
Abstract
Alterations in TP53 have been described in many cancer types including hematological neoplasms. We aimed at comparing TP53 mutations (mut) and deletions (del) in a large cohort of patients with hematological malignancies (n=3307), including AML (n=858), MDS (n=943), ALL (n=358), CLL (n=1148). Overall, alterations in TP53 were detected in 332/3307 cases (10%). The highest frequency was observed in ALL (total: 19%; mut+del: 6%; mut only: 8%; del only: 5%) and AML (total: 13%; mut+del: 5%; mut only: 7%; del only: 1%), whereas TP53 alterations occurred less frequently in CLL (total: 8%) and MDS (total: 7%). TP53 mutations were significantly more frequent in patients ⩾60 vs <60 years in AML (9% vs 2%, P<0.001) and ALL (12% vs 6%, P<0.001). TP53mut+del had a significant negative impact on overall survival in all entities, whereas differences were observed regarding TP53mut only or TP53del only: TP53mut only impacted survival in AML (36 vs 9 months, P<0.001) and MDS (65 vs 19 months, P<0.001), TP53del only in CLL (not reached vs 64 months, P=0.008) and MDS (65 vs 24 months, P=0.011). As substantial differences between the entities are observed regarding correlation to age and survival, we suggest evaluation of both TP53 deletion and mutation status.
Similar content being viewed by others


Introduction
TP53 is the most frequently mutated gene in human cancer with a frequency of ~50%.1, 2 Alterations include mutations and deletions and are generally associated with advanced stages of disease, insufficient therapy-response and poor prognosis.3, 4, 5, 6, 7 The transcription factor TP53 has a central regulatory function in various signaling pathways, such as cell cycle arrest, apoptosis and DNA repair.8, 9 Owing to its central role in conserving genome stability, the p53 protein has been described as ‘the guardian of the genome’. TP53 deletions are often found to be associated with TP53 mutations of the second allele, supporting the ‘two-hit’ hypothesis which suggests that alteration of both copies of a tumor suppressor gene is required to induce and/or drive cancer development.2, 10, 11, 12, 13 Activation of p53 occurs in response to DNA damage or other stress conditions (for example, metabolic changes, hypoxia or oncogene activation), leading to activation or repression of its target genes, mainly causing G1 cell cycle arrest and apoptosis induction, a process that is disrupted by TP53 mutation/deletion in cancer.14, 15, 16 Deletions in TP53 often result from larger deletions of the short arm of chromosome 17, where TP53 is located, which can be detected by interphase FISH (fluorescence in situ hybridization), determining the copy-number state of a gene. Thus, the TP53 function is generally preserved in the case of a TP53 deletion without accompanying TP53 mutation in the other allele. Mutations in TP53 usually result in loss of function of the p53 protein, which can include complete or partial loss of function, dependent on the site of the mutation.17 Whereas tumor suppressors are commonly inactivated by frameshift or nonsense mutations, the most frequent mutation type of TP53 in tumors is represented by missense mutations in the coding region.10, 18 Although the cancer-associated TP53 mutations are found in many different locations throughout the TP53 sequence, they usually cluster in the DNA-binding domain, disrupting the ability of p53 to bind to its target DNA sequences thus preventing transcriptional activation of the respective genes.19 About 30% of the missense mutations are located in six ‘hotspot’ residues (p.R175, p.G245, p.R248, p.R249, p.R273 and p.R282) in the DNA-binding domain of p53, with R273 and R248 being the most frequently mutated ones.20, 21 Interestingly, although TP53 mutations typically abolish the tumor suppressor activity of the protein (loss-of-function mutations), gain-of-function mutations have also been described that lead to acquisition of additional oncogenic functions that promote cell growth and offer survival advantages to the cell.21
Mutations and deletions in TP53 are found in all hematological malignancies, and were described to have crucial roles in tumorigenesis. However, the frequency varies strongly between the respective entities. Whereas TP53 mutations were described to occur quite frequently in ALL (16%)22 and AML (12%),23, 24 the frequencies were lower in CLL (7%)7, 25, 26, 27 and MDS (6%).28, 29, 30 Similar to other cancer types, TP53 mutations in hematological malignancies were found to show a negative impact on survival. Moreover, TP53 mutations were shown to be enriched in therapy-associated diseases including t-AML and t-MDS and were also found at high frequency in relapse cases where they are associated with poor outcome.31, 32 The proposed role of TP53 mutations in therapy-related cases and in relapsed disease seems to be due to the selective advantage of the respective cells caused by their resistance to therapy.10, 33
We aimed at analyzing whether differences are found regarding kind and frequency of TP53 mutations and accompanying deletions in hematological entities. For this purpose, we performed a comprehensive analysis of the TP53 mutation/deletion patterns in different hematological malignancies, including AML, MDS, ALL and CLL and analyzed (i) the frequencies of TP53 mutations and deletions, (ii) the types of mutation, (iii) the mutation load, (iv) the correlations to cytogenetic aberrations, (v) the age dependency and (vi) impact on survival.
Patients and methods
Patient cohort
A total of 3307 cases (AML: n=858, MDS: n=943, ALL: n=358, CLL: n=1148; bone marrow and/or peripheral blood samples) that were sent for diagnosis to the MLL Munich Leukemia Laboratory between August 2005 and May 2013 were analyzed. For all samples, cytogenetics and FISH analyses using probes for TP53 having been successfully performed at our laboratory. No other selection criteria were applied and selection was performed randomly otherwise. Patients agreed with the use of laboratory data for research studies. The study followed the rules of the Helsinki Declaration. The total cohort comprised 1995 male and 1312 female patients and the median age was 68 years (range: 1 month to 92 years). The ALL cohort included 27 patients <18 years (median age: 49 years, range: 1 month to 91 years), whereas the AML, MDS and CLL cohorts comprised mainly older patients (median age: AML, 67 years (range: 3 to 92 years, including only 3 patients <18 years); MDS, 73 years (range: 23 to 91 years); CLL, 67 years (range: 30 to 91 years)). In more detail, the AML cohort included exclusively patients with de novo AML at diagnosis and comprised 466 male and 392 female patients. The MDS cohort consisted of 580 male and 363 female patients. ALL cases included 207 male and 151 female patients at diagnosis, the CLL cohort comprised 742 male and 406 female patients at diagnosis (n=947) or during course of the disease prior to treatment (n=201). Totally, clinical follow-up data were available for 2708 patients. This study includes cases that were included in previous studies regarding mutational analyses in AML, MDS, ALL and CLL.22, 23, 27, 30
Cytomorphology and immunophenotype
Cytomorphologic assessment (available for 3011 patients) was based on May-Grünwald-Giemsa stains, myeloperoxidase reaction, and non-specific esterase using alpha-naphtyl-acetate as described before.34 Multiparameter flow cytometry and sample procession was carried out as described previously.35 Multiparameter flow cytometry analyses were performed using FC500 or Navios flow cytometers (Beckman Coulter, Miami, FL, USA). List mode files were analyzed using CXP Software version 2.0 and Kaluza version 1.0 (Beckman Coulter). Diagnoses were assigned according to EGIL and WHO classifications.36, 37 Detailed data on immunophenotype were available for 2503 patients.
Cytogenetics and FISH
Chromosome preparations and banding analysis were performed for all 3307 cases as previously described according to standard methods.38, 39, 40 If required, cases with an aberrant karyotype in chromosome banding analysis were additionally investigated by 24-color FISH to completely resolve the karyotype (24XCyte Human Multicolor FISH Probe Kit, Metasystems, Altlussheim, Germany). For classification of abnormalities and karyotypes, the ISCN guidelines (2013) were used.41 In all 3307 patients, interphase FISH using probes for TP53 spanning a 167 kb region in 17p13 including the complete sequence of TP53 was performed to determine the copy-number state of TP53 (Metasystems).
Mutation analysis
For the total cohort of 3307 patients, next-generation amplicon deep-sequencing was applied to identify and quantify the TP53 mutation load as previously described.22 For this, DNA was extracted according to standard methods from bone marrow and peripheral blood cells obtained at the time of diagnosis. Next-generation amplicon deep-sequencing (Illumina, San Diego, CA, USA; 454 Life Sciences, Branford, CT, USA; sensitivity: 3%) was applied to investigate the mutational hotspot regions of TP53 (ENST00000269305, exons 4–11). The template library was either generated by the ThunderStorm instrument (RainDance Technologies, Billerica, MA, USA) and sequenced with the MiSeq (Illumina, San Diego, CA, USA), or coding regions were sequenced on the 454 Life Sciences NGS platform (Roche Applied Science, Penzberg, Germany). NGS data were analyzed using the GS Amplicon Variant Analyzer Software 2.6 (454 Life Sciences) and Sequence Pilot (version 4.1.1 Build 514 for the 454 platform; version 4.1.1 Build 510 for the Illumina platform, JSI Medicalsystems, Kippenheim, Germany).
Validity of the somatic mutations was checked against the publicly accessible COSMIC v74 database (http://cancer.sanger.ac.uk/cancergenome/projects/cosmic) and functional interpretation was performed using SIFT 1.03 (http://sift.jcvi.org) and PolyPhen 2.0 (http://genetics.bwh.harvard.edu/pph2) algorithms. In addition, TP53 variants were verified using the IARC repository (r17).42 Single-nucleotide polymorphisms (SNP) were annotated according to the NCBI dbSNP (http://www.ncbi.nlm.nih.gov/snp; Build 144) database. Synonymous variants and alterations within introns with the exception of splice-site mutations at position +/− 1 or 2 were not scored. Missense variants, which did not have unique entries in the COSMIC or dbSNP databases were annotated as variants of unknown significance (VUS).
Statistical analysis
SPSS (version 19.0.0) software (IBM Corporation, Armonk, NY, USA) was used for statistical analysis. Overall survival (OS) curves were calculated according to Kaplan–Meier and compared using the two-sided log rank test. All reported P-values are two-sided and were considered significant at P⩽0.05. OS was measured from the date of diagnosis until last follow-up or death.
Results
Frequency of TP53 mutations and deletions
Overall, alterations in TP53 were detected in 332/3307 cases (10%). Of these, 173 cases showed only a TP53 mutation (mut only), 43 cases harbored a TP53 deletion only (del only) and 116 cases with TP53 mutation and an accompanying TP53 deletion (mut+del) were found. In all but one patient, in whom a TP53 deletion was detected by FISH analysis a 17p was visible by chromosome banding analyses. Regarding the respective entities, the highest frequency of TP53 alterations was observed in ALL (total: 19%; mut+del: 6%; mut only: 8%; del only: 5%) followed by AML (total: 13%; mut+del: 5%; mut only: 7%; del only: 1%). By contrast, TP53 alterations occurred less frequently in CLL (total: 8%; mut+del: 4%; mut only: 3%; del only: 1%) and MDS (total: 7%; mut+del: 1%; mut only: 5%; del only: 1%; Figures 1a and b).

Frequency of TP53 alterations in hematological malignancies. The frequency of cases with TP53 mutation only (TP53mut only, red), TP53 deletion only (TP53del only, blue) and TP53 mutation with accompanying TP53 deletion (TP53mut+del, gray) was analyzed in patients with AML (overall cases with TP53 alterations: 111/858, 13%), ALL (67/358, 19%), CLL (88/1148, 8%) and MDS (66/943, 7%). The frequency was calculated in relation to the respective total cohort sizes (a) and in relation to the number of cases with detected TP53 alteration in each cohort (b).
Number and type of TP53 mutations and mutation load
In the complete cohort, 335 TP53 mutations were detected in 289 cases. In more detail, 111 mutations in 98 cases were found in patients with AML, 56 mutations in 50 ALL cases, 89 mutations in 81 CLL cases and 79 mutations in 60 MDS cases. In all entities mainly one mutation per case was detected (AML: 85/98, 87%; ALL: 45/50, 90%; CLL: 73/81, 90%; MDS: 43/60, 72%). However, MDS cases were found to harbor a statistically increased proportion of cases with two mutations compared with the other entities (AML: 13/98, 13%; ALL: 4/50, 8%; CLL: 8/81, 10%; MDS: 16/60, 27%; P=0.003) (Supplementary Figure 1).
Missense mutations were found to be the most common mutation type in all entities with a very similar frequency ranging from 78 to 81% (AML: 78%, ALL: 80%, CLL: 80%, MDS: 81%, Supplementary Figure 2A). In most entities they were followed by frameshift mutations (AML: 9%, ALL: 7%, CLL: 10%, MDS: 13%). By contrast, in-frame, splice-site and nonsense mutations were less frequent (in-frame mutations: AML: 3%, ALL: 9%, CLL: 3%, MDS: 3%; splice-site mutations: AML: 6%, ALL: 4%, CLL: 2%, MDS: 3%; nonsense mutations: AML: 4%, ALL: 0%, CLL: 5%, MDS: 1%).
Of note, several significant correlations were observed when the number of TP53 mutations per case, the presence of a TP53 deletion, the mutation type and the presence of a complex karyotype were analyzed: in AML, the number of TP53 mutations correlated to the presence of TP53 deletions (cases with >1 mutation: 0% (0/55) with TP53 deletion vs 23% (13/56) without TP53 deletion, P<0.001). For patients with MDS, an association of a complex karyotype with the number and the type of TP53 mutations was detected (cases with >1 mutation: 37% (13/35) complex karyotype vs 13% (4/31) non-complex karyotype, P=0.047; cases with missense mutations: 43% (15/35) complex karyotype vs 71% (22/31) non-complex karyotype, P<0.027). Mutations in the six known hotspot residues (R175, G245, R248, R249, R273, and R282), which were proposed to constitute ~30% of all TP53 mutations,1 were detected with a frequency ranging from 17 to 30% (AML: 24/111, 21% of all mutations were localized in hotspot residues; ALL: 17/56, 30%; CLL: 15/89, 17%; MDS: 16/79, 20%; Supplementary Figure 2B). Interestingly, mutations in R282 were exclusively observed in patients with AML and ALL.
Analyses of the TP53 mutation loads revealed highest values in AML (median: 47%). Lower median mutation loads were found in ALL (28%), MDS (39%), and CLL (36%; Supplementary Figure 3). When the cohorts were split into cases with TP53mut only and TP53mut+del, a strong difference was detected for AML patients (median mutation load: 47% in TP53mut only vs 80% in TP53mut+del) and CLL patients (median mutation load: 21% in TP53mut only vs 39% in TP53mut+del), whereas only minor differences were observed for ALL (median mutation load: 38% in TP53mut only vs 30% in TP53mut+del) and MDS patients (median mutation load: 39% in TP53mut only vs 45% in TP53mut+del). Concerning cases with more than one mutation, mutation loads were mostly similar for all mutations, although in some cases strong differences were found, suggesting the presence of independent clones (Supplementary Table 1).
Correlation of TP53 mutations and deletions to cytogenetic aberrations
A strong correlation of TP53 alterations with a complex karyotype (⩾3 aberrations was observed in AML (of patients with TP53 alteration: 5% with normal karyotype, 67% with complex karyotype, 28% with other aberrations), ALL (16% normal, 45% complex, 39% other) and MDS (14% normal, 53% complex, 33% other). By contrast, in CLL, TP53 alterations were mainly correlated with other cytogenetic aberrations (CLL: 10% normal, 30% complex, 60% other; Figure 2). These other cytogenetic aberrations constituted typical CLL alterations mainly including deletions of chromosome 13q (del13q: 26/53 cases with other aberrations), del(6q) (6/53 cases), trisomy 12 (6/53 cases) and del(11q) (5/53 cases). When only TP53 mutations were taken into account, a similar correlation was found, although the association of TP53 mutations with other cytogenetic aberrations became more pronounced in ALL cases (AML: 5% with normal karyotype, 72% with complex karyotype, 22% with other aberrations; ALL: 16% normal, 40% complex, 44% other; CLL: 11% normal, 27% complex, 62% other; MDS: 15% normal, 57% complex, 28% other). In addition, in all entities analyzed, a correlation between a high mutation load (>50%) and a complex karyotype was observed, which was found to be statistically significant in AML, MDS and CLL cases (Supplementary Table 2).

Correlation of TP53 alteration with cytogenetic aberrations. The frequency of cases with normal karyotype (blue), complex karyotype (>3 abnormalities, red) and other aberrations (gray) is shown for all cases with TP53 alteration in each cohort.
Age dependency of TP53 alterations
The median age within the analyzed entities ranged from 49 to 73 years for patients with TP53 alterations (AML: 67 years; ALL: 49 years; CLL: 67 years; MDS: 73 years). For cases with AML, CLL and MDS, the majority of patients were in decades 6 and 7, whereas the age distribution was broader for ALL patients (Figure 3). For analyzing the age dependency of TP53 alterations, the total cohort and the respective subcohorts were split into patients ⩾60 years of age (in the total cohort: n=2308) and<60 years of age (in the total cohort: n=999). TP53mut and TP53mut+del were significantly more frequent in patients ⩾60 vs<60 years of age in AML (9% vs 2% for mut only, P<0.001; 7% vs 2% for mut+del, P=0.001) and ALL (12% vs 6% for mut only, P<0.001; 13% vs 3% for mut+del, P=0.001). By contrast, no such significant differences were observed for patients with CLL and MDS. Regarding cases harboring TP53del, no age dependency was observed in any entity. For further correlation of TP53 alteration frequency with age, the cohorts were split into decades (Figure 3): for AML and ALL patients, patients in the decades>60 years show an elevated TP53 alteration frequency compared with average alteration frequency in the total cohorts (AML: 61–70 years, 16%, 71–80 years, 19%, total alteration frequency: 13%; ALL: 61–70 years, 26%, 71–80 years, 36%, total alteration frequency: 19%).

Age distribution of patients in the respective cohorts and correlation to TP53 alterations. The number of patients with the indicated age is depicted in the total cohorts of AML (a; n=858), ALL (b; n=358), CLL (c; n=1148), MDS (d; n=943) cases. The cases in each decade are further split into cases with TP53 alteration (gray) and cases which are wild-type for TP53 (red). Numbers indicate the frequency of patients with TP53 alteration in the respective decade.
Influence of TP53 alterations on overall survival
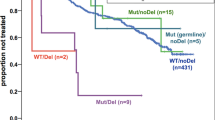
OS was significantly shorter in patients with a TP53 alteration compared with patients without TP53 alteration (TP53 wt) in all entities (TP53 wt vs TP53 alteration: AML: 36 months vs 6 months, P<0.001; ALL: not reached vs 24 months, P=0.001; CLL: not reached vs 64 months, P<0.001; MDS: 65 months vs 15 months, P<0.001; Figure 4, Table 1). Moreover, when the cohorts were split into cases with TP53mut only, TP53del only and TP53mut+del, a significant negative impact on OS was observed for TP53mut+del in all entities as well as for TP53mut only in patients with AML, CLL and MDS, whereas no significant influence was found for ALL patients. In the CLL and MDS cohorts, TP53del only was found to have a significant negative impact on OS as well, in contrast to AML and ALL cases where no adverse effect was detected for TP53del only (Figure 4; Table 1). Moreover, correlation analyses of the TP53 mutation loads with OS in the respective entities revealed a strong association of a higher mutation load with poorer outcome in MDS patients (mutation load ⩽25% vs >25%, 75 vs 12 months, P=0.010; ⩽50% vs >50%, 19 vs 6 months, P=0.001; ⩽75% vs >75%, 19 vs 6 months, P=0.002), whereas no correlation of the mutation load with OS was observed in ALL patients. In cases with AML, a significant association was only observed when the cut-off was set at 50% (⩽50% vs >50%, 9 vs 5 months, P=0.042), whereas in CLL patients very high mutation loads (>75%) correlated to a worse prognosis (⩽75% vs >75%, not reached vs 23 months, P=0.007).

Overall survival of patients with TP53 alteration. Overall survival (OS) was analyzed in patients with (gray line) and without (red line, TP53wt) TP53 alteration in patients with (a) AML (clinical follow-up data were available for n=550 patients), (b) ALL (n=268), (c) CLL (n=987), (d) MDS (n=903) (left panel, respectively). In the right panel of each entity, the OS was analyzed in patients with TP53 mutation only (TP53mut only, continuous gray line), with TP53 deletion only (TP53del only; dotted gray line) in patients that showed alterations in both alleles (TP53mut+del, broken gray line) and in patients without aberrations in TP53 (TP53wt; red line).
Discussion
TP53 is a central tumor suppressor gene, which is involved in cell cycle regulation and apoptosis induction. Our results clearly show that generally, the frequency of TP53 alterations in hematological malignancies (overall: 10%) is lower compared with solid tumors, where mutation frequencies of up to 90% have been observed for example, in uterine and ovarian cancer.43, 44 However, the frequencies clearly varied between different hematological malignancies analyzed, as the highest TP53 alteration frequency of the studied cohorts was found for ALL patients with a total alteration frequency of 19%, whereas for MDS patients only in 7% a TP53 alteration was detected. These data are in line with the TP53 alteration frequencies detected in previous studies.7, 22, 23, 24, 27, 29, 30 For CLL patients, both TP53 mutations and deletions were found to be associated with poor response to chemo(immuno)therapy, high rate of Richter transformation and thus to a poorer prognosis.45, 46 This is corroborated by our results, showing an adverse prognosis for TP53 mutations as well as deletions in patients with CLL. In patients with MDS, TP53 mutations were also found to be associated with more aggressive forms of the disease and with poorer response to treatment, also in patients with del(5q), which generally constitute a MDS subgroup with favorable prognosis. Thus, evaluation of TP53 mutation status was recommended for these patients.47, 48
Interestingly, mutations and also deletions in TP53 were found to be more frequent in acute leukemias (AML and ALL) compared with CLL and MDS. This is especially of interest for ALL patients, which were found to show a lower median age (49 years) compared with the other entities (AML, MDS and CLL cohort: 67, 73 and 67 years, respectively), corroborating previous results.22 Thus, the higher frequency of TP53 alterations in ALL cannot be explained by merely age-related occurrence. Another difference was observed for the acute leukemias compared with CLL and MDS, as—although in all entities missense mutations were found to constitute the dominant mutation type—mutations in the hotspot residue R282 were exclusively observed in patients with AML and ALL. Interestingly, although mutations in TP53 usually lead to loss-of-function of the tumor suppressor activity of the protein, the R282 site was found to belong to the gain-of function mutations.49 R282 is a structural mutation (as also for example, R175, G245, R248 und R249) causing conformational instability of the p53 protein, by contrast to the contact mutations (R273 and R248) that are localized in the p53-DNA-binding surface.50 Moreover, when overall survival was analyzed in regard to the respective TP53 mutation in a bladder cancer and colorectal cancer data set, only for R248 and R282 a significantly shorter overall survival time compared with other frequently occurring mutations (G245, R175 and R273) was detected.49 In addition, p53 R248 and R282 mutations were found to induce higher expression of the CYP3A4 protein, which is suggested to contribute to the chemoresistance associated with these p53 mutations.49 Thus, it can be speculated, as mutations in R282 were only detected in the acute diseases AML and ALL, these might contribute to the rapid proliferation of the malignant cells in these diseases. Differences were however observed between AML and ALL regarding the TP53 mutation loads, as AML patients showed the highest mutation loads of all entities analyzed (median: 47%), whereas the lowest values were detected in ALL patients (28%). As we analyzed the total cohort of n=3307 cases using next-generation amplicon deep-sequencing, the detected mutation loads can also be used to draw conclusions regarding the presence of independent clones in cases with more than one TP53 mutation: of the 43 cases with two or more TP53 mutations, 9 cases showed differences between the mutation loads of ⩾20% (Supplementary Table 1). Hence, in these cases it can be suggested that independent clones exist which each harbor a TP53 mutation.
In addition, TP53 alterations are found to be correlated to higher age in AML and ALL, whereas no such difference was observed for CLL and MDS patients. Interestingly, this effect was specifically detected for TP53 mutations but not for TP53 deletions. It can be suggested that TP53 alterations seem to have a stronger influence in acute diseases (AML, ALL) compared with MDS and CLL patients, as they are more frequent and show a clear increase with age.
Moreover, alterations in TP53 were found to be correlated to a complex karyotype in AML, ALL and MDS patients. By contrast, for CLL cases, TP53 alterations were associated with other cytogenetic aberrations, including abnormalities frequently detected in CLL such as deletions in chromosome 13q, 11q and 6q as well as trisomy 12. Thus, no specific association of TP53 alterations to a certain chromosomal aberration was found for CLL patients. An association of TP53 alterations with a complex karyotype has been described before e.g. for patients with MDS and AML, suggesting a role of TP53 alteration in a later stage of disease.24, 29, 51, 52 As no such correlation was found for CLL patients, it can be speculated that TP53 alterations already occur at an earlier phase in these patients.
In all four entities analyzed, alterations in TP53 were found to show a negative impact on survival, corroborating previous results.7, 22, 23, 24, 27, 29, 30 In addition, in all cohorts, especially the occurrence of a TP53 mutation and an accompanying TP53 deletion led to a negative impact on OS. However, some differences can be observed between the respective entities when the influence of TP53 mutation only or TP53 deletion only is analyzed in more detail: for patients with AML and MDS, the presence of a TP53 mutation without an additional deletion of the other allele is already capable of reducing overall survival drastically, whereas this effect was less pronounced in CLL patients or even not significant in cases with ALL. Hence, in ALL, the alteration of both alleles seems to be required to cause the adverse impact on prognosis. Interestingly, for cases with TP53 deletion lacking a concomitant TP53 mutation, a significant negative effect on overall survival was observed in CLL and MDS, but not in the acute diseases AML and ALL. Hence, it remains to be investigated in future studies by which mechanism the deletion of a TP53 allele in CLL and MDS patients is able to impact prognosis in these patients.
We can conclude that TP53 mutations and deletions have important roles in all hematological entities analyzed, although the general alteration frequency is clearly lower than in many solid tumors. Differences between the malignancies are observed not only regarding the alteration frequencies but also in terms of association with age and the influence on survival: although in all entities analyzed a TP53 mutation and an accompanying TP53 deletion in the second allele (‘double hit’) is always associated with an adverse outcome, differences were detected regarding OS for patients with mutation only or deletion only (‘single hit'). Moreover, analysis of the mutation loads by next-generation amplicon deep-sequencing further allows the presence of independent clones each harboring a TP53 mutation, which was found in all entities analyzed. The mutation loads were also found to have important roles regarding association to a complex karyotype and also impacted prognosis in AML, MDS and CLL patients, whereas they do not seem to show a strong influence in ALL patients. In addition, the higher sensitivity of next-generation amplicon deep-sequencing compared with Sanger sequencing allows the detection of small TP53 mutated subclones, which is of great clinical relevance, as it was shown that CLL patients harboring these small subclones showed the same clinical phenotype and poor survival as patients with clonal TP53 lesions.53 Thus, both TP53 mutations and deletions as well as the mutation loads should be evaluated and their presence needs further investigation in the future, especially regarding their clinical impact in different hematological entities.
References
Rivlin N, Brosh R, Oren M, Rotter V . Mutations in the p53 Tumor Suppressor Gene: Important Milestones at the Various Steps of Tumorigenesis. Genes Cancer 2011; 2: 466–474.
Preudhomme C, Fenaux P . The clinical significance of mutations of the P53 tumour suppressor gene in haematological malignancies. Br J Haematol 1997; 98: 502–511.
Soussi T, Legros Y, Lubin R, Ory K, Schlichtholz B . Multifactorial analysis of p53 alteration in human cancer: A review. Int J Cancer 1994; 57: 1–9.
Wickremasinghe RG, Prentice AG, Steele AJ . p53 and Notch signaling in chronic lymphocytic leukemia: clues to identifying novel therapeutic strategies. Leukemia 2011; 25: 1400–1407.
Lane DP . Cancer. p53, guardian of the genome. Nature 1992; 358: 15–16.
Fenaux P, Preudhomme C, Quiquandon I, Jonveaux P, Laï JL, Vanrumbeke M et al. Mutations of the P53 gene in acute myeloid leukaemia. Br J Haematol 1992; 80: 178–183.
Zenz T, Eichhorst B, Busch R, Denzel T, Häbe S, Winkler D et al. TP53 mutation and survival in chronic lymphocytic leukemia. J Clin Oncol 2010; 28: 4473–4479.
Rotter V, Aloni-Grinstein R, Schwartz D, Elkind NB, Simons A, Wolkowicz R et al. Does wild-type p53 play a role in normal cell differentiation? Semin Cancer Biol 1994; 5: 229–236.
Wynford-Thomas D . Cellular senescence and cancer. J Pathol 1999; 187: 100–111.
Agirre X, Novo FJ, Calasanz MJ, Larráyoz MJ, Lahortiga I, Valgañón M et al. TP53 is frequently altered by methylation, mutation, and/or deletion in acute lymphoblastic leukaemia. Mol Carcinog 2003; 38: 201–208.
Knudson AG . Mutation and Cancer: Statistical Study of Retinoblastoma. Proc Natl Acad of Sci USA 1971; 68: 820–823.
Venot C, Maratrat M, Dureuil C, Conseiller E, Bracco L, Debussche L . The requirement for the p53 proline-rich functional domain for mediation of apoptosis is correlated with specific PIG3 gene transactivation and with transcriptional repression. EMBO J 1998; 17: 4668–4679.
Harms KL, Chen X . The C terminus of p53 Family Proteins Is a Cell Fate Determinant. Mol Cell Biol 2005; 25: 2014–2030.
Shaw P, Freeman J, Bovey R, Iggo R . Regulation of specific DNA binding by p53: Evidence for a role for O-glycosylation and charged residues at the carboxy-terminus. Oncogene 1996; 12: 921–930.
Sakaguchi K, Herrera JE, Saito S, Miki T, Bustin M, Vassilev A et al. DNA damage activates p53 through a phosphorylation-acetylation cascade. Genes Dev 1998; 12: 2831–2841.
Kruse JP, Gu W . Modes of p53 regulation. Cell 2009; 137: 609–622.
Rossi D, Gaidano G . Molecular Genetics of High-risk Chronic Lymphocytic Leukemia. Expert Rev Hematol 2012; 5: 593–602.
Hollstein M, Rice K, Greenblatt MS, Soussi T, Fuchs R, Sorlie T et al. Database of p53 gene somatic mutations in human tumors and cell lines. Nucleic Acids Res 1994; 22: 3551–3555.
Bullock AN, Fersht AR . Rescuing the function of mutant p53. Nat Rev Cancer 2001; 1: 68–76.
Brosh R, Rotter V . When mutants gain new powers: news from the mutant p53 field. Nat Rev Cancer 2009; 9: 701–713.
Li J, Yang L, Gaur S, Zhang K, Wu X, Yuan YC et al. Mutants TP53 p.R273H and p.R273C but not p.R273G enhance cancer cell malignancy. Hum Mutat 2014; 35: 575–584.
Stengel A, Schnittger S, Weissmann S, Kuznia S, Kern W, Kohlmann A et al. TP53 mutations occur in 15.7% of ALL and are associated with MYC-rearrangement, low hypodiploidy, and a poor prognosis. Blood 2014; 124: 251–258.
Grossmann V, Schnittger S, Kohlmann A, Eder C, Roller A, Dicker F et al. A novel hierarchical prognostic model of AML solely based on molecular mutations. Blood 2012; 120: 2963–2972.
Rücker FG, Schlenk RF, Bullinger L, Kayser S, Teleanu V, Kett H et al. TP53 alterations in acute myeloid leukemia with complex karyotype correlate with specific copy number alterations, monosomal karyotype, and dismal outcome. Blood 2012; 119: 2114–2121.
Döhner H, Stilgenbauer S, Benner A, Leupolt E, Kröber A, Bullinger L et al. Genomic aberrations and survival in chronic lymphocytic leukemia. N Engl J Med 2000; 343: 1910–1916.
Rossi D, Cerri M, Deambrogi C, Sozzi E, Cresta S, Rasi S et al. The prognostic value of TP53 mutations in chronic lymphocytic leukemia is independent of Del17p13: implications for overall survival and chemorefractoriness. Clin Cancer Res 2009; 15: 995–1004.
Jeromin S, Weissmann S, Haferlach C, Dicker F, Bayer K, Grossmann V et al. SF3B1 mutations correlated to cytogenetics and mutations in NOTCH1, FBXW7, MYD88, XPO1 and TP53 in 1160 untreated CLL patients. Leukemia 2014; 28: 108–117.
Cazzola M, Della Porta MG, Malcovati L . The genetic basis of myelodysplasia and its clinical relevance. Blood 2013; 122: 4021–4034.
Papaemmanuil E, Gerstung M, Malcovati L, Tauro S, Gundem G, Van Loo P et al. Chronic Myeloid Disorders Working Group of the International Cancer Genome Consortium. Clinical and biological implications of driver mutations in myelodysplastic syndromes. Blood 2013; 122: 3616–3627.
Haferlach T, Nagata Y, Grossmann V, Okuno Y, Bacher U, Nagae G et al. Landscape of genetic lesions in 944 patients with myelodysplastic syndromes. Leukemia 2014; 28: 241–247.
Hof J, Krentz S, van Schewick C, Körner G, Shalapour S, Rhein P et al. Mutations and deletions of the TP53 gene predict nonresponse to treatment and poor outcome in first relapse of childhood acute lymphoblastic leukemia. J Clin Oncol 2011; 29: 3185–3193.
Wada M, Bartram CR, Nakamura H, Hachiya M, Chen DL, Borenstein J et al. Analysis of p53 mutations in a large series of lymphoid hematologic malignancies of childhood. Blood 1993; 82: 3163–3169.
Wong TN, Ramsingh G, Young AL, Miller CA, Touma W, Welch JS et al. Role of TP53 mutations in the origin and evolution of therapy-related acute myeloid leukaemia. Nature 2015; 518: 552–555.
Haferlach T, Kern W, Schoch C, Hiddemann W, Sauerland MC . Morphologic dysplasia in acute myeloid leukemia: importance of granulocytic dysplasia. J Clin Oncol 2003; 21: 3004–3005.
Kern W, Voskova D, Schoch C, Hiddemann W, Schnittger S, Haferlach T . Determination of relapse risk based on assessment of minimal residual disease during complete remission by multiparameter flow cytometry in unselected patients with acute myeloid leukemia. Blood 2004; 104: 3078–3085.
Bene MC, Castoldi G, Knapp W, Ludwig WD, Matutes E, Orfao A et al. Proposals for the immunological classification of acute leukemias. European Group for the Immunological Characterization of Leukemias (EGIL). Leukemia 1995; 9: 1783–1786.
Swerdlow SH, Campo E, Harris NL, Jaffe ES, Pileri SA, Stein H et al WHO Classification of Tumours of Haematopoietic and Lympoid Tissues. IARC: Lyon, France, 2008.
Schoch C, Schnittger S, Bursch S, Gerstner D, Hochhaus A, Berger U et al. Comparison of chromosome banding analysis, interphase- and hypermetaphase-FISH, qualitative and quantitative PCR for diagnosis and for follow-up in chronic myeloid leukemia: a study on 350 cases. Leukemia 2002; 16: 53–59.
Dicker F, Schnittger S, Haferlach T, Kern W, Schoch C . Immunostimulatory oligonucleotide-induced metaphase cytogenetics detect chromosomal aberrations in 80% of CLL patients: A study of 132 CLL cases with correlation to FISH, IgVH status, and CD38 expression. Blood 2006; 108: 3152–3160.
Haferlach C, Bacher U . Cytogenetic methods in chronic lymphocytic leukemia. Methods Mol Biol 2011; 730: 119–130.
Shaffer LG, Tommerup N . ISCN 2013: an International System for Human Cytogenetic Nomenclature. Karger: Basel, New York, 2013.
Petitjean A, Mathe E, Kato S, Ishioka C, Tavtigian SV, Hainaut P et al. Impact of mutant p53 functional properties on TP53 mutation patterns and tumor phenotype: lessons from recent developments in the IARC TP53 database. Hum Mutat 2007; 28: 622–629.
Ashour Ahmed A, Etemadmoghadam D, Temple J, Lynch AG, Riad M, Sharma R et al. Brenton Driver mutations in TP53 are ubiquitous in high grade serous carcinoma of the ovary. J Pathol 2010; 221: 49–56.
O’Hara AJ, Bell DW . The genomics and genetics of endometrial cancer. Adv Genomics Genet 2012; 2012: 33–47.
Stilgenbauer S, Schnaiter A, Paschka P, Zenz T, Rossi M, Döhner K et al. Gene mutations and treatment outcome in chronic lymphocytic leukemia: results from the CLL8 trial. Blood 2014; 123: 3247–3254.
Tam CS, Stilgenbauer S . How best to manage patients with chronic lymphocytic leuekmia with 17p deletion and/or TP53 mutation? Leuk Lymphoma 2015; 56: 587–593.
Bejar R, Stevenson KE, Caughey B, Lindsley RC, Mar BG, Stojanov P et al. Somatic mutations predict poor outcome in patients with myelodysplastic syndrome after hematopoietic stem-cell transplantation. J Clin Oncol 2014; 32: 2691–2698.
Jadersten M, Saft L, Smith A, Kulasekararaj A, Pomplun S, Göhring G et al. TP53 mutations in low-risk myelodysplastic syndromes with del(5q) predict disease progression. J Clin Oncol 2011; 29: 1971–1979.
Xu J, Wang J, Hu Y, Qian J, Xu B, Chen H et al. Unequal prognostic potentials of p53 gain-of-function mutations in human cancers associate with drug-metabolizing activity. Cell Death Dis 2014; 5: e1108.
Joerger AC, Fersht AR . The tumor suppressor p53: from structures to drug discovery. Cold Spring Harb Perspect Biol 2010; 2: a000919.
Haferlach C, Dicker F, Herholz H, Schnittger S, Kern W, Haferlach T . Mutations of the TP53 gene in acute myeloid leukemia are strongly associated with a complex aberrant karyotype. Leukemia 2008; 22: 1539–1541.
Fernandez-Mercado M, Burns A, Pellagatti A, Giagounidis A, Germing U, Agirre X et al. Targeted re-sequencing analysis of 25 genes commonly mutated in myeloid disorders in del(5q) myelodysplastic syndromes. Haematologica 2013; 98: 1856–1864.
Rossi D, Khiabanian H, Spina V, Ciardullo C, Bruscaggin A, Famà R et al. Clinical impact of small TP53 mutated subclones in chronic lymphocytic leukemia. Blood 2014; 123: 2139–2147.
Acknowledgements
We thank all co-workers at the MLL Munich Leukemia Laboratory for their technical assistance. We thank all physicians for providing and caring for patients as well as collecting the data.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
CH, WK and TH declare part ownership of MLL Munich Leukemia Laboratory. AS, MM and AF are employed by MLL Munich Leukemia Laboratory.
Additional information
Supplementary Information accompanies this paper on the Leukemia website
Rights and permissions
About this article

Cite this article
Stengel, A., Kern, W., Haferlach, T. et al. The impact of TP53 mutations and TP53 deletions on survival varies between AML, ALL, MDS and CLL: an analysis of 3307 cases. Leukemia 31, 705–711 (2017). https://doi.org/10.1038/leu.2016.263
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/leu.2016.263
This article is cited by
-
Prevalence, causes and impact of TP53-loss phenocopying events in human tumors
BMC Biology (2023)
-
Accurate interpretation of p53 immunohistochemical patterns is a surrogate biomarker for TP53 alterations in large B-cell lymphoma
BMC Cancer (2023)
-
Combined inhibition of BCL-2 and MCL-1 overcomes BAX deficiency-mediated resistance of TP53-mutant acute myeloid leukemia to individual BH3 mimetics
Blood Cancer Journal (2023)
-
Somatic TP53 single nucleotide variants, indels and copy number alterations in chronic myelomonocytic leukemia (CMML)
Leukemia (2023)
-
Deep genomic characterization highlights complexities and prognostic markers of pediatric acute myeloid leukemia
Communications Biology (2023)
