
Abstract
Sertoli–Leydig cell tumors are characterized by the presence of somatic DICER1 hotspot mutations. In this study, we sought to define the association between DICER1 hotspot mutations and different morphologic subtypes of ovarian Sertoli–Leydig cell tumors. Furthermore, we aimed to assess whether DICER1 hotspot mutations occur in other ovarian sex cord-stromal tumors, testicular sex cord-stromal tumors, or other female genital tract tumors with rhabdomyosarcomatous differentiation. We subjected a series of ovarian Sertoli–Leydig cell tumors (n=32), Sertoli cell tumors (n=5) and gynandroblastomas (n=5), testicular sex cord-stromal tumors (n=15) and a diverse group of female genital tract tumors with rhabdomyosarcomatous morphology (n=10) to DICER1 hotspot mutation analysis using Sanger sequencing. We also tested two gynandroblastomas for the presence of FOXL2 hotspot mutations (p.C134W; c.402C>G). Twenty of 32 (63%) Sertoli–Leydig cell tumors harbored a DICER1 hotspot mutation, of which 80% had the p.E1705K mutation. No association was found between DICER1 mutation status and the presence of heterologous or retiform differentiation in Sertoli–Leydig cell tumors. DICER1 mutations were found at similar frequencies in gynandroblastoma (2/5; 40%) and ovarian Sertoli cell tumors (5/8; 63%; P>0.1), and all mutated tumors harbored a p.E1705K mutation. DICER1 hotspot mutations were also identified in a single cervical rhabdomyosarcoma and in the rhabdomyosarcomatous component of a uterine carcinosarcoma. No DICER1 mutations were detected in testicular sex cord-stromal tumors. Two DICER1 wild-type gynandroblastomas harbored a p.C134W FOXL2 hotspot mutation in both tumor components. In this study we confirmed that DICER1 hotspot mutations occur in over half of ovarian Sertoli–Leydig cell tumors, and are unrelated to tumor differentiation. We also widened the spectrum of ovarian sex cord-stromal tumors with sertoliform differentiation, in which DICER1 mutations are known to occur, to include Sertoli cell tumors and gynandroblastomas. Our results suggest that DICER1 mutations may not have a role in testicular sex cord-stromal tumorigenesis.
Similar content being viewed by others

Main
Ovarian sex cord-stromal tumors are a heterogeneous group of benign or malignant neoplasms. Several subtypes of sex cord-stromal tumors are known, including adult and juvenile granulosa cell tumors, Sertoli–Leydig cell tumor, Sertoli cell tumor, gynandroblastoma and sex cord tumor with annular tubules.1 Classical Sertoli–Leydig cell tumors demonstrate a range of histologic appearances, ranging from well-differentiated with tubule formation within a delicate fibromatous stroma, to tumors with intermediate differentiation with corded or trabecular morphology, and poorly differentiated tumors that often resemble a high-grade sarcoma and can only be recognized if a well- or intermediately differentiated component is present.1, 2 Leydig cells can usually be readily identified in both well- and intermediately differentiated classical Sertoli–Leydig cell tumors, but may be scarce or absent in poorly differentiated tumors.2 In addition, a subset of Sertoli–Leydig cell tumors can demonstrate retiform or heterologous differentiation, including gastrointestinal-type (most common) or rhabdomyosarcomatous differentiation.2 Sertoli cell tumors are often well-differentiated with tubule formation but by definition lack Leydig cells in contrast to Sertoli–Leydig cell tumors.3 Gynandroblastomas are unusual ovarian sex cord tumors that demonstrate combined features of Sertoli–Leydig cell tumor and granulosa cell tumor, whereas sex cord tumors with annular tubules are frequently associated with Peutz–Jeghers syndrome and have a very classic morphology with prominent annular tubules.4 Testicular Sertoli cell tumors are rare neoplasms, constituting <1% of all testicular tumors.5 Although the majority of testicular Sertoli cell tumors are sporadic, some tumors are associated with Peutz–Jeghers syndrome and Carney syndrome.6, 7
Over recent years, massively parallel sequencing studies have unraveled the molecular underpinning of ovarian sex cord-stromal tumors. The majority of ovarian adult granulosa cell tumors have been shown to harbor somatic FOXL2 mutations,8, 9 whereas somatic DICER1 mutations have been identified in ~60% of Sertoli–Leydig cell tumors10 and, more rarely, in other tumor types including embryonal rhabdomyosarcomas and ovarian and testicular germ cell tumors, such as yolk sac tumors and teratomas.10, 11 These somatic DICER1 missense mutations affect several ‘hotspots’ in the metal-binding domain of the RNase IIIb subunit of the gene. In contrast, germline DICER1 mutations associated with the DICER1 syndrome, which confers risk for multiple tumor types including pleuropulmonary blastoma, cystic nephroma, ovarian Sertoli–Leydig cell tumor and childhood embryonal rhabdomyosarcoma,12, 13 are mainly truncating and spread across the gene. DICER1 encodes an RNA endoribonuclease that has a key role in the regulation of gene expression through the production of mature microRNAs (miRNAs).14
To date, the association between morphologic subtypes of ovarian Sertoli–Leydig cell tumor and the presence of somatic DICER1 mutations remains unclear, and it has yet to be established whether other ovarian sex cord-stromal tumors such as Sertoli cell tumor, gynandroblastoma and sex cord tumor with annular tubules may also harbor DICER1 hotspot mutations. In addition, only four testicular Sertoli cell tumors and a single testicular sex cord-stromal tumor not otherwise specified have been analyzed for the presence of somatic DICER1 mutations to date, all of which were reported to be wild-type.10
In this study, we aimed to define (i) the association between DICER1 hotspot mutations and ovarian Sertoli–Leydig cell tumor morphology, (ii) whether DICER1 hotspot mutations occur in other subtypes of ovarian sex cord-stromal tumor or in testicular sex cord-stromal tumors, (iii) whether FOXL2 mutations occur in DICER1 wild-type gynandroblastomas, and (iv) whether other female genital tract tumors with rhabdomyosarcomatous morphology harbor DICER1 hotspot mutations.
Material and methods
Case Selection
Representative formalin-fixed paraffin-embedded (FFPE) sections of ovarian Sertoli–Leydig cell tumors (n=32), gynandroblastomas (n=5), and Sertoli cell tumors (n=8, including one lipid-rich variant Sertoli cell tumor) were retrieved from the pathology files of the participating institutions, as well as 10 female genital tumors with rhabdomyosarcomatous differentiation (including 2 pediatric cervical embryonal rhabdomyosarcomas, 4 adult uterine rhabdomyosarcoma (cervix and corpus) and 4 adult uterine carcinosarcomas with heterologous rhabdomyosarcomatous differentiation) and 15 testicular sex cord-stromal tumors (including 8 classical Sertoli cell tumors, 3 Sertoli cell tumor large cell calcifying variants and 4 sex cord-stromal tumors not otherwise specified). All tumors were reviewed by two expert gynecologic pathologists (EO, RAS), who characterized the various subtypes of Sertoli–Leydig cell tumor, and the degree of differentiation of the tumors. In four Sertoli–Leydig cell tumors (two poorly differentiated with rhabdomyosarcoma, one poorly differentiated with both rhabdomyosarcoma and retiform features and one with intermediate differentiation and both gastrointestinal-type heterologous and retiform features), two separate tumor blocks were available for testing; in all remaining cases, tissue obtained from one tumor block was used for DNA extraction and Sanger sequencing analysis. This study was approved by the local ethics committees from the authors’ institutions.
Microdissection and Nucleic Acid Extraction
Eight-μm-thick sections of representative FFPE blocks of the tumor tissue were stained with nuclear fast red and microdissected using a sterile needle under a stereomicroscope to ensure >80% tumor cell content as previously described.15 In three of the five gynandroblastomas, only the sertoliform component of the tumor was available for sectioning and microdissection. In the remaining two cases, both morphologically distinct components (granulosa cell tumor-like areas and Sertoli–Leydig cell tumor-like areas) were microdissected, and the extracted DNA was subjected to DICER1 mutation testing separately. Genomic DNA was extracted from microdissected tumor tissue using the DNeasy Blood & Tissue Kit (Qiagen) and quantified using the Qubit Fluorometer assay (Invitrogen, Life Technologies) as previously described.16 DNA of sufficient quantity and quality for Sanger sequencing analysis was obtained from all cases included in this study.
PCR Amplification and Sanger Sequencing
PCR amplification of 10 ng of genomic DNA of each case was performed using the AmpliTaq 360 Master Mix Kit (Life Technologies) on a Veriti Thermal Cycler (Life Technologies) as previously described.16 Two primer pairs, one that amplifies a 200 bp fragment encompassing exon 24 of the DICER1 gene (5′-CCGCAGGTCTGTCAGGAC-3′ (forward) and 5′-CTTCTTCGGATTTGGGGATC-3′ (reverse)), and one primer pair that amplifies a 164 bp fragment, encompassing exon 25 of the DICER1 gene (5′-CTATTAGTGGCCGCATCATG-3′ (forward) and 5′-GCTTAGGAGATCTGAGGAGGATG-3′ (reverse)), were employed. In addition, two gynandroblastomas were tested for the presence of the c.402C>G (p.C134W) FOXL2 hotspot mutation utilizing a primer pair that amplifies a 279 bp fragment encompassing the site of the c.402C>G mutation of the FOXL2 gene (5′CCTCAACGAGTGCTTCATCA3′ (forward), 5′AGGAAGCCAGACTGCAGGTAC3′ (reverse)). In these two cases, the morphologically distinct tumor components were microdissected, and the extracted DNA was subjected to FOXL2 mutation testing separately.
For all 3 primer pairs used, the PCR thermocycling protocol consisted of an initial incubation step of 95 °C for 5 min, followed by 35 cycles of 95 °C for 30 s, 60 °C for 30 s, 72 °C for 30 s, and a final extension step of 72 °C for 10 min. The PCR fragments were purified with ExoSAP-IT (Affymetrix), and the sequencing reactions were performed on an ABI 3730 capillary sequencer using the ABI BigDye Terminator chemistry (v3.1) (Life Technologies) according to manufacturer’s instructions. All PCR and sequencing reactions were performed in duplicate. Sequences of the forward and reverse strands were analyzed using the MacVector software (MacVector) and DNAStarLaserGene software as previously described.16
Results
Ovarian Sertoli–Leydig Cell Tumors
Histologic review of the Sertoli–Leydig cell tumors (n=32) included in this study revealed 2 well-differentiated tumors, 20 showed intermediate differentiation (including 4 with heterologous gastrointestinal-type morphology and 6 with retiform differentiation) and 10 were poorly differentiated (including 4 with rhabdomyosarcoma). One tumor with rhabdomyosarcoma and one tumor with gastrointestinal-type heterologous morphology also demonstrated focal retiform morphology.
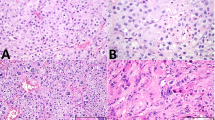
DICER1 hotspot point mutations were identified in 20 of 32 (63%) ovarian Sertoli–Leydig cell tumors (Figure 1, Table 1). DICER1 mutations were found at similar frequencies in poorly differentiated Sertoli–Leydig cell tumors (6/10, 60%) and Sertoli–Leydig cell tumors with intermediate differentiation (14/20, 70%; two-tailed Fisher’s exact test P=0.7). In contrast, both well-differentiated Sertoli–Leydig cell tumors tested were DICER1 wild-type. Furthermore, DICER1 hotspot mutations were identified at similar frequencies in tumors showing heterologous or retiform elements (9/14, 64%) and tumors without these features (11/18, 61%; two-tailed Fisher’s exact test P=1). No statistically significant difference in the prevalence of DICER1 mutations was found between tumors with heterologous (rhabdomyosarcoma or gastrointestinal epithelium) or retiform morphology (two-tailed Fisher’s exact test, P=1) (Table 1), and DICER1-mutant and wild-type examples of each of these Sertoli–Leydig cell tumor subtypes could not be differentiated on morphologic review (Figure 2).

DICER1 and FOXL1 hotspot mutations identified in ovarian Sertoli–Leydig cell tumors and gynandroblastomas. (a) Representative sequence electropherograms of the three most common DICER1 hotspot mutations identified by Sanger sequencing in the ovarian Sertoli–Leydig cell tumors analyzed in this study. (b) Representative sequence electropherogram of the FOXL2 hotspot mutations identified by Sanger sequencing in the gynandroblastomas analyzed in this study.


DICER1 mutation status and morphology in ovarian Sertoli–Leydig cell tumors. (a–d) Representative micrographs of DICER1-mutant tumors; (e–h) representative micrographs of DICER1 wild-type tumors. (a and e) Sertoli–Leydig cell tumors with heterologous gastrointestinal-like morphology, (b and f) Sertoli–Leydig cell tumors with rhabdomyosarcomatous morphology (inset), (c and g) Sertoli–Leydig cell tumors with retiform morphology, (d and h) Poorly differentiated Sertoli–Leydig cell tumors. No association between DICER1 hotspot mutation status and ovarian Sertoli–Leydig cell tumor morphology was observed.
The most frequent DICER1 hotspot mutation identified in the ovarian Sertoli–Leydig cell tumors analyzed was the p.E1705K (c.5113G>A) mutation (16/20 cases, 80%). In the subset of tumors (n=4), where more than one tissue block was available for microdissection and Sanger sequencing analysis, of which two showed exclusively rhabdomyosarcomatous heterologous morphology, the same DICER1 mutation was identified in both tested blocks. One neoplasm with both heterologous morphology and intermediately differentiated classical Sertoli–Leydig cell tumor morphology had a p.E1705K DICER1 mutation in the heterologous component only, while in the other tumor with mixed morphology, the retiform component harbored a p.E1705K DICER1 mutation whereas the rhabdomyosarcoma component was DICER1 wild-type.
Other Ovarian Tumors
Sanger sequencing analysis revealed that 5 of 8 (63%) Sertoli cell tumors and 2 of 5 (40%) gynandroblastomas harbored a DICER1 hotspot mutation (Table 1). Akin to the ovarian Sertoli–Leydig cell tumors, of which 80% harbored a p.E1705K DICER1 hotspot mutation, all 7 DICER1-mutant gynandroblastomas and Sertoli cell tumors had a p.E1705K mutation. In contrast, both a sex cord-stromal tumor not otherwise specified (0/1) and a sex cord tumor with annular tubules (0/1) were found to be DICER1 wild-type. DICER1-mutant Sertoli cell tumors could not be distinguished on morphologic grounds from DICER1 wild-type tumors (Figure 3). In the single lipid-rich variant Sertoli cell tumor tested, a DICER1 p.E1705K mutation was identified.

DICER1 mutation status and morphology in ovarian Sertoli cell tumors. (a and b) Representative micrographs of DICER1-mutant Sertoli cell tumors, (c and d) Representative micrographs of DICER1 wild-type Sertoli cell tumors. No association between DICER1 hotspot mutation status and ovarian Sertoli cell tumor morphology was observed.
FOXL2 mutation testing was performed on two of the three gynandroblastomas with wild-type DICER1; one case did not have sufficient material available for testing. For this analysis, both tumor components, the granulosa cell-like and Sertoli–Leydig cell tumor-like areas, were separately microdissected and subjected to FOXL2 mutation analysis. A p.C134W (c.402C>G) FOXL2 hotspot mutation was identified in both the granulosa cell tumor-like components and the Sertoli–Leydig cell tumor-like areas of the two DICER1 wild-type gynandroblastomas. No morphologic differences were apparent between the DICER1-mutant and wild-type gynandroblastomas, or between DICER1-mutant and FOXL2-mutant gynandroblastomas (Figure 4).

Tumor morphology and DICER1 mutation status in gynandroblastomas. (a and b) Representative micrograph of a DICER1-mutant gynandroblastoma (a, low power, b high power magnification), (c and d) Representative micrographs of the Sertoli–Leydig cell tumor-like component of DICER1 wild-type, FOXL2-mutant gynandroblastomas.
Other Female Genital Tract Tumors with a Rhabdomyosarcomatous Component
Two of the ten female genital tract tumors with rhabdomyosarcomatous differentiation (20%) studied here harbored a DICER1 hotspot mutation: a p.D1709N (c.5125G>A) mutation in a metastasis from a primary cervical rhabdomyosarcoma in an adult patient, and a p.E1813Q (c.5437G>C) mutation in a rhabdomyosarcoma arising in a uterine carcinosarcoma (Table 1). Both of these DICER1 hotspot point mutations were seen at low prevalence in the cohort of ovarian Sertoli–Leydig cell tumors analyzed. Neither of the two pediatric embryonal rhabdomyosarcomas tested was found to harbor a DICER1 hotspot mutation.
Testicular Sex Cord-Stromal Tumors
Fifteen testicular sex cord-stromal tumors including eight classical Sertoli cell tumors, three Sertoli cell tumors-large cell calcifying variant and four sex cord-stromal tumors not otherwise specified were subjected to DICER1 sequencing analysis and none harbored a DICER1 hotspot mutation.
Discussion
We have shown herein that ~60% of the Sertoli–Leydig cell tumors analyzed harbor DICER1 hotspot mutations, and demonstrated that there is no clear association between either the degree of tumor differentiation or the presence of heterologous or retiform differentiation in Sertoli–Leydig cell tumor and DICER1 hotspot mutation status. We have further shown that DICER1 hotspot mutations occur at similar frequencies in ovarian Sertoli cell tumors and gynandroblastomas, providing additional evidence to demonstrate that DICER1 hotspot mutation is a frequent genomic event across the spectrum of ovarian sex cord-stromal tumors with Sertoliform differentiation. In contrast, we have observed that testicular Sertoli cell tumors appear to lack hotspot mutations in DICER1. Finally, we have shown that DICER1 hotspot mutations do occur in female genital tract tumors with rhabdomyosarcomatous differentiation, although the true prevalence of this mutation in these tumors remains to be defined.
The overall frequency of DICER1 mutations in Sertoli–Leydig cell tumor in this cohort (63%) is similar to that reported by Heravi–Moussavi (26/43, 60%),10 but distinct from the recent findings by Goulvent et al9 who reported the presence of DICER1 mutations in only one-third of the Sertoli–Leydig cell tumors tested (6/19, 32%). It should be noted, however, that although the prevalence of DICER1 hotspot mutations in the ovarian Sertoli–Leydig cell tumors studied here and those reported by Heravi–Moussavi et al10 was similar, there was a difference in the prevalence of the residues in the Dicer RNase IIIb domain affected by mutations. Although Heravi–Moussavi et al10 found the p.D1709N (c.5125G>A) DICER1 hotpot mutation to be most common in ovarian Sertoli–Leydig cell tumors (10/26, 38%), in our study this mutation was identified in only two cases (2/20, 10%), whereas the p.E1705K (c.5113G>A) mutation was seen in the majority (80%) of DICER1-mutant tumors. To date, all three well-differentiated Sertoli–Leydig cell tumors reported, including two in this study and a single case analyzed by Goulvent et al9, were found to be DICER1 wild-type. Larger studies will be required to determine the true prevalence of DICER1 mutations in well-differentiated Sertoli–Leydig cell tumors.
The absence of a clear association between the presence of rhabdomyosarcoma within Sertoli–Leydig cell tumor and DICER1 mutation status may be somewhat unexpected given the reported frequency of rhabdomyosarcomtous tumors in the familial DICER1 mutation syndrome,12, 13 and the identification of two DICER1 hotspot mutations in the female genital tract tumors with rhabdomyosarcomatous elements (2/10, 20%), including one uterine carcinosarcoma, reported here.
Studies to date have suggested that the acquisition of somatic DICER1 mutation is a markedly rare event in epithelial cancer. Review of databases such as 1000 Genomes, The Cancer Genome Atlas (TCGA) and COSMIC by Heravi–Moussavi et al10 showed no DICER1 hotspot mutations, while Slade et al13 found DICER1 mutations in only 4 of 781 cancer cell lines, all of which occurred in microsatellite unstable tumors, where it was unlikely to constitute a driver mutation. Heravi–Moussavi et al10 identified a single somatic DICER1 mutation in only one carcinosarcoma of ovarian origin among 266 female genital tract (ovarian and endometrial) cancers. The authors did not specify, however, whether a heterologous rhabdomyosarcomatous component was present in this tumor. We performed a re-analysis of the endometrial cancer TCGA data set17 and found that among 248 endometrial cancers subjected to massively parallel sequencing, 6 (2.4%) harbored mutations affecting DICER1 hotspot residues (www.cBioPortal.org, accessed May 2015;18 Supplementary Figure 1). Of these, two were MSI-hypermutated (microsatellite unstable) endometrioid endometrial carcinomas, and two were POLE ultramutated endometrioid endometrial carcinomas, consistent with the findings by Slade et al.13 In the remaining two cases, a somatic p.E1813A DICER1 hotspot mutation was found in a copy-number high TP53-mutant serous carcinoma, and a somatic p.L539fs mutation and p.D1810A DICER1 hotspot mutation was also seen in an endometrioid carcinoma of copy-number low genomic subtype (low microsatellite instability), which also harbored characteristic PTEN and CTNNB1 mutations (Supplementary Figure 1). No DICER1 hotspot mutations were identified in the high-grade serous ovarian carcinomas reported by the TCGA.19
We have observed that several subtypes of ovarian sex cord-stromal tumors (including Sertoli cell tumor and gynandroblastoma) harbored a DICER1 hotspot mutation, which may be expected given their postulated common cell lineage. Before this study, the DICER1 mutation status of only two ovarian Sertoli cell tumors had been reported9 and both were DICER1 wild-type. In contrast, we have observed that ovarian Sertoli cell tumors and Sertoli–Leydig cell tumors harbor DICER1 mutations at similar frequencies (63% in both tumor categories). In the five gynandroblastomas tested, only two harbored a DICER1 hotspot mutation. Interestingly, however, two of the DICER1 wild-type tumors had the FOXL2 hotspot mutation previously reported to be present in the vast majority of adult-type granulosa cell tumors.8, 20 These results suggest that the gynandroblastoma category may encompass two distinct entities at the genetic level—those with a granulosa cell tumor-like genotype characterized by FOXL2 hotspot mutations and those with a Sertoli–Leydig cell tumor-like DICER1-mutant genotype. These findings are consistent with those of Goulvent et al,9 who demonstrated that DICER1 and FOXL2 mutations appear to be mutually exclusive in ovarian sex cord-stromal tumors. In the current study, however, DICER1-mutant gynandroblastomas could not be distinguished from those harboring FOXL2 mutations based on morphologic grounds, and further studies are warranted to establish the clinical significance of the molecular findings.
In murine models, Dicer has been shown to have a vital role in fertility in both ovary and testis. In the ovary, Dicer is expressed in both the oocyte and follicular granulosa cells,21 and reduced Dicer expression in granulosa cells is associated with reduced ovulation rate.22, 23, 24 Yuan et al25 demonstrated that loss of Dicer expression in the developing oocyte leads to compromised folliculogenesis, infertility and loss of ovarian function. Likewise, Dicer function is vital for both testicular Sertoli cell development and function26 and in spermatogenesis.27 In light of the importance of Dicer in gonadal function in both sexes and following the recent discoveries in ovarian sex cord-stromal tumors, we hypothesized that mutations affecting DICER1 would have a role in testicular Sertoli cell tumor oncogenesis. The results of our study do not support this hypothesis, however, but rather suggest that the pathogenesis of ovarian and testicular sex cord-stromal tumors is distinct. This conclusion is further supported by the fact that testicular Sertoli cell tumors have not been reported as part of the spectrum of the germline DICER1 mutation syndrome. Combining the results of the mutational analysis of the current 15 testicular Sertoli cell tumors with those reported by Heravi–Moussavi et al,10 20 testicular Sertoli cell tumors and sex cord-stromal tumors not otherwise specified have now been tested for the presence of DICER1 hotspot mutations, and all have been found to be DICER1 wild-type. To date, molecular studies of testicular sex cord-stromal tumors have focused on syndromic associations, such as Peutz–Jeghers syndrome, Carney Syndrome or androgen-insensitivity syndromes6, 7, 28, 29 and sporadic cases involving somatic mutations within the same genes.30 More recently, Perrone et al31 have demonstrated the presence of CTNNB1 (β-catenin) mutations in 71% (10 of 14) of testicular Sertoli cell tumors. Based on these observations, further studies are required to define the molecular underpinning of testicular Sertoli cell tumors, and to establish whether Dicer or other members of the miRNA machinery may be deregulated by mechanisms other than DICER1 hotspot mutations in this disease.
In summary, our study has confirmed the presence of DICER1 hotspot mutations in the majority of ovarian Sertoli–Leydig cell tumors. We have further demonstrated that there is no clear correlation between tumor differentiation, or the presence of retiform or heterologous elements in ovarian Sertoli–Leydig cell tumors, and DICER1 hotspot mutation status. In addition, we have shown that DICER1 hotspot mutations are also present in ovarian Sertoli cell tumors and gynandroblastomas, and that a proportion of DICER1 wild-type gynandroblastomas may harbor FOXL2 hotspot mutations. Finally, our results provide evidence to suggest that testicular sex cord-stromal tumors lack hotspot mutations in DICER1. Although the recognition of recurrent DICER1 mutation in various subtypes of ovarian sex cord-stromal tumors represents a major advance in our understanding of these tumors, the mechanisms underlying tumorigenesis in the presence of DICER1 mutations remain to be elucidated.
References
Kurman RJ, Carcangiu ML, Herrington CS (eds). Tumours of the ovary. In: WHO classification of tumours of female reproductive organs. IARC: Lyon, 2014, pp 11–86.
Young RH, Scully RE . Ovarian Sertoli-Leydig cell tumors. A clinicopathological analysis of 207 cases. Am J Surg Pathol 1985;9:543–569.
Oliva E, Alvarez T, Young RH . Sertoli cell tumors of the ovary: a clinicopathologic and immunohistochemical study of 54 cases. Am J Surg Pathol 2005;29:143–156.
Young RH, Welch WR, Dickersin R et al. Ovarian sex cord tumor with annular tubules. Review of 74 cases including 27 with Peutz-Jeghers Syndrome and 4 with adenoma malignum of the cervix. Cancer 1982;50:1384–1402.
Eble JN, Sauter G, Epstein JI (eds). Tumours of the testis and paratesticular tissue. In: World Health Organization classification of tumours. Pathology and genetics of tumours of the urinary system and male genital organs. IARC: Lyon, 2004, pp 217–278.
Young RH, Koelliker DD, Scully RE . Sertoli cell tumors of the testis, not otherwise specified: a clinicopathologic analysis of 60 cases. Am J Surg Pathol 1998;22:709–721.
Washecka RM, Mariani AJ, Honda SA . Testicular tumors in Carney’s complex. J Urol 2002;167:1299–1302.
Shah SP, Kobel M, Senz J et al. Mutation of FOXL2 in granulosa-cell tumors of the ovary. N Engl J Med 2009;360:2719–2729.
Goulvent T, Ray-Coquard I, Borel S et al. DICER1 and FOXL2 mutations in ovarian sex cord-stromal tumors: a GINECO group study. Histopathology 2015, e-pub ahead of print 1 June 2015; doi:10.1111/his.12747.
Heravi-Moussavi A, Anglesio MS, Cheng SW et al. Recurrent somatic DICER1 mutations in nonepithelial ovarian cancers. N Engl J Med 2012;366:234–242.
Witkowski L, Mattina J, Schonberger S et al. DICER1 hotspot mutations in non-epithelial gonadal tumors. Br J Cancer 2013;109:2744–2750.
Hill DA, Ivanovich J, Priest JR et al. DICER1 mutations in familial pleuropulmonary blastoma. Science 2009;325:965.
Slade I, Bacchelli C, Davies H et al. DICER1 syndrome: clarifying the diagnosis, clinical features and management implications of a pleiotropic tumor predisposition syndrome. J Med Genet 2011;48:273–278.
Foulkes WD, Priest JR, Duchaine TF . DICER1: mutations, microRNAs and mechanisms. Nat Rev Cancer 2014;14:662–672.
Geyer FC, Lacroix-Triki M, Colombo PE et al. Molecular evidence in support of the neoplastic and precursor nature of microglandular adenosis. Histopathology 2012;60:E115–E130.
Weinreb I, Piscuoglio S, Martelotto LG et al. Hotspot activating PRKD1 somatic mutations in polymorphous low-grade adenocarcinomas of the salivary glands. Nat Genet 2014;46:1166–1169.
Cancer Genome Atlas Research Network. Integrated genomic characterization of endometrial carcinoma. Nature 2013;497:67–73.
Gao J, Aksoy BA, Dogrusoz U et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal 2013;6:pl1.
Cancer Genome Atlas Research Network. Integrated genomic analyses of ovarian carcinoma. Nature 2011;474:605–615.
Oost EE, Charles A, Choong CS et al. Ovarian sex cord-stromal tumors in patients with probable or confirmed germline DICER1 mutations. Int J Gynecol Pathol 2015;34:266–274.
Lei L, Jin S, Gonzalez G et al. The regulatory role of Dicer in folliculogenesis in mice. Mol Cell Endocrinol 2010;315:63–73.
Hong X, Luense LJ, McGinnis LK et al. Dicer1 is essential for female fertility and normal development of the female reproductive system. Endocrinology 2008;149:6207–6212.
Nagaraja AK, Andreu-Vieyra C, Franco HL et al. Deletion of Dicer in somatic cells of the female reproductive tract causes sterility. Mol Endocrinol 2008;22:2336–2352.
Otsuka M, Zheng M, Hayashi M et al. Impaired microRNA processing causes corpus luteum insufficiency and infertility in mice. J Clin Invest. 2008;118:1944–1954.
Yuan S, Ortogero N, Wu Q et al. Murine follicular development requires oocyte DICER, but not DROSHA. Biol Reprod 2014;91:39.
Kim GJ, Georg I, Scherthan H et al. Dicer is required for Sertoli cell function and survival. Int J Dev Biol 2010;54:867–875.
Papaioannou MD, Pitetti JL, Ro S et al. Sertoli cell Dicer is essential for spermatogenesis in mice. Dev Biol 2009;326:250–259.
Rutgers SL, Scully RE . Pathology of the testis in intersex syndromes. Semin Diagn Pathol 1987;4:275–291.
Gourgari E, Saloustros E, Stratakis CA . Large-cell calcifying Sertoli cell tumors of the testes in pediatrics. Curr Opin Pediatr 2012;24:518–522.
Tanwar PS, Kaneko-Tarui T, Zhang L et al. Altered LKB1/AMPK/TSC1/TSC2/mTOR signaling causes disruption of Sertoli cell polarity and spermatogenesis. Hum Mol Genet 2012;21:4394–4405.
Perrone F, Bertolotti A, Montemurro G et al. Frequent mutation and nuclear localization of b-catenin in Sertoli cell tumors of the testis. Am J Surg Pathol 2014;38:66–71.
Acknowledgements
We thank Russell Towers (MSKCC) for technical support. AMS is funded by a stipend from the German Cancer Aid (Dr Mildred Scheel Stiftung) and SP by a Susan G Komen Postdoctoral Fellowship Grant (PDF14298348). Research reported in this publication was supported in part by the Cancer Center Support Grant of the National Institutes of Health/National Cancer Institute under award number P30CA008748. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on Modern Pathology website
Supplementary information
Rights and permissions
About this article

Cite this article
Conlon, N., Schultheis, A., Piscuoglio, S. et al. A survey of DICER1 hotspot mutations in ovarian and testicular sex cord-stromal tumors. Mod Pathol 28, 1603–1612 (2015). https://doi.org/10.1038/modpathol.2015.115
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/modpathol.2015.115
This article is cited by
-
DICER1 tumor predisposition syndrome: an evolving story initiated with the pleuropulmonary blastoma
Modern Pathology (2022)
-
Clinicopathologic and molecular spectrum of testicular sex cord-stromal tumors not amenable to specific histopathologic subclassification
Modern Pathology (2022)
-
Novel insights into the mixed germ cell-sex cord stromal tumor of the testis: detection of chromosomal aneuploidy and further morphological evidence supporting the neoplastic nature of the germ cell component
Virchows Archiv (2020)
-
Genomic profiling of primary and recurrent adult granulosa cell tumors of the ovary
Modern Pathology (2020)
-
Mesenteric extraovarian Sertoli-Leydig cell tumor without DICER1 hotspot mutation: a case report
Diagnostic Pathology (2019)

{kind=link}