
Abstract
Diabetics often have poor perfusion in their limbs as a result of peripheral artery disease and an impaired ability to generate collateral vessels. The receptor for advanced glycation end products (RAGE) is one protein that is thought to play a detrimental role in collateral development in diabetics due to increased levels of advanced glycation end products (AGE), one of its ligands, in diabetes. Thus, the aim of this study was to investigate the role of RAGE in both diabetic and non-diabetic settings in a model of collateral formation in mice. Streptozotocin was used to induce diabetes in both wild type and RAGE knockout mice. Increased levels of the AGE, Nɛ-(carboxymethyl) lysine (CML), were confirmed via an ELISA. A hindlimb ischemia model, in which the femoral artery is ligated, was used to drive collateral growth and reperfusion was assessed using laser Doppler perfusion imaging and histological analysis of vessels in the muscle. Both of these measurements showed impaired collateral growth in diabetic compared with wild-type mice as well as improved collateral growth in both diabetic and non-diabetic RAGE knockout mice when compared their wild-type counterparts. Distance on a freely accessed running wheel, used as a measure of perfusion recovery, showed that wild-type diabetic mice had functionally impaired recovery compared with their wild-type counterparts. Immunohistochemistry and immunoblotting showed that HMGB-1 (high-mobility group box 1), another RAGE ligand, was increased in the ischemic leg compared with the non-ischemic leg in all mice. This increase in HMGB-1 may explain improvement in animals lacking RAGE and its subsequent signaling. In conclusion, this study shows that RAGE impairs collateral growth in a diabetic setting and also in a non-diabetic setting. This demonstrates the importance of RAGE and alternate RAGE ligands in the setting of collateral vessel growth.
Similar content being viewed by others


Main
Diabetes is a major health problem in the United States, which affects over 25 million people (8.3% of the population) and results in $245 billion in disease-related expenses every year.1 Many of these costs are associated with the numerous complications of the disease, including stroke, blindness, neuropathy, kidney disease, and cardiovascular disease (CVD). Specifically, CVD is the leading cause of death in diabetic patients. The disease has a pronounced effect on the peripheral vasculature, which results in over 60% of all non-traumatic lower limb amputations occurring in diabetics.1 This high rate of amputations is due to the increased severity and extent of atherosclerotic vascular disease in diabetics. This situation is further complicated by the fact that collateral vessel formation is markedly impaired in diabetics.2, 3
The mechanism responsible for impaired collateral growth in diabetics remains unclear and is likely multifactorial. We are postulating that the accumulation of advanced glycation end products (AGEs) may be one mechanism through which collateral vessel formation is impaired in diabetics. AGEs are a post-translational modification of proteins in which amino acids and carbohydrates interact via an irreversible, nonenzymatic glycosylation process. This process is a series of related reactions termed Maillard reactions. AGEs form as part of the normal aging process; however, chronic hyperglycemia and altered redox state in diabetes increases the formation and accumulation of AGEs. One mechanism by which the effects of AGEs are elicited is through the binding of AGEs to the receptor for advance glycation end products (RAGE). RAGE is a member of the immunoglobulin superfamily of cell surface receptors that has several ligands, which include AGE motifs, HMGB-1 (high-mobility group box 1), and s100B.4 Binding of AGEs to RAGE activates several intracellular cell signaling pathways, including increased oxidative stress, activation of mitogen activated protein (MAP) kinase, p21ras, and NF-κB translocation. This results in the upregulation of many inflammatory genes, including vascular cell adhesion molecule 1 (V-CAM-1), intercellular adhesion molecule 1 (I-CAM-1), E-selectin, vascular endothelial growth factor (VEGF), tumor necrosis factor-α, interleukin-1α, interleukin-6, and heme oxygenase.5, 6, 7, 8, 9, 10, 11, 12
RAGE has been found to be expressed in a variety of tissues and cell types and is highly expressed in lung tissue in the endothelium, bronchial smooth muscles, and alveolar macrophages and leiomyocytes.13 The heart, skeletal muscle, blood vessels, kidney, brain, and several other tissues also produce RAGE.8, 13 RAGE has been shown to be elevated in tissue from diabetics especially in diseased regions of the vasculature where it is often localized with AGEs.8, 14, 15 Additionally, besides expression, activation of RAGE and subsequent downstream signaling has been observed in vitro in endothelial cells, vascular smooth muscle cells and mononuclear phagocytes9 and all three of these cell types play a role in collateral formation.
AGE formation and RAGE signaling have also been linked to a number of vascular diseases. Studies suggest that RAGE has negative effects on endothelial function, myocardial recovery in ischemia reperfusion injury, neointima formation, and angiogenesis in Matrigel plugs.16, 17, 18, 19, 20, 21, 22 Therefore, we propose that RAGE signaling likely plays an inhibitory role in vasculogenesis through promoting excess inflammation and may be one of the key mechanisms impairing vessel growth in diabetics.
The goal of this study was to investigate the role of RAGE in collateral growth in a mouse model of hindlimb ischemia. We studied collateral vessel formation in wild type and RAGE knockout mice both with and without streptozotocin (STZ) induced diabetes. The physiological impact of collateral growth was determined by measuring perfusion using laser Doppler perfusion imaging (LDPI), immunohistochemistry to provide a quantitative measure of vessel number and density, and the functional recovery of mice using running endurance.
Materials and methods
Animals
Male C567BL/6 mice were purchased from Jackson Laboratories and RAGE knockout (RAGE KO) mice11 were bred in-house at the Emory Department of Animal Resources. All animals were between 8 and 10 weeks old at the time of surgery. The animals were housed and cared for in agreement with guidelines approved by the Emory University Institutional Animal Care and Use Committee.
STZ Model of Diabetes
To induce diabetes, mice received intraperitoneal injections of 60 mg/kg STZ (Sigma) in 0.05 M sodium citrate daily for 5 days. The animals were maintained in a fasting state prior to treatment in order to optimize the effects of STZ. The STZ solution was made fresh daily. Two days after the fifth injection, blood glucose levels in blood from the tail were measured using a glucose meter (Ermaine Laboratories). If the blood glucose was less than 11 mmol/l, the mice were injected an additional two times over the next week at the same dosage. Glucose levels were tested each week to ensure that the serum glucose level is greater than 13 mmol/l. Mice with glucose levels less than 13 mmol/l were excluded from the study. Control animals were injected with the citrate buffer alone. The mice were monitored for 4 weeks after diabetes induction prior to the hindlimb ischemia study.
Quantification of AGEs
AGE levels in the plasma and muscle tissue were quantified using an ELISA to detect Nɛ-(carboxymethyl) lysine (CML), one of the most abundant AGEs and good gauge for total levels.23, 24 For the serum levels, blood was drawn via cardiac puncture following euthanasia at the end of the study. Serum samples were quantified using an ELISA kit for CML (Cell Biolabs) following the manufacturer’s instructions. To quantify CML in the muscle tissue, the adductor muscles were homogenized and lysed with a glass mortar and pestle in Hunter buffer as previously described and the tissue lysate was used in the ELISA.25 Serum samples were also used to quantify lipid levels in all of the groups.
Hindlimb Ischemia Model
Mice were anesthetized with an intraperitoneal injection of xylazine (10 mg/kg) and ketamine (80 mg/kg). Aseptic technique was employed. All hair was removed from the surgical site, thoroughly cleaned with sterile saline, and disinfected with Betadine. A unilateral incision was made over the left medial thigh of the mouse. The superficial femoral artery and vein were ligated with 6-0 silk caudally to the branching deep femoral artery, and then a second ligation was performed just proximal to the branching of the tibial arteries. The length of the artery and vein was excised between the two ligation points. The skin was closed with monofilament suture. The animals received Buprenex (0.1 mg/kg, subcutaneously) postoperatively for analgesia and were allowed to recover on a heated pad.
LDPI
Mice were anesthetized using isoflurane at 1.5% and scanned with LASER Doppler perfusion (PIM II LASER Doppler Perfusion Imager; Perimed). Perfusion was quantified by defining equivalent regions of interest on the perfusion heat map generated by the instrument and calculating the mean perfusion (in arbitrary perfusion units) in both the proximal and distal leg regions. Perfusion in the ischemic leg was normalized to flow in the non-ischemic leg (IL/NIL).
Histological Assessment of Collateral Vessel Density
Histological analysis was performed on both the ischemic and non-ischemic limbs at day 21 post hindlimb ischemia surgery. Mice were killed and the tissue was perfused with saline followed by 10% buffered formalin for fixation. The bone was then demineralized in a formic acid-based solution (Cal-Ex II; Fisher Scientific) for 48 h before processing and paraffin embedding. Sections (5- μm-thick) were immunostained for vascular smooth muscle cells or endothelial cells. Enzyme treatment was performed in proteinase K (Biolabs; 2 μg/ml) prior to incubation with primary antibodies. For smooth muscle cells, the sections were stained using a mouse monoclonal antibody to α-smooth muscle cell actin (Sigma) as the primary antibody detected using the avidin–biotin–alkaline phosphatase method (Vectastain ABC-AP; Vector Laboratories) with a hematoxylin counterstain. Images of the entire section were acquired used the Hamamatsu NanoZoomer SQ slide scanner. To visualize endothelial cells, slides were stained with biotinylated Lectin antibody (Biotinylated Griffonia simplifolia Lectin 1; Vector lab), followed by incubation with Streptavidin Qdot 655 (Invitrogen). Images were acquired using the × 20 Plan-Neo air objective on a Zeiss Axioskop microscope equipped with an AxioCam camera. ImageJ software (NIH) was used to count the number of vessels for analysis.
Running Wheel Study
As a physiologic test of ischemic limb functional recovery, experiments measuring running endurance were performed. Additional mice underwent hindlimb ischemia as described above and were used for functional testing only. On postoperative day 7, each mouse was individually housed in a cage with a running wheel attached to a sensor that counted the number of revolutions made by the wheel during a 7-day period. The distance in meters was recorded both daily and cumulatively for 7 days.
Quantification of HMGB1
HMGB1 was quantified using both immunohistochemistry and western blotting. For immunohistochemistry, histological slides were prepared as described above. Antigen retrieval was performed using a proteinase K solution and sections were then incubated with HMGB1 antibody (Abcam), followed by incubation with goat anti-rabbit antibody (Vector Labs) and incubation with Streptavadin QDot 655 (Invitrogen).
Additionally, slides were also co-stained for endothelial cells (Lectin; Vector Labs), macrophages (MAC387; Abcam), and smooth muscle cells (α-smooth muscle cell actin; Sigma) to determine which cells types were expressing HMGB1. HMGB1 antibodies from Abcam were used, followed by incubation with Goat anti-rabbit secondary antibody (Vector Labs), and finally incubated with Streptavadin QDot 605 (Invitrogen). For the double stains, Mac387 antibody (Abcam) or smooth muscle α-actin antibody (Sigma) was used followed by incubation with anti-mouse antibody (Vector Labs), and incubation with Streptavadin QDot 655 (Invitrogen) or lectin antibody (Biotinylated Griffonia simplifolia Lectin 1; Vector Labs) was used followed by incubation with Streptavidin Qdot 655 (Invitrogen). Images of the sections were collected using the × 20 and × 63 objectives (numerical aperture 0.50) on a Leica confocal microscope.
HMGB1 levels were also quantified using western bloting. Adductor muscle lysates were prepared as described above and the protein concentration in the tissue lysate was assessed using a Bradford Assay (BioRad). Proteins were separated using SDS-Page and transferred onto a nitrocellulose membrane for blotting for HMGB1 (Abcam) and β-actin (Cell Signaling Technology). Bands were detected using enhanced chemiluminescense with HRP-conjugated secondary antibodies, and densitometry using ImageJ software (NIH) was used to calculate band intensity.
Statistical Analysis
All reported results are expressed at mean±s.e.m. Graphpad Prism was used for statistical analysis. To determine significance, ANOVA with a Bonferroni post hoc test to determine differences between groups was performed using a 95% confidence interval.
Results
Two groups of mice (wild type and RAGE KO) were given a 5-day STZ course and subsequently developed diabetes with blood glucose levels in the range of 15–30 mmol/l for the duration of the study as confirmed by weekly blood glucose checks (Figure 1). We also measured a number of other physiological parameters of the mice, including body weight, blood pressure, and serum lipid levels (Supplementary Table S1), which showed no major physiologically relevant changes between the group. To further characterize the mice and presence of increased AGEs in the diabetic mice, CML levels in the serum and hindlimb muscle tissue were determined via ELISA. In both blood serum and muscle lysate, the diabetic mice of each genotype (wild type and RAGE KO) had significantly higher CML levels compared with the control mice for each genotype as seen in Figure 2a and b (P<0.001).

Weekly glucose monitoring. Glucose was measured weekly throughout the experiment to ensure the animals were diabetic. (mean and s.e.m. are plotted, *indicates P<0.001 for WT vs WT+DM, WT vs RAGE KO+DM, RAGE KO vs WT+DM, and RAGE KO vs RAGE KO+DM, ^indicates P<0.001 for WT DM vs RAGE KO+DM n=15). KO, knockout; RAGE, receptor for advanced glycation end products; WT, wild type; WT+DM, wild-type mice with diabetes.

Streptozotocin (STZ)-induced diabetes increases CML levels in blood and muscle tissue. Blood and muscle tissue were collected from the mice at the end of the study. An ELISA was used to assess CML levels in the blood serum (a) and muscle lysates (b). CML was increased in the diabetic groups of each genotype with no differences in overall levels between the genotypes. Mean and s.e.m. are plotted and *indicates P<0.05 and **indicates P<0.01 (n=8 for blood serum and n=6 for tissue lysate). CML, Nɛ-(carboxymethyl) lysine; KO, knockout; RAGE, receptor for advanced glycation end products; WT, wild type; WT+DM, wild-type mice with diabetes.
Following 4 weeks of confirmed diabetic state, the left femoral artery was ligated and excised in the hindlimb ischemia model. LDPI was performed on the mice as a non-invasive method of determining perfusion in each limb. The perfusion ratio ((ischemic limb perfusion/non-ischemic limb perfusion) × 100) was calculated to compare the groups, and the values from postoperative day 21 are shown in Figure 3. As expected, the wild-type mice with diabetes (WT+DM) had significantly less perfusion than the untreated wild-type (WT) mice (P<0.05), while there was no difference between the RAGE KO mice with and without diabetes. In addition, we saw a perfusion increase in RAGE KO+DM mice compared with the WT+DM mice (P<0.001). However, we also surprisingly found that the non-diabetic RAGE KO mice had a significant increase in the perfusion ratio when compared with the non-diabetic WT mice (P<0.001).

Diabetes impairs perfusion in WT mice but not in RAGE KO mice. Ischemia was induced in mice using the HLI procedure in either healthy or STZ-treated diabetic mice (DM). LPDI was used to noninvasively measure the reperfusion of the limb. (a) The perfusion ratio between the ischemic and non-ischemic leg on post-op day 21 shows impaired perfusion in WT+DM but not in RAGE KO mice. Mean and s.e.m. are plotted, *indicates P<0.05 vs WT, **indicates P<0.001 vs WT, and #indicates P<0.001 vs WT+DM (n=4–12). (b) Representative LDPI images of (a) WT, (b) RAGE KO, (c) WT+DM and (d) RAGE KO+DM. IL, ischemic leg; KO, knockout; NIL, non-ischemic leg; RAGE, receptor for advanced glycation end products; WT, wild type; WT+DM, wild-type mice with diabetes.
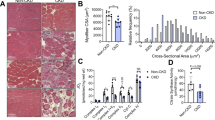
In order to determine if there was an anatomic correlate of the increase in perfusion, histology sections were prepared and stained for with antibodies for smooth-muscle α-actin or lectin. The number of vessels in each set of images was counted to determine the both the number of vessels. While the number of smooth-muscle-positive vessels did not differ between the groups, the number of capillaries/small vessels that were lectin positive showed significant difference that were in agreement with the LDPI results (Figure 4). Specifically, there were significantly less lectin-positive vessels in the WT+DM group compared with the WT group, while there was no difference in the number of vessels in the RAGE KO mice with and without diabetes. The WT+DM also had significantly less vessels than both the RAGE KO and RAGE KO+DM groups, and interestingly the WT group was also significantly less than the RAGE KO+DM group. Representative images of lectin staining for each groups are shown in Figure 4.

Diabetic wild-type mice have decreased endothelial positive vessels. Histology slides of the hindlimbs were stained for smooth-muscle α-actin or with lectin and the number of vessels with smooth muscle cells or endothelial cells was quantified. α-Actin-positive microvessels were not different between the groups. (a) Lectin-positive vessels were significantly lower in the WT+DM group vs all the other groups. Additionally, WT was significantly different than RAGE KO+DM. (b) Representative images of each group are shown where red is lectin. Mean and s.e.m. are plotted, *indicates P<0.05. **indicates P<0.01 and ***indicates P<0.001. (n=6). KO, knockout; RAGE, receptor for advanced glycation end products; WT, wild type; WT+DM, wild-type mice with diabetes.
A functional assessment of the ischemic recovery was performed with the use of voluntary running wheels. Mice were placed in the running wheel cages for 1 week on postoperative day 7. The total distance run from 7 to 14 days following HLI is shown in Figure 5. As expected the WT+DM mice had drastically impaired function when compared with the sham control mice, while none of the other groups were significantly different than the sham group or each other (P<0.05). This includes no statistically significant impairment between the RAGE KO and RAGE KO+DM mice.

WT diabetic mice have impaired function but RAGE KO diabetic mice are not statistically different. The recovered function in the leg following HLI was assessed by monitoring the distance run on a wheel in 7 days. The total distance is plotted as mean and s.e.m. The WT+DM mice ran significantly less than the Sham group, while all the other groups were not significantly different than each other (*P=0.0087 and n=3–6). KO, knockout; RAGE, receptor for advanced glycation end products; WT, wild type; WT+DM, wild-type mice with diabetes.
We quantified CML (Figure 2) to show that the mice were diabetic for a sufficient period to develop increased AGEs in both the blood and the muscle that may serve as a ligand for RAGE in the diabetic mice and result in their decreased perfusion. However, because we saw an effect in the non-diabetic animals as well, specifically the control RAGE KO animals were improved over the wild-type control mice, we looked for additional ligands for RAGE that might be present in the legs. Indeed, we found that HMGB-1, another RAGE ligand, was increased in the ischemic leg while very little was present in the non-ischemic leg as shown with immunohistochemistry and quantified via western blot in Figure 6. We also confirmed that the HMGB1 levels in the ischemic leg did not differ between the groups using both immunohistochemistry and immunoblots as well. Finally, we also performed co-staining to demonstrate that the smooth muscle cells, endothelial cells, and macrophages contain HMGB1 (Figure 6).

Ischemic legs have increased high-mobility group box 1 (HMGB-1) expression. HMGB1 was quantified both using immunohistochemistry and immunoblotting of the ischemic and non-ischemic legs. (a) Representative images of slides stained for HMGB-1 (red) so increased HMGB1 in the ischemic leg. (red=HMGB1 and blue=DAPI) (b) Expression of HMGB1 in the muscle was quantified by western blot. (n=8, *P<0.0001) (c) Representative images show that HMGB-1 expression is not different between groups. (d) Western blotting confirms no difference in HMGB1 expression between groups (n=4) (e) Co-staining reveals that HMGB1 is expressed in smooth muscle cells, endothelial cells, and macrophages (green=HMGB1, blue=DAPI, and red=α-smooth muscle actin, lectin, or MAC387). IL, ischemic leg; KO, knockout; NIL, non-ischemic leg; RAGE, receptor for advanced glycation end products; WT, wild type; WT+DM, wild-type mice with diabetes.
Discussion
In this study, the role of RAGE was assessed in regard to collateral vessel development in response to limb ischemia. The relevance of the receptor and its subsequent signaling was assessed in environments that had physiological and pathophysiologic levels of AGEs present. The RAGE knockout mice appear to have a significant improvement in blood flow as well as function. While this improvement was most dramatic in the face of chronic hyperglycemia when compared with the WT diabetic mice which had poor perfusion on LDPI, minimal positive staining for microvasculature, and poor functional capabilities as seen by the running wheel study; an additional striking finding in the study was that RAGE knockout mice also showed significant improvements in perfusion in non-diabetic animals.
Poor perfusion in diabetics has been attributed to their impaired ability to grow collateral vessels2, 3 and our hypothesis was that these changes are due to AGE-mediated signaling through RAGE. We induced diabetes with STZ and allowed the mice to remain diabetic for several weeks allowing for the formation of AGEs specifically CML (Figures 1 and 2). While AGE is the term for a number of different modifications to proteins, CML has been identified as a predominant marker of AGEs and elevated levels have been found in diabetics.9, 23, 26 CML specifically has also been shown to bind to RAGE and activate downstream signaling pathways including NF-κB.9
Using LDPI, we found that at 21 days, the RAGE knockout mice had improved perfusion ratios when compared with diabetic mice, indicating that RAGE signaling was having a negative effect on vascular growth. This improved perfusion was due to increased vascularity as evidenced by the histological analysis, which showed significantly more collateral vessels determined by staining for endothelial cells (Figures 3 and 4). These differences in perfusion ratios also translated into improved function as determined by running wheels (Figure 5). We acknowledge that the functional differences may be confounded by overall muscle atrophy associated with diabetes in the STZ-treated mice rather impaired perfusion from the HLI procedure.27, 28, 29, 30 However, when comparing the diabetic groups only to account for this possibility, the RAGE KO diabetic mice still have improved function over WT diabetic mice and the running wheel data are in line with our LDPI and histological results as well.
Indeed, these findings are in agreement with the literature, which suggests that RAGE signaling negatively effects the vasculature. Specifically, stimulation of RAGE expressed on endothelial cells has been shown to cause endothelial dysfunction of coronary arterioles in diabetic mice.31 Rats pretreated with sRAGE (the soluble form of the receptor) had improved myocardial function and decreased ischemic damage following an ischemia reperfusion injury model, and similarly, hearts from RAGE knockout mice were protected from ischemic damage in a similar model.16 In vascular smooth muscle cells, blockade of RAGE signaling inhibits cell proliferation and neointima formation in diabetic rats.17 Similarly, RAGE knockout mice had preserved luminal area and decreased neointima following denudation of the femoral artery.18, 19 Matrigel plugs containing VEGF implanted in diabetic mice had impaired angiogenesis compared with the angiogenic response in healthy mice; however, angiogenesis was preserved in RAGE knockout mice or mice that overexpress sRAGE as a decoy receptor.20 Another study in which AGE formation in diabetic mice was blocked with aminoguanidine helped restore perfusion following femoral artery ligation to levels of healthy mice.21 Similarly, a study by Tekabe et al22 using technetium-labeled RGD showed a marginally higher signal indicative of angiogenesis in RAGE knockout diabetic mice than in wild-type diabetic mice. Thus our findings of impaired perfusion by LDPI, decreased microvasculature, and reduced running in the wild-type diabetic mice compared with the diabetic RAGE KO mice further support these findings.
A surprising and novel finding of this study was the observed differences between the RAGE knockout and wild-type mice in the non-diabetic groups. The perfusion ratio as measured by LDPI and functionality of the limb from the running wheel study were both better in the RAGE knockout mice as compared with the wild-type mice. This suggests that RAGE signaling is present in even non-diabetic mice and results in negative effects on collateral growth. We hypothesize that this signaling could be the result of increased RAGE ligands in response to the ischemic conditions in the hindlimb ischemia model. It is important to note that while RAGE was originally identified for its ability to bind AGEs, as a member of the immunoglobulin superfamily of receptors, RAGE has a number of other known ligands. In addition to AGEs, RAGE can also bind to high-mobility group box 1 (HMGB1), s100, amyloid-β peptide, and Mac-1.4, 32, 33, 34 Indeed, the STZ-treated mice in this study did exhibit higher levels of CML prior to the HLI procedure (Figure 2), thus creating an environment in which CML–RAGE interactions could occur. However, the improved perfusion in the RAGE KO mice in the non-diabetic state suggests an additional RAGE ligand may be present in the ischemic leg that results in subsequent RAGE signaling. Figure 6 shows that HMGB-1 is indeed dramatically increased in the ischemic leg when compared with the non-ischemic leg, which had very little HMGB1. HMGB-1 was originally studied for its role in the nucleus as a regulator of transcription by binding to chromatin and allowing other regulatory proteins and complexes to bind to the DNA; however, it was later shown to be released from cells and act as a cytokine signaling through RAGE and Toll-like receptors 2 and 4 (TLR 2 and TLR4).35, 36, 37 Macrophages have been shown to actively release HMGB-1 in response to inflammatory stimuli while other cells release it passively during necrosis.35, 38, 39, 40, 41 The co-staining in Figure 6e shows HMGB1 in smooth muscle cells, endothelial cells, and macrophages, and while the staining only shows nuclear HMGB-1, we believe that additional HMGB-1 is released in the ischemic tissue but is too diffuse to stain. This free HMGB-1 may then act as a cytokine and bind to RAGE resulting in downstream signaling and negative effects of collateral growth. We acknowledge that the experiments in this study do not directly assess this hypothesis because a functional assessment of the contribution of HMGB1 is not possible since HMGB1 deficient mice die shortly after birth.42
The results of this study suggest that RAGE signaling appears to have negative effects on collateral vessel growth in a hindlimb ischemia model as the absence of RAGE improves recovery. Binding of ligands such as CML and HMGB1 to RAGE activates a number of intracellular signaling pathways that primarily have proinflammatory effects. In particular, NF-κB is activated and translocated to the nucleus resulting in the transcription of a number of different genetic targets. Additionally, NF-κB has been shown to regulate the expression of RAGE itself creating a positive feedback loop that further promotes the RAGE mediated signals.11, 43, 44 Depending on the cell type, a variety of different NF-κB-mediated signals, including proinflamatory genes and oxiditative stress, are upregulated.5, 6, 7, 8, 9, 10, 44 Prolonged expression of RAGE and its signaling, such as oxidative stress and inflammation, appear to have negative effects on this regenerative process. While the growth of collaterals does require inflammation and reactive oxygen species-mediated signaling, excessive oxidative stress and inflammation can also impair growth. The improvement in perfusion in the RAGE knockout animals in this paper suggests a new hypothesis that the contribution of reactive oxygen species and inflammatory signals by RAGE signaling may perturb the balance and have an inhibitory effect on collateral growth.
In conclusion, activation of RAGE by ligands including CML and HMGB-1 is likely one reason for the decreased ability to form compensatory collaterals in order to overcome issues of limb ischemia or peripheral vascular disease. A better understanding of RAGE regulation and signaling could be used to develop treatment approaches to locally decrease the receptor, impair its function, or inhibit downstream signaling resulting in improved collateral vessel formation and peripheral blood flow in diabetic patients, thus possibly decreasing limb ischemia and amputation in these patients.
References
Prevention CfDCa National Diabetes Fact Sheet: National Estimates and General Information on Diabetes and Prediabetes in the United States, 2011. Department of Health and Human Services, Centers for Disease Control and Prevention: Atlanta, GA, 2011.
Abaci A, Oguzhan A, Kahraman S et al, Effect of diabetes mellitus on formation of coronary collateral vessels. Circulation 1999; 99: 2239–2242.
Islam MM, Ali A, Khan NA et al, Comparative study of coronary collaterals in diabetic and nondiabetic patients by angiography. Mymensingh Med J 2006; 15: 170–175.
Schmidt AM, Yan SD, Yan SF, Stern DM . The biology of the receptor for advanced glycation end products and its ligands. Biochim Biophys Acta 2000; 1498: 99–111.
Schmidt AM, Hori O, Chen JX et al, Advanced glycation endproducts interacting with their endothelial receptor induce expression of vascular cell adhesion molecule-1 (VCAM-1) in cultured human endothelial cells and in mice. A potential mechanism for the accelerated vasculopathy of diabetes. J Clin Invest 1995; 96: 1395–1403.
Yan SD, Schmidt AM, Anderson GM et al, Enhanced cellular oxidant stress by the interaction of advanced glycation end products with their receptors/binding proteins. J Biol Chem 1994; 269: 9889–9897.
Lander HM, Tauras JM, Ogiste JS et al, Activation of the receptor for advanced glycation end products triggers a p21(ras)-dependent mitogen-activated protein kinase pathway regulated by oxidant stress. J Biol Chem 1997; 272: 17810–17814.
Schmidt AM, Yan SD, Wautier JL, Stern D . Activation of receptor for advanced glycation end products: a mechanism for chronic vascular dysfunction in diabetic vasculopathy and atherosclerosis. Circ Res 1999; 84: 489–497.
Kislinger T, Fu C, Huber B et al, N(epsilon)-(carboxymethyl)lysine adducts of proteins are ligands for receptor for advanced glycation end products that activate cell signaling pathways and modulate gene expression. J Biol Chem 1999; 274: 31740–31749.
Basta G, Lazzerini G, Massaro M et al, Advanced glycation end products activate endothelium through signal-transduction receptor RAGE: a mechanism for amplification of inflammatory responses. Circulation 2002; 105: 816–822.
Bierhaus A, Humpert PM, Morcos M et al, Understanding RAGE, the receptor for advanced glycation end products. J Mol Med (Berl) 2005; 83: 876–886.
Goldin A, Beckman JA, Schmidt AM, Creager MA . Advanced glycation end products: sparking the development of diabetic vascular injury. Circulation 2006; 114: 597–605.
Brett J, Schmidt AM, Yan SD et al, Survey of the distribution of a newly characterized receptor for advanced glycation end products in tissues. Am J Pathol 1993; 143: 1699–1712.
Ritthaler U, Deng Y, Zhang Y et al, Expression of receptors for advanced glycation end products in peripheral occlusive vascular disease. Am J Pathol 1995; 146: 688–694.
Soulis T, Thallas V, Youssef S et al, Advanced glycation end products and their receptors co-localise in rat organs susceptible to diabetic microvascular injury. Diabetologia 1997; 40: 619–628.
Bucciarelli LG, Kaneko M, Ananthakrishnan R et al, Receptor for advanced-glycation end products: key modulator of myocardial ischemic injury. Circulation 2006; 113: 1226–1234.
Zhou Z, Wang K, Penn MS et al, Receptor for AGE (RAGE) mediates neointimal formation in response to arterial injury. Circulation 2003; 107: 2238–2243.
Grozinger G, Schmehl J, Bantleon R et al, Decreased neointimal extracellular matrix formation in RAGE-knockout mice after microvascular denudation. Cardiovasc Intervent Radiol 2012; 35: 1439–1447.
Sakaguchi T, Yan SF, Yan SD et al, Central role of RAGE-dependent neointimal expansion in arterial restenosis. J Clin Invest 2003; 111: 959–972.
Shoji T, Koyama H, Morioka T et al, Receptor for advanced glycation end products is involved in impaired angiogenic response in diabetes. Diabetes 2006; 55: 2245–2255.
Tamarat R, Silvestre JS, Huijberts M et al, Blockade of advanced glycation end-product formation restores ischemia-induced angiogenesis in diabetic mice. Proc Natl Acad Sci USA 2003; 100: 8555–8560.
Tekabe Y, Shen X, Luma J et al, Imaging the effect of receptor for advanced glycation endproducts on angiogenic response to hindlimb ischemia in diabetes. EJNMMI Res 2011; 1: 3.
Ikeda K, Higashi T, Sano H et al, N (epsilon)-(carboxymethyl)lysine protein adduct is a major immunological epitope in proteins modified with advanced glycation end products of the Maillard reaction. Biochemistry 1996; 35: 8075–8083.
Ferreira AE, Ponces Freire AM, Voit EO . A quantitative model of the generation of N(epsilon)-(carboxymethyl)lysine in the Maillard reaction between collagen and glucose. Biochem J 2003; 376: 109–121.
Lyle AN, Joseph G, Fan AE et al, Reactive oxygen species regulate osteopontin expression in a murine model of postischemic neovascularization. Arterioscler Thromb Vasc Biol 2012; 32: 1383–1391.
Reddy S, Bichler J, Wells-Knecht KJ, Thorpe SR, Baynes JW . N (epsilon)-(carboxymethyl)lysine is a dominant advanced glycation end product (AGE) antigen in tissue proteins. Biochemistry 1995; 34: 10872–10878.
Ostler JE, Maurya SK, Dials J et al, Effects of insulin resistance on skeletal muscle growth and exercise capacity in type 2 diabetic mouse models. Am J Physiol Endocrinol Metab 2014; 306: E592–E605.
Pepato MT, Migliorini RH, Goldberg AL, Kettelhut IC . Role of different proteolytic pathways in degradation of muscle protein from streptozotocin-diabetic rats. Am J Physiol 1996; 271: E340–E347.
Park SW, Goodpaster BH, Lee JS et al, Excessive loss of skeletal muscle mass in older adults with type 2 diabetes. Diabetes Care 2009; 32: 1993–1997.
Park SW, Goodpaster BH, Strotmeyer ES et al, Accelerated loss of skeletal muscle strength in older adults with type 2 diabetes: the Health, Aging, and Body Composition Study. Diabetes Care 2007; 30: 1507–1512.
Gao X, Zhang H, Schmidt AM, Zhang C . AGE/RAGE produces endothelial dysfunction in coronary arterioles in type 2 diabeticmice. Am J Physiol Heart Circ Physiol 2008; 295: H491–H498.
Hofmann MA, Drury S, Fu C et al, RAGE mediates a novel proinflammatory axis: a central cell surface receptor for S100/calgranulin polypeptides. Cell 1999; 97: 889–901.
Yan SD, Chen X, Fu J et al, RAGE and amyloid-beta peptide neurotoxicity in Alzheimer's disease. Nature 1996; 382: 685–691.
Hori O, Brett J, Slattery T et al, The receptor for advanced glycation end products (RAGE) is a cellular binding site for amphoterin. Mediation of neurite outgrowth and co-expression of rage and amphoterin in the developing nervous system. J Biol Chem 1995; 270: 25752–25761.
Lotze MT, Tracey KJ . High-mobility group box 1 protein (HMGB1): nuclear weapon in the immune arsenal. Nat Rev Immunol 2005; 5: 331–342.
Park JS, Svetkauskaite D, He Q et al, Involvement of toll-like receptors 2 and 4 in cellular activation by high mobility group box 1 protein. J Biol Chem 2004; 279: 7370–7377.
Kokkola R, Andersson A, Mullins G et al, RAGE is the major receptor for the proinflammatory activity of HMGB1 in rodent macrophages. Scand J Immunol 2005; 61: 1–9.
Bonaldi T, Talamo F, Scaffidi P et al, Monocytic cells hyperacetylate chromatin protein HMGB1 to redirect it towards secretion. EMBO J 2003; 22: 5551–5560.
Scaffidi P, Misteli T, Bianchi ME . Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature 2002; 418: 191–195.
Vezzoli M, Castellani P, Corna G et al, High-mobility group box 1 release and redox regulation accompany regeneration and remodeling of skeletal muscle. Antioxid Redox Signal 2011; 15: 2161–2174.
Wang H, Bloom O, Zhang M et al, HMG-1 as a late mediator of endotoxin lethality in mice. Science 1999; 285: 248–251.
Calogero S, Grassi F, Aguzzi A et al, The lack of chromosomal protein Hmg1 does not disrupt cell growth but causes lethal hypoglycaemia in newborn mice. Nat Genet 1999; 22: 276–280.
Tanaka N, Yonekura H, Yamagishi S et al, The receptor for advanced glycation end products is induced by the glycation products themselves and tumor necrosis factor-alpha through nuclear factor-kappa B, and by 17beta-estradiol through Sp-1 in human vascular endothelial cells. J Biol Chem 2000; 275: 25781–25790.
Bierhaus A, Schiekofer S, Schwaninger M et al, Diabetes-associated sustained activation of the transcription factor nuclear factor-kappaB. Diabetes 2001; 50: 2792–2808.
Acknowledgements
This work was supported in part by NIH P01HL095070 (to WRT), T32HL007745 (to DG), and NIH F32HL124974 (to LH).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on the Laboratory Investigation website
Diabetics often have poor perfusion in their limbs as a result of peripheral artery disease and impaired ability to generate collateral vessels. This study reveals a role of the receptor for advanced glycation end products (RAGE) in collateral formation using a mouse model. Mechanistically, activation of RAGE by ligands, including AGE and the chromatin protein high mobility group box 1, decreases the ability to form compensatory collaterals.
Supplementary information
Rights and permissions
About this article

Cite this article
Hansen, L., Gupta, D., Joseph, G. et al. The receptor for advanced glycation end products impairs collateral formation in both diabetic and non-diabetic mice. Lab Invest 97, 34–42 (2017). https://doi.org/10.1038/labinvest.2016.113
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/labinvest.2016.113
