
Abstract
The majority of penile squamous cell carcinomas is caused by transforming human papilloma virus (HPV) infection. The etiology of HPV-negative cancers is unclear, but TP53 mutations have been implicated. Archival tissues of 108 invasive squamous cell carcinoma from a single pathology institution in a low-incidence area were analyzed for HPV-DNA and p16ink4a overexpression and for TP53 mutations by ion torrent next-generation sequencing. Library preparation failed in 32/108 squamous cell carcinomas. Institutional review board approval was obtained. Thirty of 76 squamous cell carcinomas (43%; average 63 years) were HPV-negative with 8/33 squamous cell carcinomas being TP53 wild-type (24%; average 63 years). Twenty-five of 33 squamous cell carcinomas (76%; average 65 years) showed 32 different somatic TP53 mutations (23 missense mutations in exons 5–8, 6 nonsense, 1 frameshift and 2 splice-site mutations). Several hotspot mutations were detected multiple times (R175H, R248, R282, and R273). Eighteen of 19 squamous cell carcinomas with TP53 expression in immunohistochemistry had TP53 mutations. Fifty percent of TP53-negative squamous cell carcinomas showed mostly truncating loss-of-function TP53 mutations. Patients without mutations had longer survival (5 years: 86% vs 61%; 10 years: 60% vs 22%), but valid clinically relevant conclusions cannot be drawn due to different tumor stages and heterogeneous treatment of the cases presented in this study. Somatic TP53 mutations are a common feature in HPV-negative penile squamous cell carcinomas and offer an explanation for HPV-independent penile carcinogenesis. About half of HPV-negative penile cancers are driven by oncogenic activation of TP53, while a quarter is induced by loss of TP53 tumor suppressor function. Detection of TP53 mutations should be carried out by sequencing, as immunohistochemical TP53 staining could not identify all squamous cell carcinomas with TP53 mutations.
Similar content being viewed by others


Main
Penile cancers are mostly squamous cell carcinomas. They are common in South American and African countries, the so-called high-incidence countries, and rare in the North American and European countries, referred to as low-incidence countries for penile cancers. Initial publications dealt with penile cancers from high-incidence countries and concentrated on histological features of penile squamous cell carcinomas.1, 2 Subsequent investigations focused on HPV-induced penile carcinogenesis.3, 4, 5 Only during the past years were HPV-negative penile cancers recognized and cancers from low-incidence countries investigated.6 Among these two mutually exclusive etiologic groups HPV-induced penile carcinogenesis with the precursor lesion penile intraepithelial neoplasia is well understood. Interaction of oncogenes E6 and E7 with cell cycle proteins leads to autonomous cell proliferation and nuclear accumulation of cyclin-dependent kinase inhibitor p16ink4a.7, 8 Immunohistochemical demonstration of p16ink4a overexpression is considered an indirect marker for a transcriptionally active (transforming) HPV-high-risk infection.5, 9, 10 In contrast, HPV-negative penile carcinogenesis is poorly understood. These cancers arise via the precursor lesion differentiated penile intraepithelial neoplasia11, 12, 13, 14, 15 and often are associated with the inflammatory skin diseases lichen sclerosus and lichen planus. HPV-negative penile squamous cell carcinomas and differentiated penile intraepithelial neoplasias lack p16ink4a overexpression but commonly show nuclear p53 expression in proliferating tumor cells.16, 17 Immunohistochemical p53 positivity has been linked to TP53 mutations18 similar to vulvar carcinogenesis.19, 20 Treatment for penile squamous cell carcinomas is presently surgical, followed by adjuvant chemo- and/or radiation therapy or primary chemoradiation in large inoperable squamous cell carcinomas. Owing to the rarity of these tumors, treatment schemes do not differ according to etiololgy. Prognosis of HPV-negative penile squamous cell carcinomas has been described to be worse than that of HPV-induced penile squamous cell carcinomas.18 Radiation and chemotherapy resulted in better regression, longer disease-free survival, and better overall survival not only in HPV-induced penile squamous cell carcinomas but also in HPV-induced squamous cell carcinomas of the head and neck area.21 There are few publications on TP53 mutations in penile cancers in the literature and data from a Japanese study are conflicting with respect to correlation of p53 immunohistochemistry and TP53 mutations.22 We were interested if and what type of mutations in the TP53 gene are present in penile cancers from a low-incidence country and if immunohistochemical TP53 staining correlated with the mutational status.
Materials and methods
Squamous cell carcinomas were divided into (i) HPV-induced squamous cell carcinomas based on demonstration of HPV-DNA along with concomitant p16ink4a overexpression and (ii) squamous cell carcinomas arising independent of transforming HPV infections (no p16ink4a overexpression and typically no HPV-DNA; detection of HPV-DNA only in the absence of p16ink4a overexpression is not indicative of transformation), referred to as HPV-negative cancers in this paper. HPV genotyping was performed on formalin-fixed and paraffin-embedded tumor tissue and is based on the principle of reverse hybridization and the SPF10 primer set, which amplifies a 65-bp region in the L1 open reading frame of the HPV genome (INNO-LiPA HPV GENOTYPING EXTRA; Innogenetics Diagnostic, Heiden, Germany and CHIPRON GmbH, Berlin, Germany). Multiplex PCR and duplex hybridization allows detection of DNA of 32 HPV subtypes including HPV-high risk subtypes 16, 18, 31, 33, 35, 39, 45, 51, 52, 56, 58, and 59; HPV-low risk subtypes 6 and 11; and potentially carcinogenic HPV subtypes 26, 30, 34, 53, 66, 67, 68, 69, 70, 73, 82, 85, and 97. DNA was extracted on a Maxwell, MDx Research System (Promega, Fitchburg, WI, USA). Next-generation sequencing libraries were prepared using the AmpliSeq Library Kit 2.0 (Thermo Fisher Scientific) and the Ion Ampliseq TP53 primer pool covering all exons of the TP53 gene. Sequencing was performed on an Ion Proton benchtop sequencer (Thermo Fisher Scientific) to a length of 200 bp. Initial data analysis was performed using the Ion Torrent Suite Software (Thermo Fisher Scientific, open source, GPL, https://github.com/iontorrent/). Briefly, this included base calling, alignment to the reference genome (hg19) using the TMAP mapper and variant calling by a modified diBayes approach taking into account the flow space information. Called variants were annotated using open source software ANNOVAR23 and SnpEff.24 All coding, nonsynonymous mutations were further evaluated and visually inspected in IGV (http://www.broadinstitute.org/igv/) and variant calls resulting from technical read errors or sequence effects were excluded from the analysis. All annotations of TP53 mutations are based on reference transcript NM_000546.
p16ink4a overexpression was analyzed immunohistochemically with antibody to p16ink4a (Roche MTM Laboratories, Heidelberg, Germany) and defined as homogeneous continuous strong nuclear and cytoplasmic staining of the entire dysplastic epithelium, beginning in the basal and parabasal cells and extending continuously to the superficial layers of the dysplastic epithelium.9 Discontinuous and patchy staining—independent of the percentage of positivity—was interpreted as negative.25 All squamous cell carcinomas were evaluated for p53 expression with antibody clone DO-7 (Dako, Denmark). P53 staining was interpreted as negative when none or only individual basal keratinocyte nuclei showed a reaction. A continuous strong nuclear staining of basal keratinocyte nuclei in differentiated penile intraepithelial neoplasia was interpreted as positive. In invasive squamous cell carcinomas, uniform nuclear staining of proliferating invasive cancer cells was interpreted as homogeneous staining. If a tumor showed focal areas without TP53 expression staining was determined to be heterogeneous.
Results
HPV Genotyping
A total of 108 penile squamous cell carcinomas were genotyped for HPV but 32/108 squamous cell carcinomas could not be analyzed for TP53 mutations by next-generation sequencing due to poor DNA quality and/or low tumor cell content and were thus excluded from the study. Forty-three of 76 squamous cell carcinomas (57%) were classified as HPV-induced squamous cell carcinomas based on detection of HPV-DNA and immunohistochemical p16ink4a overexpression in the tumor tissue (Figures 1a and b). Thirty-three of 76 squamous cell carcinomas (43%) were classified as non-HPV-induced cancers. All non-HPV-induced squamous cell carcinomas lacked p16ink4a overexpression and 32/33 squamous cell carcinomas were devoid of HPV-DNA. One squamous cell carcinoma contained HPV-DNA but in the absence of p16ink4a overexpression (as indirect maker of a transforming infection), this squamous cell carcinomas was classified as non-HPV-induced. Histologically, 10/33 HPV-negative squamous cell carcinomas corresponded to a highly differentiated and/or verrucous G1 tumor, whereas 15/33 squamous cell carcinomas were moderately or poorly differentiated (G2–3). Three squamous cell carcinomas revealed papillary differentiation and one squamous cell carcinomas each showed focal clear-cell, adenomatous, or basaloid differentiation. The overall average age of the patients with HPV-negative squamous cell carcinomas was 63 years at diagnosis (Table 1).



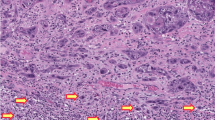
Immunohistochemical staining of penile cancer. (a and b) Hematoxylin and eosin (HE) (a) and p16ink4a (b) staining of a HPV16-positive high-grade penile intraepithelial neoplasia; (c and d) HE (c) and p53 (d) staining of HPV-negative differentiated penile intraepithelial neoplasia; (e and f) HE (e) and p53 (f) staining of invasive HPV-negative squamous cell carcinoma with d-PeIN (g and h) HE (g) and p53 (h) staining of the tumor invasion front of a basaloid HPV-negative squamous cell carcinoma. HPV, human papilloma virus.
TP53 Mutation Status
Next-generaton sequencing revealed that 8/33 squamous cell carcinomas had TP53 wild-type and amino-acid changing mutations were present in the coding region of the TP53 gene in 25/33 HPV-negative squamous cell carcinomas (76%). Nineteen of 25 squamous cell carcinomas revealed only one mutation, and 6/25 squamous cell carcinomas had multiple mutations. A total of 32 mutations were detected of which 23/32 were substitution missense variants, 6/32 were substitution nonsense (ie, stop-gain) variants, 1/32 was a frameshift variant, and 2/32 were located in the splice site of intron 3 or intron 6, respectively (Table 1). All substitution missense mutations were located in exons 5–8 coding for the DNA binding domain of TP53 (Figure 2a). Six of 25 mutated HPV-negative squamous cell carcinomas showed a structural mutation (a frameshift or stop-gain mutation). A stop-gain and a frameshift mutation were found in the transactivation domain and another stop-gain mutation was present in the tetramerization domain (Figure 2a). Among the six squamous cell carcinomas with multiple TP53 mutations, 2/6 squamous cell carcinomas had two different TP53 missense mutations, 2/6 squamous cell carcinomas had a missense and a stop-gain mutation, 1/6 squamous cell carcinoma had a stop-gain mutation and a splice-site mutation in intron 6, and 1/6 squamous cell carcinoma had two missense mutations and an additional mutation in the splice-site acceptor of intron 3 (Table 1). The ‘hotspot’ R175H mutation was the most common mutation and detected five times, mutations at R248 and at R282 were detected three times each and mutations at R273 twice. The distribution of mutation types in penile squamous cell carcinomas is similar to the distribution of TP53 mutations in a cross-section of tumor types (Figure 2b) listed in the COSMIC database.

TP53 mutations in human papilloma virus (HPV)-negative penile squamous cell carcinomas. (a) TP53 cDNA structure and localization of missense (dash) and structural (asterisk) mutations detected in HPV-negative penile squamous cell carcinomas. (b) Distribution of the types of TP53 mutations found in HPV-negative penile squamous cell carcinomas (left) and all cancers (right, data from COSMIC database).
The mean age at diagnosis of men with squamous cell carcinomas harboring TP53 mutation was 63 and 65 years in patients with squamous cell carcinomas lacking TP53 mutations (Table 1). Patients without mutations had longer disease-free survival (5 years: 86% vs 61%; 10 years: 60% vs 22%) but valid clinically relevant conclusions cannot be drawn due to different tumor stages and heterogeneous treatment of the cases presented in this study.
Immunohistochemical p53 Analysis
Immunohistochemistry with antibody to p53 revealed nuclear staining in 19/33 HPV-negative squamous cell carcinomas (Figures 1c–h and Table 1). Twelve of 19 squamous cell carcinomas showed uniform nuclear staining of >90% proliferating tumor cells and 7/19 squamous cell carcinomas showed heterogeneous staining with strongly positive tumor areas alternating with larger areas negative for p53 expression. Eighteen of 19 squamous cell carcinomas harbored TP53 mutations with 17/18 squamous cell carcinomas carrying at least one missense mutation and 1/18 squamous cell carcinoma harboring a loss-of-function mutation (case 8). Among the 14 squamous cell carcinomas without p53 staining, 7/14 squamous cell carcinomas revealed TP53 mutations with 3/7 squamous cell carcinomas being missense mutations and 4/7 squamous cell carcinomas harboring frameshift mutations including two gain-of-function mutations (case 23 and 26). Seven of 14 p53-negative squamous cell carcinomas were devoid of TP53 mutations.
Discussion
In this study, we demonstrate a high incidence (76%) of TP53 mutations in a cohort of HPV-negative penile squamous cell carcinomas from the low-incidence area of southern Austria. This high rate of TP53 mutation in penile cancers strongly suggests a functional role of TP53 in the etiology of penile squamous cell carcinomas. About half of HPV-negative penile cancers were driven by oncogenic activation of TP53, whereas a quarter was induced by loss of TP53 tumor suppressor function possibly in cooperation with additional oncogenic mutations, possibly in PIK3CA, KRAS, and HRAS.26 In our cohort, the etiology of a quarter of HPV-negative penile squamous cell carcinomas was unrelated to TP53 mutations. In HPV-induced carcinogenesis the binding of E6 protein to TP53 inhibits its tumor suppressor function while concurrent activation of RB1 by binding of E7 drives epithelial neoplasia. TP53 function, however, can be restored by radiation and chemotherapy. On the other hand, chemo- and radioresistance are mediated by permanent inactivation of TP53, for example, by mutations of the TP53 coding region since the effectiveness of DNA-damaging anticancer chemotherapeutic agents, such as 5-FU, doxorubicin, bleomycin, and cisplatin, and radiation depend—at least in part—on p53-mediated reponses. This may explain the worse prognosis of HPV-negative squamous cell carcinomas after radiation and chemotherapy with respect to overall survival not only in HPV-negative penile squamous cell carcinomas but also in HPV-negative squamous cell carcinomas of the head and neck area.21
TP53 is the most commonly mutated gene in cancer and has been at the focus of research for several decades. Initially described as a tumor suppressor gene deactivated by loss-of-function mutations, a wealth of evidence has been mounting that mutant TP53 proteins also harbor oncogenic potential.27 The R175H mutation, which is the most common hotspot mutation in TP53 in general and was the most common mutation in this study of penile squamous cell carcinomas, has been described as a conformational mutation leading to cell cycle progression and activation of genes that promote cell survival. R248Q is the most prominent example of a contact mutation, when the amino-acid residues make direct contact with DNA. Arginine 248 is the most commonly mutated amino acid in the TP53 gene and the R248Q mutant has been shown to confer invasive ability when overexpressed in TP53-null cells.28 R248 was mutated three times in our cohort, twice as R248Q and one time as R248W. Mutations at R282, present three times in our cohort, have been associated with higher activity of drug-metabolizing enzymes like CYP3A4,29 whereas R273C, present twice in our cohort, has been implicated in multiple biological pathways like proliferation, inhibition of apoptosis, and metabolism.30 Mutation types in penile squamous cell carcinomas are similar to the distribution of TP53 mutations in a cross-section of different tumor types listed in the COSMIC database. This database also lists 25 penile carcinomas, of which only 10 squamous cell carcinomas (40%) are TP53 mutated with 9 missense mutations and one in-frame codon insertion. The lower incidence of TP53 mutations in this collection of tumors suggests analysis of a mixed group of samples including both HPV-induced and HPV-negative squamous cell carcinomas. The COSMIC database, however, does not include information on HPV status.
Future efforts in individualized medicine and oncology focus on pharmacological therapy targeting tumor cells with TP53 mutations. These may involve agents that activate the remaining WT TP53 allele, degrade or reconstitute mutant p53 protein or reactivate mutant p53 protein and convert it to wild-type p53. Promising candidates are the synthetic lethal drug paclitaxel. Recent advances in research allow targeting p53 pathways at several levels of downstream signaling pathways and metabolic pathways affected by oncogenic p53. Two interesting candidate drugs are the widely prescribed and easily available drug metformin, an antidiabetic drug that increases TAp63 levels, and statins, which anticancer activity are based on inhibition of HMG-Co-A reductase. Other interesting approaches include SAHA, an inhibitor of the histone deacethylase, which triggers degradation of mutated p53, thus inhibiting most of its gain of function.31, 32
Identification of TP53 mutations via immunohistochemical demonstration of p53 staining would be a cost-effective and fast method; however, in our cohort, immunohistochemical staining results of squamous cell carcinomas with antibody to p53 correlated with the presence and absence of TP53 mutations in only 75% of specimens. Immunohistochemistry failed to identify seven cases with TP53 mutations including two gain-of-function mutations (cases 23 and 26).
Some blocks were stored for a long time, and some of the tissue sections were completely negative, even in repeat stains. This was most likely due to loss of antigenicity, particularly in cancers with missence mutations where nuclear p53 accumulation would have been expected. In other cases with completely p53-negative cancers and a positive internal control staining in surrounding differentiated penile intraepithelial neoplasia or associated dermatosis, complete lack of p53 nuclear staining could indeed be considered evidence of abnormality and correlated with disruptive p53 mutations.
Interestingly, loss-of-function mutations were overrepresented in squamous cell carcinomas without p53 staining (4/7) vs 1/19 p53-positive squamous cell carcinoma (Table 1). Failure to detect 53 protein overexpression also can be explained by fixation problems and/or loss of antigenicity in samples stored for prolonged periods of time, which was the case in our patient cohort. Presently, a reliable detection of TP53 mutations and alterations in squamous cell carcinomas can only be achieved by sequencing of the TP53 gene in addition to immunohistochemistry. Larger studies are needed to learn if TP53 mutational status can be correlated with a more aggressive tumor behavior and if it may become a treatment-relevant parameter in the future. Consequently, it is plausible that all patients with penile cancer that are negative for both p16 and TP53 by immunohistochemistry might still carry a TP53 mutation. These patients should thus be referred for TP53 sequencing as the presence of a TP53 mutation could influence therapy and these data will add to the body of evidence pointing to TP53 mutations as a causative event behind HPV-negative penile cancer.
In summary, we demonstrate a high prevalence of TP53 mutations in HPV-negative penile squamous cell carcinomas including several of the well described gain-of-function mutations of TP53 and offers a plausible etiology for the majority of HPV-independent penile squamous cell carcinomas in a representative cohort of penile cancers from a low-incidence country leaving only 10% of penile squamous cell carcinomas with unclear etiology. TP53 mutations are associated with a wide range of biological functions such as confering invasive properties and possibly resistance to treatment. Further studies into the role of oncogenic gain-of-function mutations of TP53 and the possible cooperation of oncogenic mutations in other genes with deleterious TP53 mutations should be performed to further elucidate the network of molecular changes responsible for neoplastic transformation in penile cancer.
References
Velazquez EF, Ayala G, Liu H et al, Histologic grade and perineural invasion are more important than tumor thickness as predictor of nodal metastasis in penile squamous cell carcinoma invading 5 to 10 mm. Am J Surg Pathol 2008; 32: 974–979.
Cubilla AL . The role of pathologic prognostic factors in squamous cell carcinoma of the penis. World J Urol 2009; 27: 169–177.
Rubin MA, Kleter B, Zhou M et al, Detection and typing of human papillomavirus DNA in penile carcinoma: evidence for multiple independent pathways of penile carcinogenesis. Am J Pathol 2001; 159: 1211–1218.
Heideman DAM, Waterboer T, Pawlita M et al, Human papillomavirus-16 is the predominant type etiologically involved in penile squamous cell carcinoma. J Clin Oncol 2007; 25: 4550–4556.
Cubilla AL, Lloveras B, Alejo M et al, Value of p16(INK)4(a) in the pathology of invasive penile squamous cell carcinomas: a report of 202 cases. Am J Surg Pathol 2011; 35: 253–261.
Mannweiler S, Sygulla S, Winter E et al, Two major pathways of penile carcinogenesis: HPV-induced penile cancers overexpress p16ink4a, HPV-negative cancers associated with dermatoses express p53, but lack p16ink4a overexpression. J Am Acad Dermatol 2013; 69: 73–81.
Stankiewicz E, Kudahetti SC, Prowse DM et al, HPV infection and immunochemical detection of cell-cycle markers in verrucous carcinoma of the penis. Mod Pathol 2009; 22: 1160–1168.
Stankiewicz E, Prowse DM, Ktori E et al, The retinoblastoma protein/p16INK4A pathway but not p53 is disrupted by human papillomavirus in penile squamous cell carcinoma: HPV and protein expression in penile SCC. Histopathology 2011; 58: 433–439.
Reuschenbach M, Seiz M, von Knebel Doeberitz C et al, Evaluation of cervical cone biopsies for coexpression of p16INK4a and Ki-67 in epithelial cells. Int J Cancer 2012; 130: 388–394.
Snow AN, Laudadio J . Human papillomavirus detection in head and neck squamous cell carcinomas. Adv Anat Pathol 2010; 17: 394–403.
Chaux A, Velazquez EF, Barreto JE et al, New pathologic entities in penile carcinomas: an update of the 2004 World Health Organization classification. Semin Diagn Pathol 2012; 29: 59–66.
Mannweiler S, Sygulla S, Beham-Schmid C et al, Penile carcinogenesis in a low-incidence area: a clinicopathologic and molecular analysis of 115 invasive carcinomas with special emphasis on chronic inflammatory skin diseases. Am J Surg Pathol 2011; 35: 998–1006.
Oertell J, Caballero C, Iglesias M et al, Differentiated precursor lesions and low-grade variants of squamous cell carcinomas are frequent findings in foreskins of patients from a region of high penile cancer incidence: preputial neoplastic precursor lesions. Histopathology 2011; 58: 925–933.
Velazquez EF, Chaux A, Cubilla AL . Histologic classification of penile intraepithelial neoplasia. Semin Diagn Pathol 2012; 29: 96–102.
Moch H, Cubilla AL, Humphrey PA et al, The 2016 WHO classification of tumours of the urinary system and male genital organs—part a: renal, penile, and testicular tumours. Eur Urol 2016; 70: 93–105.
Yang B, Hart WR . Vulvar intraepithelial neoplasia of the simplex (differentiated) type: a clinicopathologic study including analysis of HPV and p53 expression. Am J Surg Pathol 2000; 24: 429–441.
Chaux A, Pfannl R, Rodríguez IM et al, Distinctive immunohistochemical profile of penile intraepithelial lesions: a study of 74 cases. Am J Surg Pathol 2011; 35: 553–562.
Rocha RM, Ignácio JA, Jordán J et al, A clinical, pathologic, and molecular study of p53 and murine double minute 2 in penile carcinogenesis and its relation to prognosis. Hum Pathol 2012; 43: 481–488.
Pinto AP, Miron A, Yassin Y et al, Differentiated vulvar intraepithelial neoplasia contains Tp53 mutations and is genetically linked to vulvar squamous cell carcinoma. Mod Pathol 2010; 23: 404–412.
Choschzick M, Hantaredja W, Tennstedt P et al, Role of TP53 mutations in vulvar carcinomas. Int J Gynecol Pathol 2011; 30: 497–504.
Wiest T, Schwarz E, Enders C et al, Involvement of intact HPV16 E6/E7 gene expression in head and neck cancers with unaltered p53 status and perturbed pRb cell cycle control. Oncogene 2002; 21: 1510–1517.
Yanagawa N, Osakabe M, Hayashi M et al, Detection of HPV-DNA, p53 alterations, and methylation in penile squamous cell carcinoma in Japanese men. Pathol Int 2008; 58: 477–482.
Wang K, Li M, Hakonarson H . ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res 2010; 38: e164.
Cingolani P, Platts A, Wang LL et al, A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly (Austin) 2012; 6: 80–92.
Muñoz N, Bosch FX, de Sanjosé S et al, Epidemiologic classification of human papillomavirus types associated with cervical cancer. N Engl J Med 2003; 348: 518–527.
Ferrandiz-Pulido C, Masferrer E, Toll A et al, mTOR signaling pathway in penile squamous cell carcinoma: pmTOR and peIF4E over expression correlate with aggressive tumor behavior. J Urol 2013; 190: 2288–2295.
Freed-Pastor WA, Prives C . Mutant p53: one name, many proteins. Genes Dev 2012; 26: 1268–1286.
Yoshikawa K, Hamada J, Tada M et al, Mutant p53 R248Q but not R248W enhances in vitro invasiveness of human lung cancer NCI-H1299 cells. Biomed Res 2010; 31: 401–411.
Xu J, Wang J, Hu Y et al, Unequal prognostic potentials of p53 gain-of-function mutations in human cancers associate with drug-metabolizing activity. Cell Death Dis 2014; 5: e1108.
Freed-Pastor WA, Mizuno H, Zhao X et al, Mutant p53 disrupts mammary tissue architecture via the mevalonate pathway. Cell 2012; 148: 244–258.
Wang Z, Sun Y . Targeting p53 for novel anticancer therapy. Transl Oncol 2010; 3: 1–12.
Zawacka-Pankau J, Selivanova G . Pharmacological reactivation of p53 as a strategy to treat cancer. J Intern Med 2015; 277: 248–259.
Acknowledgements
We thank the entire staff of the Molecular Diagnostics and the Immunohistochemistry lab at the Institute of Pathology, Medical University of Graz for HPV genotyping, immunohistochemical staining, and related services. This research was, in part, funded by the Human Technology Interface Grant of the County of Styria, GZ: ABT08-22-T-7/2013-23.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article

Cite this article
Kashofer, K., Winter, E., Halbwedl, I. et al. HPV-negative penile squamous cell carcinoma: disruptive mutations in the TP53 gene are common. Mod Pathol 30, 1013–1020 (2017). https://doi.org/10.1038/modpathol.2017.26
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/modpathol.2017.26
This article is cited by
-
Pathologie und histopathologische Begutachtung des Peniskarzinoms
Der Urologe (2018)
