
Abstract
Ischemia-reperfusion injury (IRI) is the main cause of morbidity and mortality due to graft rejection after liver transplantation. During IRI, an intense inflammatory process occurs in the liver. This hepatic inflammation is initiated by the ischemic period but occurs mainly during the reperfusion phase, and is characterized by a large neutrophil recruitment to the liver. Production of cytokines, chemokines, and danger signals results in activation of resident hepatocytes, leukocytes, and Kupffer cells. The role of neutrophils as the main amplifiers of liver injury in IRI has been recognized in many publications. Several studies have shown that elimination of excessive neutrophils or inhibition of their function leads to reduction of liver injury and inflammation. However, the mechanisms involved in neutrophil recruitment during liver IRI are not well known. In addition, the molecules necessary for this type of migration are poorly defined, as the liver presents an atypical sinusoidal vasculature in which the classical leukocyte migration paradigm only partially applies. This review summarizes recent advances in neutrophil-mediated liver damage, and its application to liver IRI. Basic mechanisms of activation of neutrophils and their unique mechanisms of recruitment into the liver vasculature are discussed. In particular, the role of danger signals, adhesion molecules, chemokines, glycosaminoglycans (GAGs), and metalloproteinases is explored. The precise definition of the molecular events that govern the recruitment of neutrophils and their movement into inflamed tissue may offer new therapeutic alternatives for hepatic injury by IRI and other inflammatory diseases of the liver.
Similar content being viewed by others
Main
The interruption of blood flow (ischemia) with consequent lack of oxygen and nutrient supply is an inherent phenomenon during various surgical procedures.1 In liver surgery, clinical situations exist in which periods of ischemia can be particularly long, such as during removal of liver tumors, trauma, vascular reconstruction, and transplantation.2, 3, 4 Once the blood flow, oxygen tension, and nutrients are restored (reperfusion), there is an increase in injury initiated by ischemia, aggravating the overall damage. This phenomenon is known as ischemia-reperfusion injury (IRI), which affects liver viability and directly correlates to graft rejection.5, 6, 7 IRI remains the major problem in clinical transplantation. IRI causes up to 10% of early transplant failures and can lead to a higher incidence of both acute and chronic rejection.8, 9, 10 In an ischemic liver, when the oxygen levels fall, adenosine triphosphate (ATP) production is impaired.11 The deleterious effects of ATP reduction are further enhanced by the production of reactive oxygen species (ROS), cytokines, vasoactive agents, and increased expression of adhesion molecules.5 Generation of ROS results in an increase in the intracellular calcium concentration and causes intracellular pH changes, resulting in organelle damage and apoptotic or necrotic cell death.12
However, liver damage and failure are caused mainly during the reperfusion period, when there is a shift from metabolic distress caused by ischemia to an excessive innate immune response triggered by reperfusion. Liver IRI shows a significant inflammatory component, and neutrophils are considered central factors in the events leading to injury after reperfusion.13, 14 Indeed, excessive numbers of neutrophils are recruited and activated during reperfusion and, as discussed below, overwhelming evidence suggests that this excessive neutrophil influx contributes to the pathogenesis of IRI, including in the liver. Neutrophil-induced liver injury is a multistep process that includes neutrophil activation, recruitment of these cells from the vasculature, transendothelial neutrophil migration, and contact to parenchymal cells. Chemokines are potent chemoattractants for neutrophils in this context and have been shown to contribute to hepatic neutrophil recruitment and liver IRI.15, 16 Moreover, neutrophil recruitment depends on the interaction of chemokines with G protein-coupled receptors (GPCRs) on leukocytes and glycosaminoglycans (GAGs) expressed on endothelial vessels, creating a concentration-dependent gradient of chemokines produced at the inflammatory site.17 For over 30 years, experimental models of hepatic IRI have provided significant knowledge of the disease pathogenesis, and have led to significant improvement of our understanding of the molecular mechanisms underlying reperfusion lesions.18, 19, 20 However, this complex process has led to several controversies in the field and needs better analysis. Here, we provide an overview of recent progress in the role of innate immunity on hepatic IRI, with a focus on the inflammatory mechanisms triggered by neutrophils that contribute to the pathogenesis of liver IRI.
Pathophysiology of liver iri: innate immune response
As any reperfusion-associated injury, liver IRI is characterized by intense neutrophil infiltration in post-ischemic tissues. However, before this stage, resident cells act to create an inflammatory milieu, which contributes to neutrophil recruitment. First, IRI of the liver induces cell damage. This damage results in the release of endogenous molecules named danger-associated molecular patterns (DAMPs), in analogy to the term pathogen-associated molecular patterns.21 The list of DAMPs includes high mobility group box 1 (HMGB1), deoxyribonucleic acid (DNA), ATP, urate, mitochondrial formyl peptides, and S100 proteins. These molecules are detected by a variety of immune receptors, and have an important role in the initial (over)activation of the immune response.22 During reperfusion, DAMPs released from apoptotic and necrotic cells stimulate Kupffer cells (liver-resident macrophages) to produce inflammatory mediators, such as chemokines, cytokines, and ROS. This process incites the next phase of reperfusion injury by orchestrating the homing, activation, and adhesion of neutrophils.22
All cells of the innate immune system, including neutrophils, express pattern recognition receptors, which can recognize DAMPs. These DAMP receptors include Toll-like receptors (TLRs), Retinoic Acid-Inducible Gene I-like receptors, nucleotide-binding oligomerization domain-like receptors, and C-type lectin receptors. By sensing DAMPs, these receptors promote an inflammatory response following reperfusion.23 TLRs are expressed in all cell types in the liver and their signaling leads to production of cytokines and chemokines through the activation of nuclear factor-kappa B, promoting the recruitment of leukocytes.21 One of these endogenous ligands, DNA, is released from damaged hepatocytes and binds to TLR9 expressed on neutrophils, promoting further neutrophil recruitment and activation and subsequent hepatotoxicity. TLR9 expression on neutrophils is upregulated upon liver damage and these cells sense and react to extracellular DNA by activating the TLR9/nuclear-factor kappa B pathway.24, 25 In vitro experiments show that DNA released from necrotic hepatocytes increases cytokine expression by neutrophils through a TLR9-dependent mechanism. In addition, TLR9−/− mice subjected to 1 h of ischemia and 12 h of reperfusion show reduced hepatocellular necrosis, serum alanine aminotransferase levels, and lower production of inflammatory cytokines, showing that TLR9 signaling in neutrophils increases hepatic injury induced by IRI.25 Following liver IRI, damaged hepatocytes have been shown to release the DAMP HMGB1, which exacerbates the hepatic injury through activation of TLR4.26 The result of this activation creates an inflammatory environment that further incites the influx of inflammatory cells, including neutrophils.
In addition, complement activation has a key role in post-ischemic inflammation and injury and complement inhibition is recognized as a potential therapeutic strategy for reducing IRI.27 The complement system consists of ~30 soluble and membrane-bound proteins that are activated by one of three pathways: the antibody-dependent classical pathway, the alternative pathway, and the mannose-binding lectin pathway.28 Activation of complement leads to the sequential production of the effector molecules complement protein 3a (C3a), complement protein 5a (C5a), and the membrane attack complex (MAC). C3a and C5a are soluble bioactive peptides that are cleaved from their parent proteins by C3 and C5 enzymatic convertases, and the membrane attack complex is a terminal cytolytic protein complex assembled in cell membranes after cleavage of C5.29 Activation of this cascade system of soluble plasma proteins results in the recruitment of a large number of inflammatory cells, ultimately leading to cell injury and death.27 Jaeschke et al30 have shown that depletion of serum complement before liver ischemia prevents neutrophil accumulation during reperfusion. Furthermore, in partial hepatic IRI in rats, treatment with the C5a receptor antagonist attenuates the increases in liver enzymes, serum, and tissue tumor necrosis factor-alpha, infiltrating neutrophils, and also reduces liver histopathology.31
Neutrophils
In liver inflammation, the defense against initial environmental challenges and injury is driven by a complex set of leukocytes, including natural killer cells, natural killer T cells, dendritic cells, neutrophils, eosinophils, and complement components.32 Of these cell subsets, neutrophils are known to induce liver injury, and are the largest circulating fraction of leukocytes and the first cells to arrive at the site of injury. The process of neutrophil recruitment into the liver during IRI-induced sterile inflammation is particularly different from the mechanism in other organs and to the one induced by pathogens. The multistep process for neutrophils to reach the liver parenchyma and resulting in tissue damage includes neutrophil activation, adherence within the hepatic vasculature, neutrophil transmigration to the liver parenchyma, and release of granule enzymes.33, 34
Role of chemokines and gags in liver iri
The role of neutrophils in liver IRI has been shown in several publications.35, 36 A range of inflammatory mediators and GPCR agonists, including chemokines, the complement proteins C3a and C5a, mitochondrial-derived formyl peptides, and leukotriene B4, can induce neutrophil influx in the hepatic microvasculature.15, 16, 30, 31, 37, 38 Among these mediators, the role of chemokines is worth highlighting. Chemokines constitute a group of small structurally related chemotactic proteins indispensable for the coordination of leukocyte migration during inflammation.39 Previous studies have demonstrated that chemokines with a Cys-Xaa-Cys motif (CXC chemokines) mediate neutrophil infiltration during liver IRI and other inflammatory conditions.40, 41, 42
After tissue damage, chemokines are locally secreted by parenchymal cells and resident leukocytes, thereby creating a gradient along which neutrophils can migrate from the blood vessels to the site of inflammation. In mice, CC chemokine ligand 1 (CXCL1) and CXCL2, homologs of human interleukin-8, or CXCL8 according to the systematic chemokine nomenclature, are important chemoattractants of neutrophils. By recruiting and activating neutrophils, human CXCL8 has been implicated in a wide range of diseases, including liver IRI.43, 44 These chemokine ligands are recognized by the receptor CXC chemokine receptor 2 (CXCR2) expressed on the neutrophil surface and mediate neutrophil recruitment.45 Human CXCL6 and CXCL8 also bind to human CXC chemokine receptor 1 (CXCR1), and murine CXCL6 is the only ligand for murine CXCR1.46, 47, 48, 49 It was demonstrated that after liver IRI, expression of CXCL1 and CXCL2 in the ischemic lobes increases 100–1000-fold.50 Consistent with these observations, CXCL1 and CXCL2 are expressed on the luminal surface of liver sinusoids around sites of necrosis. They form a chemokine gradient that guides neutrophil migration in the direction of the injury.51 In mice, the hepatic expression of CXCL1 and CXCL2 increases in temporally distinct patterns after hepatic IR. The expression of CXCL2 increases in the early phase of reperfusion, before any detectable increase in neutrophil accumulation, suggesting that CXCL2 may be involved in the initial recruitment of neutrophils to the ischemic lobe.15 Neutralization of the rat homolog of human CXCL5 and CXCL6 in an IRI model resulted in reduced neutrophil sequestration in the liver and serum ALT levels.16 Moreover, Repertaxin (DF1681B), an allosteric antagonist of CXCR2, is able to prevent neutrophil influx and liver damage.52 Wengner et al53 demonstrated the process of neutrophil response to chemokines in a murine model of peritonitis. After 2 h of a single intraperitoneal (i.p.) injection of thioglycolate, a significant increase in the circulating numbers of neutrophils is noted. This is inhibited by 84% when mice are pretreated with neutralizing monoclonal antibodies to CXCL1 and CXCL2. Indeed, when chemokine is administered i.p. an increase in both circulating neutrophils in blood and neutrophils in the peritoneum is observed, consistent with the concept that these chemokines have a dual action, acting locally to stimulate recruitment and systemically to promote mobilization.53 When these chemokines are produced locally, they are retained on the sinusoidal wall, creating a chemotactic gradient for neutrophils.51
Chemokine production only is not sufficient to lead to neutrophil recruitment. For chemokines to have an effect on leukocyte trafficking, besides being produced, they need to be present at a defined concentration range and in the right place to be able to induce chemotaxis. This process requires interaction of chemokines with GAGs expressed on the vasculature.54 Thus, to create a gradient and to induce neutrophil migration in vivo, it is necessary for chemokines to interact with two essential receptors. First, they need to bind to their particular GPCRs, which are expressed by specific leukocytes. Chemokine–GPCR interactions induce the endothelial adhesion of leukocyte subtypes followed by leukocyte extravasation and directional migration toward the site of inflammation.55 In addition to binding to specific GPCRs, chemokines also bind cell surface GAGs at the vascular endothelium or GAGs in the extracellular matrix (ECM).56, 57 Endothelial cells present GAGs such as heparan sulfate on their cell surface to which chemokines bind via their GAG-binding site. Because of the chemokine–GAG interaction, a stable chemokine gradient is formed on the surface of the endothelial cells and activated neutrophils migrate along this gradient toward the site of inflammation.56 This interaction and formation of immobilized chemical concentration gradients is also called haptotaxis.58 During recent years, the importance of GAG–chemokine interactions in leukocyte migration has been investigated in more detail. Several GAGs, such as hyaluronan, dermatan sulfate, and heparan sulfate have been implicated in events associated with inflammation, such as cytokine/chemokine production and presentation and leukocyte recruitment.59, 60 More recently, in vivo, chemokines have been shown to localize within capillary venules in a GAG-dependent way. In these studies, an intravascular gradient of chemokines was sequestered by heparan sulfate and immobilized on the endothelium.61 This retention of chemokines on the endothelial layers occurs by interactions between sulfated domains of GAGs and basic amino-acid motifs on chemokines. Chemokine presentation by the GAGs on the endothelial cell wall prevents their diffusion and degradation and retains high local concentrations of the produced chemokines.62
Chemokine binding to GAGs has been proven to be indispensable for chemokine activity and neutrophil recruitment in vivo.61, 63, 64, 65 The local concentration of chemokines induces crawling of neutrophils toward an inflammation site.61 Chemokine production at sites of inflammation also results in the generation of GAG-mediated chemokine gradients in the ECM. Reduced chemokine-induced neutrophil migration is seen in mice with disturbed heparan sulfate.66 Moreover, a study by Vanheule et al. 67 shows that the blockade of GAG–chemokine interactions is able to inhibit neutrophil extravasation in a murine model of monosodium urate-induced gout. Importantly, removing hyaluronan from the sinusoidal endothelium, or blocking its interaction with its principal receptor (CD44), reduces neutrophil recruitment as well.68 Although therapeutic intervention in the chemokine system has long been focused on the development of chemokine and chemokine receptor antagonists,69 more recently, it has been suggested that interruption of chemokine–GAG interactions might represent an innovative and useful way of interfering with chemokine action that may result in a decrease in inflammation.17
Intrahepatic neutrophil migration
In sterile inflammation, there is a multistep hierarchy of directional cues that guides neutrophil localization to sites of damage. Neutrophils migrate intravascularly through the sinusoid channels toward the stressed tissue, ultimately infiltrating directly into the area of necrosis. These findings suggest that there is an intravascular gradient that guides neutrophils to the site of inflammation.51 During liver IRI, direct cellular damage from oxidative stress during ischemia results in the passive release of DAMPs from necrotic cells. DAMPs induce sterile inflammation through stimulation of chemokine production by resident cells and, consequently, neutrophil recruitment. When injected into mice, purified DAMPs or necrotic cells mobilize neutrophils to the site of inoculation.70 Using an in vivo imaging to reveal the multistep events that guide the recruitment of neutrophils to locations of sterile injury, McDonald et al. demonstrated CXCL2 expression on the luminal surface of the liver sinusoids that was maximal at ~150 μm from the injury and gradually decreased out to 650 μm, demonstrating the presence of an intravascular gradient that leads toward the injured area. However, despite the intravascular gradient of CXCL2 was consistently observed to abruptly end ~100–150 μm proximal to the border of necrotic tissue, neutrophils continue to migrate into the area of necrosis, beyond the maximum expression of CXCL2. Furthermore, within this proximal 150 μm surrounding the injury, directional neutrophil migration was independent of CXCR2. This indicates that, from a certain point, neutrophils migrate to the area of necrosis guided by other factors, which are independent of chemokines and CXCR2 (Figure 1). This chemotactic stimulus was identified as mitochondrial N-formyl peptides.38 During necrosis, the mitochondrial content is spilled out into the extracellular milieu and mitochondrial N-formyl peptides can attract and activate neutrophils through the specific receptor formyl peptide receptor 1 (FPR1), which is as chemokine receptors a GPCR.71 In this way, neutrophils migrate first toward an intravascular chemokine gradient, and then switch to a formyl peptide gradient near the injury site, where the FPR1 signal works in the absence of a chemokine gradient.72 Studies in other animal models such as acetaminophen-induced liver injury have shown that antagonization of FPR1 causes partial reduction of neutrophil recruitment. However, the strategy to block both FPR1 and CXCR2 simultaneously significantly reduces neutrophil migration to the liver and liver damage.73 This shows that chemokines and formyl peptides work together in a sequential manner to guide neutrophils to sites of sterile inflammation.51

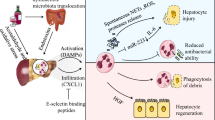
Chemokine-, GAG-, and MMP-dependent liver damage after ischemia-reperfusion injury. Because of the unusual hepatic microvasculature, which has a reduced diameter and blood flow, neutrophils are in intimate contact with the liver sinusoidal endothelial cells (LSEC), and may be mechanically trapped in the sinusoids independent of selectins. Cell death by necrosis releases multiple danger-associated molecular patterns (DAMPs). Kupffer cells are the primary sentinel cells that sense cell death and generate pro-inflammatory cytokines and chemokines. Chemokines bind to GAGs on the surface of endothelial cells, forming a chemotactic gradient in the inflamed tissue. They guide intravascular neutrophil recruitment to the injury area. Interaction of leukocytes with chemokines on the endothelium leads to integrin activation, firm adhesion, transmigration across the endothelial barrier, and accumulation of leukocytes at the site of inflammation. Neutrophils migrate directly into the area of cell death, where DAMPs induce direct neutrophil chemotaxis via mitochondrial formyl peptide-signaling through FPR1. In parallel, MMPs, particularly MMP-9, facilitate local matrix degradation and neutrophil extravasation across vascular barriers. Besides neutrophil recruitment, MMP-mediated loss of endothelial barrier integrity interferes with the liver’s ability to regenerate after hepatic ischemia-reperfusion. In addition, MMPs contribute to tissue injury by promoting parenchyma cell detachment from ECM, resulting in additional tissue necrosis.
Role of matrix metalloproteases in liver iri
The ECM, formed by the complex network of proteins and glycans surrounding cells in all solid tissues, is among the most important regulators of cellular and tissue functions in the body. The ECM regulates various cellular functions, such as adhesion, migration, differentiation, proliferation, and survival. Degradation of ECM is essential in many physiological processes, including angiogenesis and wound healing. However, dysregulation of ECM production and proteolysis is often associated with the development of liver pathology.74 The alterations of the ECM are in agreement with reports describing the potential implications of matrix metalloproteinases (MMPs) in liver diseases.75, 76 Using a model of partial liver IRI in rats, Cursio et al77 showed high expression of MMPs in a specific time-dependent pattern, which was associated with the release of liver enzymes and large area of tissue necrosis by histology. In the same work, in rats treated with a specific MMP inhibitor, intensity of liver necrosis was significantly reduced. Leukocyte migration across endothelial and ECM barriers is dependent on cellular adhesion/release and focal matrix degradation mechanisms.78 Although adhesion molecules are critical for the successful promotion of neutrophil transmigration by providing attachment to the vascular endothelium, there is a growing body of evidence suggesting that MMPs are critical for facilitating leukocyte movement across vascular barriers.78, 79
The MMPs are a family of 24 proteases using zinc-dependent catalysis to breakdown ECM components, allowing cell movement and tissue reorganization.80 Although this step is important in physiological processes such as tissue repair, the inappropriate, prolonged, or excessive expression or release of these enzymes has harmful consequences for the liver.81 The role of MMPs has been investigated in several pathological conditions, including cancer,82, 83 autoimmune diseases such as rheumatoid arthritis, lupus erythematosus, and multiple sclerosis,84, 85, 86, 87, 88 and chronic inflammation including inflammatory bowel diseases,89, 90 lung disease,91 and liver IRI.92, 93, 94
MMPs have been categorized into five major groups according to their ECM substrate specificity: collagenases, gelatinases, membrane-type enzymes, stromelysins, and matrilysins.95 Among the different MMPs, gelatinases (MMP-2 and MMP-9) are within the most prominent MMPs detected during liver IRI.96, 97 MMP-2 and MMP-9 have gelatin-binding domains that resemble a similar motif in fibronectin. This motif is involved in the binding of fibronectin to denatured collagen.98 Fibronectin is a large ECM glycoprotein with a recognized central role in cellular adhesion and migration, and it is likely an important ECM protein involved in leukocyte adhesion during liver IRI. The expression of cellular (EIIIA+) fibronectin by liver sinusoidal endothelial cells is an early feature after injury, and it precedes leukocyte recruitment in hepatic IRI.99 A study of Moore et al100 shows that the interactions between fibronectin and its integrin receptor α4β1, expressed on leukocytes, specifically upregulate the expression and activation of MMP-9 by infiltrating leukocytes in steatotic livers subjected to IRI. The high expression of MMP-9 is accompanied by massive neutrophil infiltration, increased levels of proinflammatory cytokines, and impaired liver function. Moreover, the blockade of fibronectin-α4β1–integrin interactions inhibits the activation of MMP-9 by leukocytes and prevents cytokine release and cellular migration to the liver after IRI. In healthy tissues, MMP-2 is constitutively expressed in fibroblasts, endothelial cells, and epithelial cells, and may be modestly up- or downregulated by inflammatory stimuli.101 In contrast, MMP-9 is inducible and its expression is observed primarily in leukocytes including monocytes, macrophages, and neutrophils.100 MMP-9 is released by neutrophils, and, once produced, this enzyme is capable of digesting components of connective tissue matrix, such as fibronectin and collagen type IV, facilitating the movement of neutrophils toward the site of damage.84, 94
The role of MMP-9 in the mechanism of neutrophil infiltration has been examined using MMP-9−/− mice and mice treated with MMP-9-blocking antibodies or less specific small-molecule MMP inhibitors.80 In a model of liver IRI, Hamada et al93 show significantly reduced liver damage by histological assessment with lowered circulating levels of aminotransferases, reduced hepatocyte necrosis, and improved sinusoidal blood flow. Improved liver function correlates with reduced parenchymal infiltration of neutrophils and reduced liver content of neutrophil myeloperoxidase (MPO). In addition, MMP-9−/− neutrophils have an impaired ability to migrate across fibronectin relative to normal cells. MMP-9 is mostly detected in Ly-6G and macrophage antigen-1 (MAC-1) leukocytes adherent to the vessel walls and infiltrating the damaged livers of wild-type mice after liver IRI. These findings have not been limited to the liver, but also to other organs such as the heart and the brain.102, 103
Although the release of MMP-9 by neutrophils seems to have a harmful effect, MMP-2 seems to have an opposite role. Recent reports have shown direct evidence of a protective role for MMP-2 expression in the liver. For instance, mice treated with an anti-MMP-2-neutralizing antibody are characterized by intensified liver damage after IRI.93 Moreover, MMP-2−/− mice subjected to hepatic IRI show exacerbated liver damage compared with WT mice. Furthermore, MMP-2 deficiency results in upregulation of MMP-9 activity, spontaneous neutrophil infiltration in naive livers, and amplifies MMP-9-dependent transmigration of neutrophils in vitro and after hepatic IRI.97 Tissue inhibitors of metalloproteinases (TIMPs) regulate the activity of MMPs. Alterations in the MMP–TIMP balance have been linked to pathological conditions that require disruption of the basement membrane, such as tumor invasion, angiogenesis, and wound healing.104 There are at least four identified members (TIMP 1–4) in the TIMP family, varying in tissue-specific expression and in their ability to inhibit various MMPs.105
Among the different TIMPs, TIMP-1 is of particular interest. TIMP-1 expression is very low in naive livers and it is induced after liver IRI. However, it is still insufficient to prevent an elevated MMP activity in liver IRI.100 Duarte et al have shown that animals lacking TIMP-1 show further impaired liver function and histological preservation after IRI. In this model, TIMP-1 deficiency leads to lethal liver IRI, as over 60% of the TIMP-1−/− mice died post reperfusion, whereas all TIMP-1+/+ mice recovered and survived surgery. In addition, lack of TIMP-1 expression is accompanied by markedly high levels of MMP-9 activity, which facilitates leukocyte transmigration across vascular barriers in hepatic IRI. Indeed, TIMP-1−/− livers are characterized by massive neutrophil infiltration and by upregulation of proinflammatory mediators, including tumor necrosis factor-alpha, IFN-γ, and inducible nitric oxide synthase post IRI.106 Altogether, MMPs and TIMPs seem to have important roles in the preservation of liver homeostasis, and it is suggested that they might be targeted to ameliorate liver damage.
Neutrophil adhesion and migration
As the inflammatory process develops, leukocytes are recruited to the liver. In hepatic IRI and many other acute injuries, neutrophils are the first to arrive and usually in large numbers. Neutrophil recruitment during inflammation is normally attributed to a multistep cascade involving initial tethering and rolling by selectins along the vessel wall, followed by firm adhesion to the vascular endothelium and emigration out of the vasculature.107 This classical paradigm is well characterized for a number of organ microvasculatures including mesentery, peritoneum, skeletal muscle, and skin (Figure 2).108

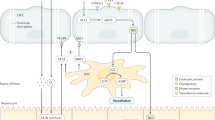
Classical mechanism of leukocyte recruitment. In most tissues, leukocytes (eg, neutrophils) initially tether to and roll on the endothelium adjacent to the inflammatory focus through selectin interactions with mucin-like selectin receptors. Engagement of chemokine receptors with their ligands presented on the endothelium stimulates activation of integrins on the leukocyte cell surface, enabling their interaction with their respective receptors to facilitate arrest, adhesion, and subsequent transmigration by paracellular or transcellular routes. Following a chemotactic gradient, they reach the extravascular compartment to accumulate in areas rich in chemoattractants and release lytic enzymes through degranulation.
Most tissues require selectin-mediated tethering and rolling in post-capillary venules, followed by firm adhesion mediated by integrins for proper neutrophil recruitment. Even though neutrophils follow the intravascular recruitment paradigm in most tissues, as skin and muscle, they use a very different strategy in the liver. For example, there is little evidence for the requirement of selectin-mediated rolling or β2 integrin-mediated adhesion for neutrophil migration in the liver sinusoids. In the liver sinusoids, the adhesive response to N-formyl-Met-Leu-Phe (fMLF) is not dependent upon selectins as adhesion is not reduced in the sinusoidal vessels of P-selectin-deficient mice or E-selectin/P-selectin-deficient mice treated with L-selectin antibody.109, 110 Because of the unusual hepatic microvasculature, which has a reduced diameter and blood flow, it is likely that neutrophils would be mechanically trapped in the inflamed sinusoids.51, 111 Moreover, hepatic sinusoids are devoid of selectins and neutrophils move directly to integrin-mediated adhesion.22 Thus, in these narrow capillaries, the rolling process is likely to be unnecessary.112 This may explain why clinical trials of anti-adhesion therapy, in an attempt to reduce liver injury associated with traumatic shock and reperfusion injury, fail to show a significant benefit.113
Using intravital microscopy to visualize the liver microvasculature, Wong et al109 show that the majority of leukocytes (80%) adhere within the sinusoids in response to a chemotactic stimulus such as fMLF, whereas 20% adhere to post-sinusoidal venules. This is in marked contrast to leukocytes adhering exclusively to post-capillary venules in tissues as the mouse cremaster muscle. In hepatic IRI, the main sinusoidal ligand for neutrophils is intercellular adhesion molecule-1 (ICAM-1).114, 115 Around sites of focal hepatic necrosis, neutrophil adhesion to the sinusoidal endothelium is mediated by the binding of the integrin Mac-1 (also known as CD11b/CD18) to ICAM-1. The absence of ICAM-1 reduces neutrophil recruitment to the liver following acute hepatocellular necrosis induced by IRI.116, 117 Subsequently, neutrophils migrate through the intravascular channels toward the site of injury, penetrating directly into the area of damage. Menezes et al118show that fMLF induces recruitment of neutrophils similar to focal necrosis by adhesion and crawling dependent on Mac-1 and ICAM-1. Furthermore, ICAM-1 has been shown upregulated after hepatic IRI, and blocking of Mac-1 has protective effects against hepatic reperfusion injury in vivo.119, 120, 121
Once outside the vessel, individual neutrophils often show extremely coordinated chemotaxis and cluster formation reminiscent of the swarming behavior of insects. This behavior was shown recently by Lammermann and co-workers122, 123, 124 in a model of focal ear skin damage and infected lymph nodes. They observed that once neutrophils have crossed the endothelium, arriving at the interstitial space, these cells adopt an amoeboid migration strategy, which relies mainly on contraction and protrusion of the cytoplasm, independently of adhesion molecules. This phenomenon happens in the 3D environment of the interstitium, usually composed of a meshwork of fibrillar ECM, such as collagen fibers. In this scenario, leukocyte migration is completely dependent on the acto-myosin cytoskeleton, in which leukocyte migration would happen by contraction of the cell posterior coupled to frontal protrusion of lamellipodia, propelling the cell through the fibrillar ECM at high speed and without integrin requirement.124, 125 Interestingly, these concepts have not been applied to the liver so far, a tissue in which the reduced extravascular space is tightly packed with hepatocytes and non-parenchymal cells.
Mechanisms of neutrophil-mediated liver injury
As we described above, during organ transplantation, the liver is exposed to prolonged times of ischemia, which results in cell stress and cell death. This early damage is essential to liver injury during the reperfusion. Neutrophils are considered crucial effector cells in the pathophysiology of liver IRI. Because of their high mobility and the capability to release potent cytotoxic mediators, neutrophils primarily work to eliminate invading microorganisms and/or remove dead cells at the site of inflammation.126 However, in the liver and other organs, an excessive inflammatory response carries the risk of additional tissue damage as demonstrated during liver IRI and other conditions.127, 128
Interestingly, activation and accumulation of neutrophils in the sinusoids do not cause tissue damage to the epithelium, as it does in other organs. Only after migrating across the endothelium and in close proximity to the hepatocytes can neutrophils cause damage by oxidative stress, triggered by two major cytotoxic mediators, ie, ROS and proteases.129 During the first hours of reperfusion, Kupffer cells, the resident macrophages of the liver, release ROS and proinflammatory cytokines, aggravating the initial damage caused by ischemia.126 The later phase includes a cascade of inflammatory events culminating in excessive neutrophil infiltration into the liver, and consequently amplification of the tissue lesion.36, 72 The adhesion of neutrophils to the targets induces their degranulation with release of proteases and ROS, which can diffuse into hepatocytes, inducing an intracellular oxidant stress, mitochondrial dysfunction, and damage.130 Neutrophil extravasation into the parenchyma is a prerequisite for cytotoxicity, as neutrophils are recruited into the sinusoids without causing damage.128
A critical role for ROS is observed in the pathophysiology of the liver IRI. Normally, ROS production is useful to eliminate circulating pathogens and is the mechanism responsible for the ‘respiratory burst’ observed when cells are activated. However, excessive ROS after an ischemic insult is detrimental.131 ROS formation is dangerous for cells because of the presence of polyunsaturated fatty acids in cellular membranes, the substantial number of unprotected protein sulfhydryl groups and DNA bases.132 Despite this, hepatic cells have sophisticated defense systems, such as expression of superoxide dismutases (SOD 1 in the cytosol and SOD 2 in the mitochondria), and reduced glutathione (GSH), which are responsible for the detoxification of organic and inorganic peroxides, forming part of the cellular enzymatic antioxidant defense system.133, 134, 135, 136 However, increased ROS production causes disturbances of the cellular homeostasis and, if not counteracted, this can lead to cell death. Excessive ROS formation results in its diffusion into hepatocytes, where ROS can lead to lysosomal iron mobilization and translocation to the mitochondria, which triggers a mitochondrial oxidant stress and eventually necrotic cell death.132, 137 Consistent with this, inhibition of oxidant stress through blocking nicotinamide adenine dinucleotide phosphate oxidase reduces liver injury induced by neutrophils.138 Cahova et al139 show that the antioxidant metformin is able to reduce mitochondrial damage (markers: cytochrome C release, citrate synthase activity, mitochondrial DNA copy number, and mitochondrial respiration), mitochondrial ROS production, and liver inflammation in rats subjected to IRI. In addition, Xue et al140 show an example where the combination of increased ROS formation and impaired defense systems causes cell death. In this work, in rats subjected to liver IRI, administration of GSH by means of the portal vein before ischemia increases the 7-day survival rates of rats after liver IRI from 38 to 75%. In addition, GSH pretreatment decreases intrahepatic neutrophil accumulation. ROS from Kupffer cells also contributes to the activation of inflammatory pathways that lead to neutrophil accumulation in the liver, resulting in additional, prolonged injury. Thus, Kupffer cell-derived ROS are involved in the pathogenesis of liver IRI injury through direct oxidant-mediated damage and by augmenting the local activation of proinflammatory pathways.141
Neutrophil-derived proteases also seem to have an important role in the cause of direct cell death of hepatocytes. Once neutrophils adhere to hepatocytes, full degranulation occurs, with consequent release into the area around the cell of several proteases, such elastases, MMP-9, cathepsin G, MPO, and proteinase-3.141 Neutrophil elastase is a serine protease found in the azurophil granules of neutrophils. The requirement for neutrophils to migrate out of the vasculature and through the basement membrane, as well as the potent proteolytic function of neutrophil elastase, have led to the theory that neutrophil elastase might be involved in the pathogenesis of inflammatory tissue injury such as that exemplified by liver IRI.142
Neutrophil elastase is stored in azurophil granules in its active form until it is released following neutrophil exposure to the inflammatory stimuli. As a result, excessive release of neutrophil elastase degrades elastin, collagens, laminins, and other ECM components, thereby leading to subsequent organ damage through endothelial cell injury.142, 143 Consistent with this, a study of human liver damage identified a neutrophil elastase inhibitor that has therapeutic potential, and this inhibitor is associated with a reduced release of HMGB1 and reduced interleukin-6 levels.144 In addition, in mice subjected to 90 min of ischemia and 6 and 24 h of reperfusion, treatment with Sivelestat, a neutrophil elastase inhibitor, ameliorates the hepatocellular damage and decreases local neutrophil activity and infiltration.142
One of the main molecules released after neutrophil recruitment and activation is MPO. This enzyme is used as a neutrophil marker, because of its high expression in these cells. Thus, its activity correlates with neutrophil accumulation in tissues. In the presence of physiological chloride concentrations, MPO reacts with hydrogen peroxide to catalyze formation of hypochlorous acid/hypochlorite and other oxidizing species.145 The participation of MPO has been shown in a range of pathophysiological conditions, including liver IRI.20, 146 Kato et al147 have shown an increased MPO activity, which was consistent with increased neutrophil accumulation and hepatocyte necrosis.
Although it is clear that neutrophils can cause cell death and necrosis, their phagocytic actions are believed to be relevant for proper tissue repair and protection. Therefore, it is not simply to say that the effects of the function of neutrophils are entirely harmful or may contribute to resolution of inflammatory responses.148, 149 The relevance of neutrophils in any particular setting is clearly context-dependent. For example, Reber et al150 have shown that neutrophils can contribute to optimal host protection against endotoxemia, as neutrophil-depleted mice show increased mortality. This beneficial role was induced through MPO activity. On the other hand, Marques et al showed that, in contrast to depletion of Kupffer cells, depletion of neutrophils with anti-Ly-6G reduced acetaminophen-mediated toxicity and liver injury.24 In this liver toxicity model, a limited neutrophil influx may assist to clear cell debris, as suggested by Williams et al,151 but a high influx of neutrophils enhanced liver injury and inflammation.24 Thus, the amount of toxic compounds in the liver and consequently the number of infiltrating neutrophils may determine whether primarily the beneficial or rather the pathological effects of neutrophils predominate. In the context of liver IRI, there is overwhelming evidence for a deleterious role of neutrophil migration and function, as discussed above, and the activity of the neutrophil proteases together with ROS production appears to mediate most of the deleterious actions of neutrophils.
Conclusions
In summary, IRI triggers an inflammatory process in the liver with recruitment of neutrophils into the vasculature, mainly in the sinusoids. Neutrophil recruitment involves a multistep process, which includes participation of DAMPs released from necrotic cells, chemokine production, chemokine binding to GAGs, expression of adhesion molecules by endothelial cells and leukocytes, and release of effector molecules by neutrophils, such as ROS and MMPs. These steps comprise mechanical trapping of cells in the sinusoids, chemotaxis, extravasation, and adherence of neutrophils to target cells. Pharmacological manipulation of the recruitment of excessive neutrophil numbers and their movement into tissue still hold promise as strategies to treat IRI and may improve overall graft success in transplantation. Understanding the mechanisms that allow neutrophils to respond to sterile tissue injury and cell death is fundamental to our understanding of both homeostatic innate immune functions and immune responses in disease.
References
Bellanti F . Ischemia-reperfusion injury: evidences for translational research. Ann Transl Med 2016;4 (Suppl 1):S55.
Chouillard EK, Gumbs AA, Cherqui D . Vascular clamping in liver surgery: physiology, indications and techniques. Ann Surg Innov Res 2010;4:2.
Zhang B, Liu QH, Zhou CJ et al. Protective effect of eNOS overexpression against ischemia/reperfusion injury in small-for-size liver transplantation. Exp Ther Med 2016;12:3181–3188.
Zhai Y, Petrowsky H, Hong JC et al. Ischaemia-reperfusion injury in liver transplantation—from bench to bedside. Nat Rev Gastroenterol Hepatol 2013;10:79–89.
Montalvo-Jave EE, Escalante-Tattersfield T, Ortega-Salgado JA et al. Factors in the pathophysiology of the liver ischemia-reperfusion injury. J Surg Res 2008;147:153–159.
Serracino-Inglott F, Habib NA, Mathie RT . Hepatic ischemia-reperfusion injury. Am J Surg 2001;181:160–166.
Romanque UP, Uribe MM, Videla LA . Molecular mechanisms in liver ischemic-reperfusion injury and ischemic preconditioning. Rev Med Chil 2005;133:469–476.
Busuttil RW, Tanaka K . The utility of marginal donors in liver transplantation. Liver Transpl 2003;9:651–663.
Mendes-Braz M, Elias-Miro M, Jimenez-Castro MB et al. The current state of knowledge of hepatic ischemia-reperfusion injury based on its study in experimental models. J Biomed Biotechnol 2012;2012:298657.
Papadopoulos D, Siempis T, Theodorakou E et al. Hepatic ischemia and reperfusion injury and trauma: current concepts. Arch Trauma Res 2013;2:63–70.
Selzner M, Selzner N, Jochum W et al. Increased ischemic injury in old mouse liver: an ATP-dependent mechanism. Liver Transpl 2007;13:382–390.
Guan LY, Fu PY, Li PD et al. Mechanisms of hepatic ischemia-reperfusion injury and protective effects of nitric oxide. World J Gastrointest Surg 2014;6:122–128.
Honda M, Takeichi T, Asonuma K et al. Intravital imaging of neutrophil recruitment in hepatic ischemia-reperfusion injury in mice. Transplantation 2013;95:551–558.
Vedder NB, Fouty BW, Winn RK et al. Role of neutrophils in generalized reperfusion injury associated with resuscitation from shock. Surgery 1989;106:509–516.
Lentsch AB, Yoshidome H, Cheadle WG et al. Chemokine involvement in hepatic ischemia/reperfusion injury in mice: roles for macrophage inflammatory protein-2 and Kupffer cells. Hepatology 1998;27:507–512.
Colletti LM, Kunkel SL, Walz A et al. The role of cytokine networks in the local liver injury following hepatic ischemia/reperfusion in the rat. Hepatology 1996;23:506–514.
Adage T, Piccinini AM, Falsone A et al. Structure-based design of decoy chemokines as a way to explore the pharmacological potential of glycosaminoglycans. Br J Pharmacol 2012;167:1195–1205.
Frederiks WM, James J, Bosch KS et al. A model for provoking ischemic necrosis in rat liver parenchyma and its quantitative analysis. Exp Pathol 1982;22:245–252.
Colletti LM, Remick DG, Burtch GD et al. Role of tumor necrosis factor-alpha in the pathophysiologic alterations after hepatic ischemia/reperfusion injury in the rat. J Clin Invest 1990;85:1936–1943.
Jaeschke H, Farhood A, Smith CW . Neutrophils contribute to ischemia/reperfusion injury in rat liver in vivo. FASEB J 1990;4:3355–3359.
Land WG . The role of damage-associated molecular patterns in human diseases: part i—promoting inflammation and immunity. Sultan Qaboos Univ Med J 2015;15:e9–e21.
van Golen RF, van Gulik TM, Heger M . The sterile immune response during hepatic ischemia/reperfusion. Cytokine Growth Factor Rev 2012;23:69–84.
Takeuchi O, Akira S . Pattern recognition receptors and inflammation. Cell 2010;140:805–820.
Marques PE, Oliveira AG, Pereira RV et al. Hepatic DNA deposition drives drug-induced liver injury and inflammation in mice. Hepatology 2015;61:348–360.
Bamboat ZM, Balachandran VP, Ocuin LM et al. Toll-like receptor 9 inhibition confers protection from liver ischemia-reperfusion injury. Hepatology 2010;51:621–632.
Tsung A, Sahai R, Tanaka H et al. The nuclear factor HMGB1 mediates hepatic injury after murine liver ischemia-reperfusion. J Exp Med 2005;201:1135–1143.
Diepenhorst GM, van Gulik TM, Hack CE . Complement-mediated ischemia-reperfusion injury: lessons learned from animal and clinical studies. Ann Surg 2009;249:889–899.
Datta G, Fuller BJ, Davidson BR . Molecular mechanisms of liver ischemia reperfusion injury: insights from transgenic knockout models. World J Gastroenterol 2013;19:1683–1698.
Marshall KM, He S, Zhong Z et al. Dissecting the complement pathway in hepatic injury and regeneration with a novel protective strategy. J Exp Med 2014;211:1793–1805.
Jaeschke H, Farhood A, Bautista AP et al. Complement activates Kupffer cells and neutrophils during reperfusion after hepatic ischemia. Am J Physiol 1993;264 (4 Pt 1):G801–G809.
Arumugam TV, Woodruff TM, Stocks SZ et al. Protective effect of a human C5a receptor antagonist against hepatic ischaemia-reperfusion injury in rats. J Hepatol 2004;40:934–941.
Xu R, Huang H, Zhang Z et al. The role of neutrophils in the development of liver diseases. Cell Mol Immunol 2014;11:224–231.
Adams DH, Ju C, Ramaiah SK et al. Mechanisms of immune-mediated liver injury. Toxicol Sci 2010;115:307–321.
Lee WY, Kubes P . Leukocyte adhesion in the liver: distinct adhesion paradigm from other organs. J Hepatol 2008;48:504–512.
Kubes P, Mehal WZ . Sterile inflammation in the liver. Gastroenterology 2012;143:1158–1172.
Ramaiah SK, Jaeschke H . Role of neutrophils in the pathogenesis of acute inflammatory liver injury. Toxicol Pathol 2007;35:757–766.
Hughes H, Farhood A, Jaeschke H . Role of leukotriene B4 in the pathogenesis of hepatic ischemia-reperfusion injury in the rat. Prostaglandins Leukot Essent Fatty Acids 1992;45:113–119.
Zhang Q, Raoof M, Chen Y et al. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature 2010;464:104–107.
Rot A, von Andrian UH . Chemokines in innate and adaptive host defense: basic chemokinese grammar for immune cells. Annu Rev Immunol 2004;22:891–928.
Su Y, Richmond A . Chemokine regulation of neutrophil infiltration of skin wounds. Adv Wound Care 2015;4:631–640.
Roh YS, Zhang B, Loomba R et al. TLR2 and TLR9 contribute to alcohol-mediated liver injury through induction of CXCL1 and neutrophil infiltration. Am J Physiol Gastrointest Liver Physiol 2015;309:G30–G41.
Wilson GC, Freeman CM, Kuethe JW et al. CXC chemokine receptor-4 signaling limits hepatocyte proliferation after hepatic ischemia-reperfusion in mice. Am J Physiol Gastrointest Liver Physiol 2015;308:G702–G709.
Mosher B, Dean R, Harkema J et al. Inhibition of Kupffer cells reduced CXC chemokine production and liver injury. J Surg Res 2001;99:201–210.
Li X, Klintman D, Liu Q et al. Critical role of CXC chemokines in endotoxemic liver injury in mice. J Leukoc Biol 2004;75:443–452.
Kuboki S, Shin T, Huber N et al. Hepatocyte signaling through CXC chemokine receptor-2 is detrimental to liver recovery after ischemia/reperfusion in mice. Hepatology 2008;48:1213–1223.
Fan X, Patera AC, Pong-Kennedy A et al. Murine CXCR1 is a functional receptor for GCP-2/CXCL6 and interleukin-8/CXCL8. J Biol Chem 2007;282:11658–11666.
Wuyts A, Van Osselaer N, Haelens A et al. Characterization of synthetic human granulocyte chemotactic protein 2: usage of chemokine receptors CXCR1 and CXCR2 and in vivo inflammatory properties. Biochemistry 1997;36:2716–2723.
Murphy PM, Tiffany HL . Cloning of complementary DNA encoding a functional human interleukin-8 receptor. Science 1991;253:1280–1283.
Holmes WE, Lee J, Kuang WJ et al. Structure and functional expression of a human interleukin-8 receptor. Science 1991;253:1278–1280.
Wilson GC, Kuboki S, Freeman CM et al. CXC chemokines function as a rheostat for hepatocyte proliferation and liver regeneration. PLoS ONE 2015;10:e0120092.
McDonald B, Pittman K, Menezes GB et al. Intravascular danger signals guide neutrophils to sites of sterile inflammation. Science 2010;330:362–366.
Bertini R, Allegretti M, Bizzarri C et al. Noncompetitive allosteric inhibitors of the inflammatory chemokine receptors CXCR1 and CXCR2: prevention of reperfusion injury. Proc Natl Acad Sci UA 2004;101:11791–11796.
Wengner AM, Pitchford SC, Furze RC et al. The coordinated action of G-CSF and ELR+CXC chemokines in neutrophil mobilization during acute inflammation. Blood 2008;111:42–49.
Verkaar F, van Offenbeek J, van der Lee MM et al. Chemokine cooperativity is caused by competitive glycosaminoglycan binding. J Immunol 2014;192:3908–3914.
Ley K, Laudanna C, Cybulsky MI et al. Getting to the site of inflammation: the leukocyte adhesion cascade updated. Nat Rev Immunol 2007;7:678–689.
Handel TM, Johnson Z, Crown SE et al. Regulation of protein function by glycosaminoglycans—as exemplified by chemokines. Annu Rev Biochem 2005;74:385–410.
Johnson Z, Proudfoot AE, Handel TM . Interaction of chemokines and glycosaminoglycans: a new twist in the regulation of chemokine function with opportunities for therapeutic intervention. Cytokine Growth Factor Rev 2005;16:625–636.
Rink I, Rink J, Helmer D et al. A haptotaxis assay for leukocytes based on surface-bound chemokine gradients. J Immunol 2015;194:5549–5558.
Taylor KR, Gallo RL . Glycosaminoglycans and their proteoglycans: host-associated molecular patterns for initiation and modulation of inflammation. FASEB J 2006;20:9–22.
Trowbridge JM, Gallo RL . Dermatan sulfate: new functions from an old glycosaminoglycan. Glycobiology 2002;12:117R–125R.
Massena S, Christoffersson G, Hjertstrom E et al. A chemotactic gradient sequestered on endothelial heparan sulfate induces directional intraluminal crawling of neutrophils. Blood 2010;116:1924–1931.
Middleton J, Patterson AM, Gardner L et al. Leukocyte extravasation: chemokine transport and presentation by the endothelium. Blood 2002;100:3853–3860.
Severin IC, Gaudry JP, Johnson Z et al. Characterization of the chemokine CXCL11-heparin interaction suggests two different affinities for glycosaminoglycans. J Biol Chem 2010;285:17713–17724.
Sarris M, Masson JB, Maurin D et al. Inflammatory chemokines direct and restrict leukocyte migration within live tissues as glycan-bound gradients. Curr Biol 2012;22:2375–2382.
Dyer DP, Thomson JM, Hermant A et al. TSG-6 inhibits neutrophil migration via direct interaction with the chemokine CXCL8. J Immunol 2014;192:2177–2185.
Bao X, Moseman EA, Saito H et al. Endothelial heparan sulfate controls chemokine presentation in recruitment of lymphocytes and dendritic cells to lymph nodes. Immunity 2010;33:817–829.
Vanheule V, Janssens R, Boff D et al. The positively charged COOH-terminal glycosaminoglycan-binding CXCL9(74-103) peptide inhibits CXCL8-induced neutrophil extravasation and monosodium urate crystal-induced gout in mice. J Biol Chem 2015;290:21292–21304.
McDonald B, McAvoy EF, Lam F et al. Interaction of CD44 and hyaluronan is the dominant mechanism for neutrophil sequestration in inflamed liver sinusoids. J Exp Med 2008;205:915–927.
Allegretti M, Cesta MC, Garin A et al. Current status of chemokine receptor inhibitors in development. Immunol Lett 2012;145:68–78.
Chen CJ, Kono H, Golenbock D et al. Identification of a key pathway required for the sterile inflammatory response triggered by dying cells. Nat Med 2007;13:851–856.
Chen GY, Nunez G . Sterile inflammation: sensing and reacting to damage. Nat Rev Immunol 2010;10:826–837.
Kolaczkowska E, Kubes P . Neutrophil recruitment and function in health and inflammation. Nat Rev Immunol 2013;13:159–175.
Marques PE, Amaral SS, Pires DA et al. Chemokines and mitochondrial products activate neutrophils to amplify organ injury during mouse acute liver failure. Hepatology 2012;56:1971–1982.
Cox TR, Erler JT . Remodeling and homeostasis of the extracellular matrix: implications for fibrotic diseases and cancer. Dis Model Mech 2011;4:165–178.
Kuyvenhoven JP, Verspaget HW, Gao Q et al. Assessment of serum matrix metalloproteinases MMP-2 and MMP-9 after human liver transplantation: increased serum MMP-9 level in acute rejection. Transplantation 2004;77:1646–1652.
Dechene A, Sowa JP, Gieseler RK et al. Acute liver failure is associated with elevated liver stiffness and hepatic stellate cell activation. Hepatology 2010;52:1008–1016.
Cursio R, Mari B, Louis K et al. Rat liver injury after normothermic ischemia is prevented by a phosphinic matrix metalloproteinase inhibitor. FASEB J 2002;16:93–95.
Coito AJ . Leukocyte transmigration across endothelial and extracellular matrix protein barriers in liver ischemia/reperfusion injury. Curr Opin Organ Transplant 2011;16:34–40.
Keck T, Balcom JHt, Fernandez-del Castillo C et al. Matrix metalloproteinase-9 promotes neutrophil migration and alveolar capillary leakage in pancreatitis-associated lung injury in the rat. Gastroenterology 2002;122:188–201.
Hu J, Van den Steen PE, Sang QX et al. Matrix metalloproteinase inhibitors as therapy for inflammatory and vascular diseases. Nat Rev Drug Discov 2007;6:480–498.
Palladini G, Ferrigno A, Richelmi P et al. Role of matrix metalloproteinases in cholestasis and hepatic ischemia/reperfusion injury: a review. World J Gastroenterol 2015;21:12114–12124.
Bjorklund M, Koivunen E . Gelatinase-mediated migration and invasion of cancer cells. Biochim Biophys Acta 2005;1755:37–69.
Morini M, Mottolese M, Ferrari N et al. The alpha 3 beta 1 integrin is associated with mammary carcinoma cell metastasis, invasion, and gelatinase B (MMP-9) activity. Int J Cancer 2000;87:336–342.
Ram M, Sherer Y, Shoenfeld Y . Matrix metalloproteinase-9 and autoimmune diseases. J Clin Immunol 2006;26:299–307.
Posthumus MD, Limburg PC, Westra J et al. Serum levels of matrix metalloproteinase-3 in relation to the development of radiological damage in patients with early rheumatoid arthritis. Rheumatology 1999;38:1081–1087.
Yong VW, Zabad RK, Agrawal S et al. Elevation of matrix metalloproteinases (MMPs) in multiple sclerosis and impact of immunomodulators. J Neurol Sci 2007;259:79–84.
Faber-Elmann A, Sthoeger Z, Tcherniack A et al. Activity of matrix metalloproteinase-9 is elevated in sera of patients with systemic lupus erythematosus. Clin Exp Immunol 2002;127:393–398.
Gerwien H, Hermann S, Zhang X et al. Imaging matrix metalloproteinase activity in multiple sclerosis as a specific marker of leukocyte penetration of the blood-brain barrier. Sci Transl Med 2016;8:364ra152.
Garg P, Vijay-Kumar M, Wang L et al. Matrix metalloproteinase-9-mediated tissue injury overrides the protective effect of matrix metalloproteinase-2 during colitis. Am J Physiol Gastrointest Liver Physiol 2009;296:G175–G184.
de Bruyn M, Vandooren J, Ugarte-Berzal E et al. The molecular biology of matrix metalloproteinases and tissue inhibitors of metalloproteinases in inflammatory bowel diseases. Crit Rev Biochem Mol Biol 2016;51:295–358.
Craig VJ, Zhang L, Hagood JS et al. Matrix metalloproteinases as therapeutic targets for idiopathic pulmonary fibrosis. Am J Respir Cell Mol Biol 2015;53:585–600.
Kato H, Kuriyama N, Duarte S et al. MMP-9 deficiency shelters endothelial PECAM-1 expression and enhances regeneration of steatotic livers after ischemia and reperfusion injury. J Hepatol 2014;60:1032–1039.
Hamada T, Fondevila C, Busuttil RW et al. Metalloproteinase-9 deficiency protects against hepatic ischemia/reperfusion injury. Hepatology 2008;47:186–198.
Kuyvenhoven JP, Ringers J, Verspaget HW et al. Serum matrix metalloproteinase MMP-2 and MMP-9 in the late phase of ischemia and reperfusion injury in human orthotopic liver transplantation. Transplant Proc 2003;35:2967–2969.
Bourboulia D, Stetler-Stevenson WG . Matrix metalloproteinases (MMPs) and tissue inhibitors of metalloproteinases (TIMPs): positive and negative regulators in tumor cell adhesion. Semin Cancer Biol 2010;20:161–168.
Nagase H, Woessner JF Jr . Matrix metalloproteinases. J Biol Chem 1999;274:21491–21494.
Kato H, Duarte S, Liu D et al. Matrix metalloproteinase-2 (MMP-2) gene deletion enhances MMP-9 activity, impairs PARP-1 degradation, and exacerbates hepatic ischemia and reperfusion injury in mice. PLoS ONE 2015;10:e0137642.
Parks WC, Wilson CL, Lopez-Boado YS . Matrix metalloproteinases as modulators of inflammation and innate immunity. Nat Rev Immunol 2004;4:617–629.
Amersi F, Shen XD, Moore C et al. Fibronectin-alpha 4 beta 1 integrin-mediated blockade protects genetically fat Zucker rat livers from ischemia/reperfusion injury. Am J Pathol 2003;162:1229–1239.
Moore C, Shen XD, Gao F et al. Fibronectin-alpha4beta1 integrin interactions regulate metalloproteinase-9 expression in steatotic liver ischemia and reperfusion injury. Am J Pathol 2007;170:567–577.
Nascimento GC, Rizzi E, Gerlach RF et al. Expression of MMP-2 and MMP-9 in the rat trigeminal ganglion during the development of temporomandibular joint inflammation. Braz J Med Biol Res 2013;46:956–967.
Cheung PY, Sawicki G, Wozniak M et al. Matrix metalloproteinase-2 contributes to ischemia-reperfusion injury in the heart. Circulation 2000;101:1833–1839.
Martin A, Garofalakis A, Tavitian B . In vivo evidence that the increase in matrix metalloproteinase activity occurs early after cerebral ischemia. Mol Imaging 2012;11:22–26.
Chirco R, Liu XW, Jung KK et al. Novel functions of TIMPs in cell signaling. Cancer Metastasis Rev 2006;25:99–113.
Egeblad M, Werb Z . New functions for the matrix metalloproteinases in cancer progression. Nat Rev Cancer 2002;2:161–174.
Duarte S, Hamada T, Kuriyama N et al. TIMP-1 deficiency leads to lethal partial hepatic ischemia and reperfusion injury. Hepatology 2012;56:1074–1085.
Sanz MJ, Kubes P . Neutrophil-active chemokines in in vivo imaging of neutrophil trafficking. Eur J Immunol 2012;42:278–283.
Kim MS, Lee KH, Lee WM et al. CD44 disruption attenuates murine hepatic ischemia/reperfusion injury. J Korean Med Sci 2011;26:919–926.
Wong J, Johnston B, Lee SS et al. A minimal role for selectins in the recruitment of leukocytes into the inflamed liver microvasculature. J Clin Invest 1997;99:2782–2790.
Jaeschke H, Hasegawa T . Role of neutrophils in acute inflammatory liver injury. Liver Int 2006;26:912–919.
Marques PE, Oliveira AG, Chang L et al. Understanding liver immunology using intravital microscopy. J Hepatol 2015;63:733–742.
Hickey MJ, Westhorpe CL . Imaging inflammatory leukocyte recruitment in kidney, lung and liver—challenges to the multi-step paradigm. Immunol Cell Biol 2013;91:281–289.
Harlan JM, Winn RK . Leukocyte-endothelial interactions: clinical trials of anti-adhesion therapy. Crit Care Med 2002;30:S214–S219.
Benkoel L, Dodero F, Hardwigsen J et al. Expression of intercellular adhesion molecule-1 (ICAM- 1) during ischemia-reperfusion in human liver tissue allograft: image analysis by confocal laser scanning microscopy. Dig Dis Sci 2003;48:2167–2172.
Farhood A, McGuire GM, Manning AM et al. Intercellular adhesion molecule 1 (ICAM-1) expression and its role in neutrophil-induced ischemia-reperfusion injury in rat liver. J Leukoc Biol 1995;57:368–374.
Nakano H, Kuzume M, Namatame K et al. Efficacy of intraportal injection of anti-ICAM-1 monoclonal antibody against liver cell injury following warm ischemia in the rat. Am J Surg 1995;170:64–66.
Nishimura Y, Takei Y, Kawano S et al. The F(ab’)2 fragment of an anti-ICAM-1 monoclonal antibody attenuates liver injury after orthotopic liver transplantation. Transplantation 1996;61:99–104.
Menezes GB, Lee WY, Zhou H et al. Selective down-regulation of neutrophil Mac-1 in endotoxemic hepatic microcirculation via IL-10. J Immunol 2009;183:7557–7568.
Meyer K, Brown MF, Zibari G et al. ICAM-1 upregulation in distant tissues after hepatic ischemia/reperfusion: a clue to the mechanism of multiple organ failure. J Pediatr Surg 1998;33:350–353.
Jaeschke H, Farhood A, Bautista AP et al. Functional inactivation of neutrophils with a Mac-1 (CD11b/CD18) monoclonal antibody protects against ischemia-reperfusion injury in rat liver. Hepatology 1993;17:915–923.
Marubayashi S, Oshiro Y, Maeda T et al. Protective effect of monoclonal antibodies to adhesion molecules on rat liver ischemia-reperfusion injury. Surgery 1997;122:45–52.
Lammermann T, Afonso PV, Angermann BR et al. Neutrophil swarms require LTB4 and integrins at sites of cell death in vivo. Nature 2013;498:371–375.
Lammermann T . In the eye of the neutrophil swarm-navigation signals that bring neutrophils together in inflamed and infected tissues. J Leukoc Biol 2016;100:55–63.
Lammermann T, Sixt M . Mechanical modes of ‘amoeboid’ cell migration. Curr Opin Cell Biol 2009;21:636–644.
Lammermann T, Germain RN . The multiple faces of leukocyte interstitial migration. Semin Immunopathol 2014;36:227–251.
Jaeschke H . Molecular mechanisms of hepatic ischemia-reperfusion injury and preconditioning. Am J Physiol Gastrointest Liver Physiol 2003;284:G15–G26.
Gujral JS, Farhood A, Bajt ML et al. Neutrophils aggravate acute liver injury during obstructive cholestasis in bile duct-ligated mice. Hepatology 2003;38:355–363.
Jaeschke H, Smith CW . Mechanisms of neutrophil-induced parenchymal cell injury. J Leukoc Biol 1997;61:647–653.
Schofield ZV, Woodruff TM, Halai R et al. Neutrophils—a key component of ischemia-reperfusion injury. Shock 2013;40:463–470.
Jaeschke H . Mechanisms of Liver Injury. II. Mechanisms of neutrophil-induced liver cell injury during hepatic ischemia-reperfusion and other acute inflammatory conditions. Am J Physiol Gastrointest Liver Physiol 2006;290:G1083–G1088.
Evankovich J, Billiar T, Tsung A . Toll-like receptors in hepatic ischemia/reperfusion and transplantation. Gastroenterol Res Pract 2010;2010:pii537263.
Jaeschke H, Ramachandran A . Reactive oxygen species in the normal and acutely injured liver. J Hepatol 2011;55:227–228.
Jaeschke H . Reactive oxygen and mechanisms of inflammatory liver injury: present concepts. J Gastroenterol Hepatol 2011;26 (Suppl 1):173–179.
Bilzer M, Paumgartner G, Gerbes AL . Glutathione protects the rat liver against reperfusion injury after hypothermic preservation. Gastroenterology 1999;117:200–210.
Bilzer M, Baron A, Schauer R et al. Glutathione treatment protects the rat liver against injury after warm ischemia and Kupffer cell activation. Digestion 2002;66:49–57.
Schauer RJ, Gerbes AL, Vonier D et al. Glutathione protects the rat liver against reperfusion injury after prolonged warm ischemia. Ann Surg 2004;239:220–231.
Uchiyama A, Kim JS, Kon K et al. Translocation of iron from lysosomes into mitochondria is a key event during oxidative stress-induced hepatocellular injury. Hepatology 2008;48:1644–1654.
Hasegawa T, Malle E, Farhood A et al. Generation of hypochlorite-modified proteins by neutrophils during ischemia-reperfusion injury in rat liver: attenuation by ischemic preconditioning. Am J Physiol Gastrointest Liver Physiol 2005;289:G760–G767.
Cahova M, Palenickova E, Dankova H et al. Metformin prevents ischemia reperfusion-induced oxidative stress in the fatty liver by attenuation of reactive oxygen species formation. Am J Physiol Gastrointest Liver Physiol 2015;309:G100–G111.
Xue F, Wang G, Pang Z et al. Protective effect of glutathione against liver warm ischemia-reperfusion injury in rats is associated with regulation of P-selectin and neutrophil infiltration. Anat Rec 2008;291:1016–1022.
Faurschou M, Borregaard N . Neutrophil granules and secretory vesicles in inflammation. Microbes Infect 2003;5:1317–1327.
Uchida Y, Freitas MC, Zhao D et al. The protective function of neutrophil elastase inhibitor in liver ischemia/reperfusion injury. Transplantation 2010;89:1050–1056.
Mainardi CL, Dixit SN, Kang AH . Degradation of type IV (basement membrane) collagen by a proteinase isolated from human polymorphonuclear leukocyte granules. J Biol Chem 1980;255:5435–5441.
Tsujii S, Okabayashi T, Shiga M et al. The effect of the neutrophil elastase inhibitor sivelestat on early injury after liver resection. World J Surg 2012;36:1122–1127.
Klebanoff SJ . Myeloperoxidase: friend and foe. J Leukoc Biol 2005;77:598–625.
Kawachi S, Hines IN, Laroux FS et al. Nitric oxide synthase and postischemic liver injury. Biochem Biophys Res Commun 2000;276:851–854.
Kato A, Edwards MJ, Lentsch AB . Gene deletion of NF-kappa B p50 does not alter the hepatic inflammatory response to ischemia/reperfusion. J Hepatol 2002;37:48–55.
Borregaard N . Neutrophils, from marrow to microbes. Immunity 2010;33:657–670.
Mayadas TN, Cullere X, Lowell CA . The multifaceted functions of neutrophils. Annu Rev Pathol 2014;9:181–218.
Reber LL, Gillis CM, Starkl P et al. Neutrophil myeloperoxidase diminishes the toxic effects and mortality induced by lipopolysaccharide. J Exp Med 2017;214:1249–1258.
Williams CD, Bajt ML, Sharpe MR et al. Neutrophil activation during acetaminophen hepatotoxicity and repair in mice and humans. Toxicol Appl Pharmacol 2014;275:122–133.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
This review discusses the important role of neutrophils in ischemia and reperfusion injury in the liver. The interactions between major molecules that play a role in this pathology, including danger signals, adhesion molecules, cytokines and chemokines, glycosaminoglycans and neutrophil-derived enzymes and their target cells are discussed in detail.
Rights and permissions
About this article

Cite this article
Oliveira, T., Marques, P., Proost, P. et al. Neutrophils: a cornerstone of liver ischemia and reperfusion injury. Lab Invest 98, 51–62 (2018). https://doi.org/10.1038/labinvest.2017.90
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/labinvest.2017.90
This article is cited by
-
Formyl peptide receptor 2 activation by mitochondrial formyl peptides stimulates the neutrophil proinflammatory response via the ERK pathway and exacerbates ischemia–reperfusion injury
Cellular & Molecular Biology Letters (2023)
-
Key m6A regulators mediated methylation modification pattern and immune infiltration characterization in hepatic ischemia-reperfusion injury
BMC Medical Genomics (2023)
-
Targeting NF-κB in Hepatic Ischemia–Reperfusion Alleviation: from Signaling Networks to Therapeutic Targeting
Molecular Neurobiology (2023)
-
Implication of Hypotension in the Pathogenesis of Cognitive Impairment and Brain Injury in Chronic Liver Disease
Neurochemical Research (2023)
-
Diminazene aceturate attenuates hepatic ischemia/reperfusion injury in mice
Scientific Reports (2022)
