary and non-pigmentary defects and their pathogenesis. Pigment Cell Melanoma Res. 26, 176–192 (2012).
Witkop, C. J., Nunez Babcock, M., Rao, G. H., Gaudier, F., Summers, C. G., Shanahan, F. et al. Albinism and Hermansky-Pudlak syndrome in Puerto Rico. Bol. Asoc. Med. P. R. 82, 333–339 (1990).
Anikster, Y., Huizing, M., White, J., Shevchenko, Y. O., Fitzpatrick, D. L., Touchman, J. W. et al. Mutation of a new gene causes a unique form of Hermansky−Pudlak syndrome in a genetic isolate of central Puerto Rico. Nat. Genet. 28, 376–378 (2001).
Dell’Angelica, E. C., Shotelersuk, V., Aguilar, R. C., Gahl, W. A. & Bonifacino, J. S. Altered trafficking of lysosomal proteins in Hermansky-Pudlak syndrome due to mutations in the beta 3A subunit of the AP-3 adaptor. Mol. Cell 3, 11–21 (1999).
Suzuki, T., Li, W., Zhang, Q., Novak, E. K., Sviderskaya, E. V., Hill, S. P. et al. Hermansky-Pudlak syndrome is caused by mutations in HPS4, the human homolog of the mouse light-ear gene. Nat. Genet. 30, 321–324 (2002).
Zhang, Q., Zhao, B., Li, W., Oiso, N., Novak, E. K., Rusiniak, M.E. et al. Ru2 and Ru encode mouse orthologs of the genes mutated in human Hermansky-Pudlak syndrome types 5 and 6. Nat. Genet. 33, 145–153 (2003).
Li, W., Zhang, Q., Oiso, N., Novak, E. K., Gautam, R., O’Brien, E. P. et al. Hermansky-Pudlak syndrome type 7 (HPS-7) results from mutant dysbindin, a member of the biogenesis of lysosome-related organelles complex 1 (BLOC-1). Nat. Genet. 35, 84–89 (2003).
Morgan, N. V., Pasha, S., Johnson, C. A., Ainsworth, J. R., Eady, R. A., Dawood, B. et al. A germline mutation in BLOC1S3/reduced pigmentation causes a novel variant of Hermansky-Pudlak Syndrome (HPS8). Am. J. Hum. Genet. 78, 160–166 (2006).
Cullinane, A. R., Curry, J. A., Carmona-Rivera, C., Summers, C. G., Ciccone, C., Cardillo, N. D. et al. A BLOC-1 mutation screen reveals that PLDN is mutated in Hermansky-Pudlak Syndrome Type 9. Am. J. Hum. Genet. 88, 778–787 (2011).
Ito, S., Suzuki, T., Inagaki, K., Takamori, K., Yamada, T., Nakazawa, M et al. High frequency of Hermansky-Pudlak sundrome type1 (HPS1) among Japanese albinism patients and functional analysis of HPS1 mutant protein. J. Invest. Dermatol. 125, 715–720 (2005).
Nomura, T., Sato, M., Hosono, K., Hikoya, A., Negishi, T., Sawada, M. et al. Two sisters with suspected ocular albinism. Folia Japonica Ophthalmol. Clin. 5, 367–372 (2012).
Huang, X. F., Xiang, P., Chen, J., Xing, D. J., Huang, N., Min, Q. et al. Targeted exome sequencing identified novel USH2A mutations in Usher syndrome families. PLoS ONE 8, e63832 (2013).
Huang, X. F., Huang, F., Wu, K. C., Chen, J., Pang, C. P., Lu, F. et al. Genotype-phenotype correlation and mutation spectrum in a large cohort of patients with inherited retinal dystrophy revealed by next-generation sequencing. Genet. Med. 17, 271–278 (2015).
Jin, Z. B., Huang, X. F., Lv, J. N., Xiang, L., Li, D. Q., Chen, J. et al. SLC7A14 linked to autosomal recessive retinitis pigmentosa. Nat. Commun. 5, 3517 (2014).
Miyake, N., Tsukaguchi, H., Koshimizu, E., Shono, A., Matsunaga, S., Shiina, M. et al. Biallelic mutations in nuclear pore complex subunit NUP107 cause early-childhood-onset steroid-resistant nephrotic syndrome. Am. J. Hum. Genet. 97, 555–566 (2015).
Factor, S.A., Ortof, E., Dentinger, M. P., Mankes, R. & Barron, K. D. Platelet morphology in Parkinson's disease: an electron microscopic study. J. Neurol. Sci. 122, 84–89 (1994).
Huizing, M., Helip-Wooley, A., Westbroek, W., Gunay-Aygun, M. & Gahl, W. A. Disorders of lysosome-related organelle biogenesis: clinical and molecular genetics. Annu. Rev. Genomics Hum. Genet 9, 359–386 (2008).
Sitaram, A. & Marks, M. S. Mechanisms of protein delivery to melanosomes in pigment cells. Physiology 27, 85–99 (2015).
Schreyer-Shafir, N., Huizing, M., Anikster, Y., Nusinker, Z., Bejarano-Achache, I., Maftzir, G. et al. A new genetic isolate with a unique phenotype of syndromic oculocutaneous albinism: clinical, molecular, and cellular characteristics. Hum. Mutat. 27, 1158 (2006).
Huizing, M., Pederson, B., Hess, R. A., Griffin, A., Helip-Wooley, A., Westbroek, W. et al. Clinical and cellular characterization of Hermansky-Pudlak syndrome type-6. J. Med. Genet. 46, 803–810 (2009).
Marks, M. S. Organelle biogenesis: En BLOC exchange for RAB32 and RAB38. Curr. Biol. 22, R963–R965 (2012).
Acknowledgements
We thank the patients and their parents who participated in this study. This work was supported by a grant for Research on Measures for Intractable Diseases (no. 14525125) and a grant for Comprehensive Research on Disability Health and Welfare (no. 13802019) from the Ministry of Health, Labour and Welfare, Tokyo, Japan; and the Strategic Research Program for Brain Science (SRPBS) (no. 11105137), the Initiative on Rare and Undiagnosed Diseases in Pediatrics (no. 15gk0110012h0101), and Initiative on Rare and Undiagnosed Diseases for Adults (no. 15ek0109001h0101) from the Japan Agency for Medical Research and Development (AMED), Tokyo, Japan.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on Journal of Human Genetics website
Rights and permissions
About this article

Cite this article
Miyamichi, D., Asahina, M., Nakajima, J. et al. Novel HPS6 mutations identified by whole-exome sequencing in two Japanese sisters with suspected ocular albinism. J Hum Genet 61, 839–842 (2016). https://doi.org/10.1038/jhg.2016.56
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/jhg.2016.56
This article is cited by
-
Clinical characteristics of a Japanese patient with Bardet-Biedl syndrome caused by BBS10 mutations
Japanese Journal of Ophthalmology (2018)

{kind=link}