
Abstract
Aggrecan is a critical proteoglycan component of the extracellular matrix of the growth plates and articular cartilage and has a key role in the biophysical and biomechanical properties of cartilage. Recently, heterozygous mutations in the ACAN gene, which encodes aggrecan, have been identified in patients with short stature and accelerated bone age. We herein report another family with a heterozygous ACAN mutation associated with idiopathic short stature along with accelerated bone age and early-onset herniation of the lumbar discs at the levels of L1/2 through L5/S1. Whole-exome sequencing identified a novel heterozygous frameshift mutation in the ACAN gene (c.1744delT; p.Phe582fs*69) in all of the affected family members but not in the unaffected one, providing further evidence that ACAN haploinsufficiency causes short stature with advanced bone maturation. In addition, we advocate early-onset multiple disc herniation as a novel phenotype associated with ACAN haploinsufficiency.
Similar content being viewed by others
Introduction
Idiopathic short stature (ISS) is a condition in which the height of the individual is more than 2 s.d. score below the corresponding mean height for a given age, sex and population and with no identifiable disorder present.1 Recently, mutations in the SHOX gene or the NPR2 gene have been found to account for a minor portion of patients with ISS; however, the genetic background of ISS remains largely unexplored.2, 3
Aggrecan, encoded by the ACAN gene, is a major proteoglycan component of the extracellular matrix of the growth plates and the articular cartilage. Aggrecan provides the hydrated gel structure, which is important for the load-bearing properties of joints, and also has a key role in chondrocyte and bone morphogenesis.4 To date, at least 24 pathological ACAN mutations have been identified in patients with highly variable phenotypes, such as spondyloepimetaphyseal dysplasia aggrecan type, spondyloepiphyseal dysplasia Kimberley type, familial osteochondritis dissecans, early-onset osteoarthritis and short stature with advanced bone maturation.5, 6, 7, 8, 9, 10, 11, 12
We herein report a Japanese family with ISS with a novel heterozygous frameshift mutation in the ACAN gene (c.1744delT; p.Phe582fs*69), providing further evidence that ACAN haploinsufficiency causes short stature with advanced bone maturation. Furthermore, we advocate multiple lumbar disc herniation as a novel phenotype associated with ACAN haploinsufficiency.
Materials and methods
Case reports
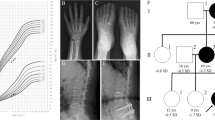
A Japanese male patient was born at 39 weeks gestation after an uncomplicated pregnancy and delivery. At birth, his length was 50 cm (−0.3 s.d.) and weight 3.17 kg (+0.6 s.d.). At 10 years of age, the patient was referred to us because of short stature. His height was 121.2 cm (−2.5 s.d.), weight 38 kg (+0.9 s.d.) and arm span 125 cm. He was prepubertal (Tanner stage P1; bilateral testicular volume: 2 ml each) and had no dysmorphic features (Figure 1a). He showed advanced bone age (11 years and 6 months). At 11.0 years of age, his bone age was assessed as 12 years and 4 months, and his bilateral testicular volume had increased to 4 ml each. Following these assessments, he was started on a long-acting gonadotropin-releasing hormone (GnRH) analog (leuprolide acetate) and discontinued the therapy at 13 years and 6 months of age. Endocrine studies indicated partial growth hormone (GH) deficiency. Thus, recombinant human GH therapy (0.175 mg kg−1 per week) was started at 11 years and 6 months of age. The combinational treatment seems to have been effective in slowing the bone age acceleration and accelerating his height growth, although he has not yet reached his adult height (Figure 2).

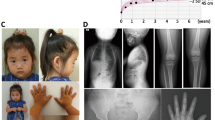
Clinical phenotype of the patients with the ACAN mutation. The younger brother, but not the elder brother (a), has mild mid-facial hypoplasia (b) and brachydactyly (c). Lumbar magnetic resonance imaging of the affected mother (d) shows multiple lumbar disc herniation (white arrows).

Growth charts of the siblings with ACAN haploinsufficiency. The elder brother (III:1) has been treated with a combination of growth hormone (GH) and gonadotropin-releasing hormone (GnRH) analog, and the younger brother (III:2) has been treated with GnRH analog alone. A full color version of this figure is available at the Journal of Human Genetics journal online.
The patient’s younger brother also had short stature. He was born at 38 weeks gestation after an uncomplicated pregnancy and delivery. At birth, his length was 47.5 cm (−0.3 s.d.) and weight 3.12 kg (+0.6 s.d.). At the time studied, he was 8 years of age. His height was 110.4 cm (−2.7 s.d.), weight 21.2 kg (−1.8 s.d.), occipitofrontal circumference 54.5 cm (+2.0 s.d.) and arm span 115 cm. He had dysmorphic features with mild mid-facial hypoplasia, mild lordosis and brachydactyly (Figures 1b and c). He exhibited normal mental and motor development. His bone age was advanced at 8 years and 6 months, although he was prepubertal (Tanner stage P1; bilateral testicular volume: 2 ml each). Endocrinological studies indicated normal growth hormone secretion and thyroid functions. A Skeletal survey showed no evidence of skeletal dysplasia. At 9.0 years of age, his bone age was markedly advanced at 10 years and 6 months, although he remained prepubertal. GnRH analog therapy was started because a standard GnRH loading test showed a pubertal gonadotropin response (luteinizing hormone, basal <0.1→peak 7.32 IU l−1; follicle-stimulating hormone, 1.32→8.67 IU l−1); however, the GnRH analog therapy failed to show marked effects in height growth (Figure 2). At 12 years and 7 months of age, his height was 126.8 cm (−2.5 s.d.), and his bone age was still advanced at 13.0 years.
Their mother’s height was 141.5 cm (−3.0 s.d.), and the maternal aunt’s height was 140 cm (−3.1 s.d.). Both had mildly dysmorphic features with mid-face hypoplasia. The mother had suffered from severe lower back pain and had been diagnosed with multiple lumbar disc herniation at the levels of L1/2 through L5/S1 since 10 years of age, although she had no symptoms in or around other joints (Figure 1d).
Whole-exome sequencing
This study was approved by the Institutional Review Board at Nagasaki University Graduate School of Biomedical Sciences. Written informed consent was obtained from the parents and the maternal aunt. DNA was obtained from peripheral blood samples from the patients with short stature and the phenotypically normal father (Figure 3a). Whole-exome sequencing using a SureSelect Human All Exon V5 (Agilent Technologies, Santa Clara, CA, USA) ran on a HiSeq 2500 platform (Illumina, San Diego, CA, USA). The mutation was confirmed via Sanger sequencing using a BigDye terminator and 3130 × l genetic analyzer (Applied Biosystems, Carlsbad, CA, USA). To confirm a heterozygous mutation, the corresponding PCR products were subcloned with TOPO TA Cloning Kit (Invitrogen, Carlsbad, CA, USA), and normal and mutant alleles were sequenced separately. Based on the pedigree (Figure 3a), a dominant mode of inheritance was assumed for the analysis. The Online Mendelian Inheritance of Man (http://www.omim.org) database was used to determine any known disease associations.

Genomic analyses in the family. (a) Pedigree of the family. (b) Electrochromatograms of the ACAN gene in the family from direct sequences and the subcloned normal and mutant alleles are shown. The deleted sequence is shaded in gray. (c) The structure of ACAN and the position of the mutation. The black and white boxes on the genomic DNA (gDNA) denote the coding regions on exons 1–18 and the untranslated regions, respectively. CS, chondroitin sulfate attachment domain; G1, globular domain 1; G2, globular domain 2; G3, globular domain 3; IGD, interglobular domain; KS, keratan sulfate attachment domain. A full color version of this figure is available at the Journal of Human Genetics journal online.
Results
In the overlap strategy, only four heterozygous mutations in known disease-associated genes (ACAN, PRPF31, CACNA1B and SMAD6) were identified in all of the affected individuals. Of these, a heterozygous frameshift mutation in ACAN (NM_013227.3; a 1-bp deletion at exon 10) was proposed as the sole candidate in association with the previously reported phenotypes. This ACAN mutation is predicted to cause a frameshift at codon 582 for ACAN and the resultant termination at codon 651 (c.1744delT; p.Phe582fs*69) (NP_037359.3) (Figures 3b and c).
Discussion
We identified a heterozygous frameshift mutation in the ACAN gene in a Japanese family with mild disproportionate ISS. The mutation was predicted to cause early truncation of the aggrecan protein and undergo nonsense-mediated mRNA decay.13 To date, at least 25 pathological ACAN mutations including our case have been reported.5, 6, 7, 8, 9, 10, 11, 12 The autosomal recessive condition in a single family was characterized by extreme short stature (66–71 cm final height) with short necks, barrel chests and craniofacial abnormalities, including macrocephaly, brachydactyly and mid-face hypoplasia, referred as spondyloepimetaphyseal dysplasia aggrecan type.5 On the other hand, the heterozygous mutations exhibit a broad phenotypic spectrum of short stature associated with advanced bone maturation, early-onset osteoarthritis and mild dysmorphic features including mid-facial hypoplasia, brachydactyly, broad great toes and lumbar lordosis, with no apparent genotype–phenotype correlations.6, 7, 8, 9 The phenotypes are highly variable, even among patients within the same family. Locations and types of the mutations in the literatures and this study are shown in Table 1 and Figure 4. Seventeen (68%) of the 25 mutations lead to premature stop codons, which result in early truncation of the aggrecan protein and probably nonsense-mediated mRNA decay, implying that haploinsufficiency of ACAN is the main mechanism for the heterozygous aggrecan-related diseases.

The positions and types of the ACAN mutations. The mutations identified in the previously reported and present studies (NM_013227.3, NP_037359.3) are indicated in the structure of ACAN gene. Superscript notations indicate mutations associated with spondyloepiphyseal dysplasia Kimberley type (a), osteochondritis dissecans (b) and spondyloepimetaphyseal dysplasia aggrecan type (c), respectively. gDNA, genomic DNA.
To date, only two mutations in ACAN (p.Leu2355Pro and p.Val2417Met) have been associated with osteochondritis dissecans, which is characterized by the separation of an articular cartilage and subchondral bone fragment from the articular surface.11, 12 As both missense mutations were located at the C-terminal C-type lectin domain in the ACAN gene, dominant-negative effects of the mutant proteins have been proposed for the specific articular phenotype (Table 1).
The mother with the ACAN mutation in the present study exhibited early-onset severe herniation of multiple lumbar discs. The phenotype is likely associated with haploinsufficiency of ACAN, for the following reasons: First, since significant reduction in aggrecan mRNA levels has been reported in cartilaginous tissue of homozygous and heterozygous aggrecan-deficient mice with a 7-bp deletion in exon 5 of the aggrecan gene, the ACAN expression should be reduced similarly in the present patients with ACAN haploinsufficiency.14 Second, aggrecan levels in the cartilage decrease with age in humans. The degradation and reduction of aggrecan with age result in the degeneration of discs and the impairment of the disc function, leading to disc herniation.15 Third, while homozygous aggrecan-deficient mice are characterized by dwarfism and cleft palate and die shortly after birth because of respiratory failure, the heterozygotes exhibit mild dwarfism and develop spinal misalignment with age. A histological examination revealed a high incidence of herniation and degeneration of vertebral discs in the mice.14 The spinal phenotype of the aggrecan-deficient mice is very similar to that of the mother in the present report. Taken together, the clinical findings of the mother imply that an ACAN mutation, which leads to reduced aggrecan expression in the cartilage, can lead to degeneration of the disc cartilage and cause early-onset multiple spinal disc herniation not only in mice but also in humans. As such, it is noteworthy that several patients with ACAN mutations have been recently reported to have back pain and intervertebral disc disease, although the details have not yet been described.9
As seen in our patients, individuals with ACAN haploinsufficiency have mild short stature with advanced bone age at the prepubertal stage; however, premature growth cessation after the start of puberty results in severe short stature in adulthood. Premature hypertrophic chondrocyte maturation followed by the decreased expression of aggrecan in the extrachondrocyte matrix and early invasion of blood vessels and osteoblasts into the growth plates have been proposed as the mechanisms for the advanced bone age, early epiphyseal fusion and premature growth cessation.16, 17 Recently, the effectiveness of combined GH and GnRH analog treatment for adult height has been reported in several cases of ACAN mutations.6, 8, 9 van der Steen et al.8 reported that patients with ACAN mutation who had received GH treatment in combination with GnRH analog treatment for 2 years from the onset of puberty achieved 5 to 8 cm greater adult height than their family members of the same sex with an ACAN mutation. In our study, the estimated final height of the elder brother (158.5 cm), who has received combined GH and GnRH analog treatment, is higher than that of the younger brother (145.6 cm), who has received GnRH analog only.18 However, since these patients have not yet reached their adult heights, the effectiveness of the combined treatment is still uncertain. Further studies are needed to clarify the effectiveness of GH and GnRH analog treatment for patients with ACAN mutations.
In conclusion, our study has provided further evidence that ACAN haploinsufficiency causes ISS with accelerated bone maturation and has also expanded the clinical spectrum of aggrecan-related bone disorder. Further studies are needed to determine the degree of risk patients with ACAN mutation have for multiple disc herniation.
References
Cohen, P., Rogol, A. D., Deal, C. L., Saenger, P., Reiter, E. O., Ross, J. L. et al. Consensus statement on the diagnosis and treatment of children with idiopathic short stature: a summary of the Growth Hormone Research Society, the Lawson Wilkins Pediatric Endocrine Society, and the European Society for Paediatric Endocrinology Workshop. J. Clin. Endocrinol. Metab. 93, 4210–4217 (2008).
Shima, H., Tanaka, T., Kamimaki, T., Dateki, S., Muroya, K., Horikawa, R. et al. Systematic molecular analyses of SHOX in Japanese patients with idiopathic short stature and Leri–Weill dyschondrosteosis. J. Hum. Genet. 61, 585–591 (2016).
Wang, S. R., Jacobsen, C. M., Carmichael, H., Edmund, A. B., Robinson, J. W., Olney, R. C. et al. Heterozygous mutations in natriuretic peptide receptor-B (NPR2) gene as a cause of short stature. Hum. Mutat. 36, 474–481 (2015).
Sivan, S. S., Wachtel, E. & Roughley, P. Structure, function, aging and turnover of aggrecan in the intervertebral disc. Biochim. Biophys. Acta 1840, 3181–3189 (2014).
Tompson, S. W., Merriman, B., Funari, V. A., Fresquet, M., Lachman, R. S., Rimoin, D. L. et al. A recessive skeletal dysplasia, SEMD aggrecan type, results from a missense mutation affecting the C-type lectin domain of aggrecan. Am. J. Hum. Genet. 84, 72–79 (2009).
Nilsson, O., Guo, M. H., Dunbar, N., Popovic, J., Flynn, D., Jacobsen, C. et al. Short stature, accelerated bone maturation, and early growth cessation due to heterozygous aggrecan mutations. J. Clin. Endocrinol. Metab. 99, 1510–1518 (2014).
Quintos, J. B., Guo, M. H. & Dauber, A. Idiopathic short stature due to novel heterozygous mutation of the aggrecan gene. J. Pediatr. Endocrinol. Metab. 28, 927–932 (2015).
van der Steen, M., Pfundt, R., Maas, S. J., Bakker-vanWaarde, W. M., Odink, R. J. & Hokken-Koelega, A. C. ACAN gene mutations in short children born SGA and response to growth hormone treatment. J. Clin. Endocrinol. Metab. (e-pub ahead of print 6 October 2016; doi:10.1210/jc.2016-2941).
Gkourogianni, A., Andrew, M., Tyzinski, L., Crocker, M., Douglas, J., Dunbar, N. et al. Clinical characterization of patients with autosomal dominant short stature due to aggrecan mutations. J. Clin. Endocrinol. Metab. (e-pub ahead of print 21 November 2016; doi:10.1210/jc.2016-3313).
Gleghorn, L., Ramesar, R., Beighton, P. & Wallis, G. A mutation in the variable repeat region of the aggrecan gene (AGC1) causes a form of spondyloepiphyseal dysplasia associated with severe, premature osteoarthritis. Am. J. Hum. Genet. 77, 484–490 (2005).
Stattin, E. L., Wiklund, F., Lindblom, K., Onnerfjord, P., Jonsson, B. A., Tegner, Y. et al. A missense mutation in the aggrecan C-type lectin domain disrupts extracellular matrix interactions and causes dominant familial osteochondritis dissecans. Am. J. Hum. Genet. 86, 126–137 (2010).
Stattin, E. L., Tegner, Y., Domellöf, M. & Dahl, N. Familial osteochondritis dissecans associated with early osteoarthritis and disproportionate short stature. Osteoarthritis Cartilage 16, 890–896 (2008).
Holbrook, J. A., Neu-Yilik, G., Hentze, M. W. & Kulozik, A. E. Nonsense-mediated decay approaches the clinic. Nat. Genet. 36, 801–808 (2004).
Watanabe, H., Nakata, K., Kimata, K., Nakanishi, I. & Yamada, Y. Dwarfism and age-associated spinal degeneration of heterozygote cmd mice defective in aggrecan. Proc. Natl Acad. Sci. USA 94, 6943–6947 (1997).
Roughley, P. J. Biology of intervertebral disc aging and degeneration: involvement of the extracellular matrix. Spine 29, 2691–2699 (2004).
Domowicz, M. S., Cortes, M., Henry, J. G. & Schwartz, N. B. Aggrecan modulation of growth plate morphogenesis. Dev. Biol. 329, 242–257 (2009).
Lauing, K. L., Cortes, M., Domowicz, M. S., Henry, J. G., Baria, A. T. & Schwartz, N. B. Aggrecan is required for growth plate cytoarchitecture and differentiation. Dev. Biol. 396, 224–236 (2014).
Tanaka, T., Naiki, Y., Horikawa, R. & Sato, M. Adult height prediction method for pubertal short children (Growth Potential 2 Method). J. Jpn Assoc. Hum. Auxol. (Japanese) 15, 17–22 (2009).
Acknowledgements
We thank the patients and their family who participated in this study. This work was supported by a grant for the Initiative on Rare and Undiagnosed Diseases in Pediatrics (no 15gk0110012h0101) from the Japan Agency for Medical Research and Development (AMED), Tokyo, Japan.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article

Cite this article
Dateki, S., Nakatomi, A., Watanabe, S. et al. Identification of a novel heterozygous mutation of the Aggrecan gene in a family with idiopathic short stature and multiple intervertebral disc herniation. J Hum Genet 62, 717–721 (2017). https://doi.org/10.1038/jhg.2017.33
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/jhg.2017.33
This article is cited by
-
Growth plate gene involment and isolated short stature
Endocrine (2021)
-
Novel aggrecan variant, p. Gln2364Pro, causes severe familial nonsyndromic adult short stature and poor growth hormone response in Chinese children
BMC Medical Genetics (2018)
-
Ablation of protein phosphatase 5 (PP5) leads to enhanced both bone and cartilage development in mice
Cell Death & Disease (2018)
-
A balanced reciprocal translocation t(10;15)(q22.3;q26.1) interrupting ACAN gene in a family with proportionate short stature
Journal of Endocrinological Investigation (2018)
-
Genetic screening confirms heterozygous mutations in ACAN as a major cause of idiopathic short stature
Scientific Reports (2017)
