
Abstract
The recent discovery of natural immunity to the hepatitis C virus and vaccine efficacy in the chimpanzee challenge model has allowed optimism about the development of at least a partly effective vaccine against this heterogeneous pathogen that is responsible for much of the chronic liver disease around the world. The immune systems of some infected individuals can spontaneously clear the virus, whereas other people need treatment with antivirals that work partly by stimulating humoral and cellular immune responses. Therefore, therapeutic vaccine strategies are also being pursued to improve treatment outcome.
Similar content being viewed by others

Main
A decade ago, an effective vaccination against the hepatitis C virus (HCV) was considered only a remote possibility. Three factors contributed to this: the high propensity of HCV to promote chronic persistent infections1; evidence that convalescent humans and chimpanzees could be readily reinfected following re-exposure2; and the considerable genetic heterogeneity of this positive-stranded RNA virus3.
The situation today is more positive for two reasons. First, we now know that spontaneous eradication of the virus occurs in up to 50% of acute infections4 and that this viral clearance is associated with specific immune responses to the virus. Recapitulation of such immune responses by appropriate vaccination is therefore a realistic option. Second, clear evidence for at least some natural immunity has emerged recently in both humans5 and chimpanzees6,7,8. (Chimpanzees are the only animal model available and develop only mild clinical sequelae.) Convalescent humans and chimpanzees are protected against re-exposure to the virus in the majority of cases, even against very divergent viral strains. Importantly, protection is usually at the level of prevention of progression to chronic, persistent infection following re-exposure rather than prevention of acute reinfection but this could translate to effective prophylaxis because, in humans, it is the chronic, persistent nature of HCV infection that is mainly associated with viral pathogenicity1,4. Although some re-exposed individuals develop chronic infection9, most do not5,6,7,8. This suggests that the generation of at least a partly effective vaccine against HCV is feasible. Indeed, emerging vaccine efficacy data from the chimpanzee challenge model indicate that it is possible to impede the progression to chronic infection in vaccinees. Until very recently10,11,12, it was not possible to grow HCV efficiently in cell culture, and so the use of inactivated or live, attenuated viral vaccines has not yet been evaluated. Vaccine approaches have therefore included the use of adjuvanted recombinant polypeptide subunits of the virus in attempts to prime viral-neutralizing antibodies to the envelope glycoproteins 1 and 2 (gpE1 and gpE2), as well as priming MHC class-II-restricted CD4+ T helper (TH) and MHC class-I-restricted CD8+ cytotoxic lymphocyte (CTL) responses to these and other viral proteins. Both types of T cell can secrete antiviral cytokines such as interferon-γ (IFN-γ), and CD8+ CTLs have the potential to kill infected cells.
It is difficult to prime CD8+ CTLs using polypeptide subunit vaccines, although certain adjuvants are capable of eliciting such responses13,14. Various forms of plasmid DNA vaccine are also being explored to elicit HCV-specific humoral and cellular immune responses to encoded antigens which, by virtue of being newly synthesized in the cytosol of transfected cells, can be particularly effective at priming CD8+ CTLs. DNA vaccines also include immunostimulatory deoxycytosine-deoxyguanosine (CpG)-containing motifs capable of activating antigen-presenting dendritic cells. This would lead to stimulation of innate immune responses (such as the synthesis of type 1 interferons and natural killer (NK) cells) as well as adaptive B- and T-cell responses to vaccine antigens. Various live attenuated or defective viral or bacterial vectors expressing HCV genes are also being investigated because improved vaccine immunogenicity can result from more efficient expression and delivery of HCV antigens. This may include the targeting of antigen-presenting cells in some cases. The use of various prime/boost immunization modes and regimens are also being explored to optimize vaccine immunogenicity and potency.
In this review, we will summarize current knowledge regarding the correlates of immunity to HCV as well as the results of pre-clinical studies using vaccine candidates designed to recapitulate protective immunity. Emerging results from the chimpanzee challenge model suggest that successful vaccination against homologous and at least some heterologous HCV strains may be feasible, although the relative roles of humoral and cellular immunity in protection need to be better defined. The status and issues surrounding clinical development will be discussed as well as the rationale and prospect for immunotherapeutic vaccination strategies.
Correlates of immunity
Infected humans and chimpanzees who mount an early, multi-specific CD4+ TH and CD8+ T-cell response to HCV proteins can eradicate the virus (see the review in this issue by Bowen and Walker, page 946, and refs 15–24). These activated T cells secrete pro-inflammatory cytokines (TH1-type) such as IFN-γ, which is directly antiviral for HCV replicons in cell culture25 and temporally associated with large reductions in viral load during acute infection20. Specific CD8+ CTLs can kill HCV-infected cells, although large reductions in viral load during acute infection were not associated with an increase in acute hepatitis, suggesting that cytolytic activity may not be the main factor in viral control20. Further work is required, however, to better define these cellular correlates of immunity. It is clear that these cellular immune responses to the virus can occur in the absence of antibody to gpE1 and gpE2 (ref. 19), indicating that such antibodies are not absolutely required for recovery from acute infection.
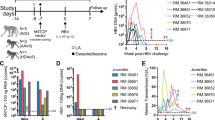
Until very recently10,11,12, HCV has not been propagated efficiently in cell culture, meaning that a direct assay for viral-neutralizing antibody has not been available. However, the recent production of lentiviral/HCV pseudoparticles (HCVpp) bearing HCV envelope glycoproteins on the particle surface have been used to show that patients not only have antibodies that can neutralize the infectivity of such pseudoparticles but that such antibodies cross-neutralize pseudoparticles derived from many different HCV genotypes26,27,28. This suggests that a broad cross-neutralizing antibody to HCV may exist and could be exploited in vaccine strategies. Furthermore, the recent application of these pseudoparticle infectivity assays to the investigation of immune correlates of protection are beginning to indicate that such ‘neutralizing’ antibodies, when present, may be associated with recovery from acute infection, at least in some cases29 (for an example, see Fig. 1). The relative roles of humoral and cellular immunity in recovery remain unclear.

Association between circulating viral RNA load (blue) and antibodies that neutralize infectivity of HCV pseudoparticles (HCVpp; purple, expressed as percentage of neutralization). The percentage of neutralization of control pseudoparticles (Controlpp) is shown in red. Serum alanine aminotransferase (ALT) levels indicative of hepatitis are shown in green. Adapted from ref. 29.
Studies in the past several years have helped to define the sophisticated battle initiated in the infected host. This RNA virus (which cannot integrate into the host genome) has evolved mechanisms to persist and to evade the host's innate and adaptive immune mechanisms (see the reviews in this issue by Gale and Foy, page 939, and Bowen and Walker, page 946). The virus inhibits the induction of type-1 interferons30,31, inhibits NK cells32,33, readily produces escape mutants to CTLs34 and neutralizing antibodies directed to the amino-terminal region of gpE235,36,37,38 HCV may also inhibit the binding of virion-neutralizing antibodies by masking with lipoproteins28,39. Effector T cells specific to the virus also seem to be downregulated in some way as a consequence of persistent HCV infection40,41. Other mechanisms of viral persistence are likely to emerge in the future. By contrast, if the host has the ability to elicit early and broad TH1-type CD4+ and CD8+ T-cell responses to the virus15,16,17,18,19,20,21,22,23,24 and also has a NK receptor repertoire that facilitates innate immune clearance of virus42, then eradication of virus can occur. It is also possible that the presence of viral-neutralizing antibody may enhance this process29.
Prevention strategies
Results from our recent studies have made us optimistic about successfully vaccinating against HCV. These studies involved the use of the recombinant HCV envelope glycoproteins gpE1 and gpE2 as vaccine antigens. Derived from mammalian cells, the two glycoproteins associate together to from a non-disulphide linked gpE1–gpE2 heterodimer that is thought to resemble the pre-virion envelope structure43. When combined with oil/water-based adjuvants and used to vaccinate naive chimpanzees, this vaccine candidate elicits anti-envelope antibodies as well as TH cell responses to gpE1 and gpE2. Our earlier work showed that when these vaccinated animals were challenged experimentally with homologous viral inocula, the highest responding animals (in terms of anti-gpE1/gpE2 antibody titres) were completely protected against infection44. Using sensitive RT–PCR assays, no viraemia was detected in blood or liver samples at any time after challenge in these seemingly ‘sterilized’ animals. This apparent sterilizing immunity correlated directly with anti-gpE2 antibody titres that prevent the binding of gpE2 (or the virus itself) to CD81 (ref. 45), which has been shown to be an important receptor component for binding of infectious HCV10,11,12,46 and for cell entry of lentiviral/HCV pseudoparticles47. Furthermore, although lower-responding animals became infected, the majority underwent an abortive acute infection that did not result in the persistently infected carrier state44,48 that in humans can be associated with chronic liver disease1,4. Overall, these data showed that the carrier rate in vaccinees was significantly lower than in unimmunized controls44,48 (Table 1).
A crucial question that remained was whether the vaccine derived from strain HCV-1 would protect against heterologous strains of the virus. Recently, we have challenged nine chimpanzee vaccinees with the HCV-H strain that, like the vaccine strain HCV-1, is of the 1a genotype that predominates in the United States3. Although none of the vaccinated animals was protected against acute infection, all but one vaccinee resolved the acute infection and failed to progress to the carrier state49 (as demonstrated by the persistent absence of detectable viraemia in follow-up blood samples using sensitive RT–PCR assays). By contrast, the majority of control animals became carriers when challenged with HCV-H, indicating that the vaccine significantly reduced chronic, persistent infection49 (Table 1). Although the viral challenge doses were small (10–100 chimpanzee infectious doses50 (CID50)), such doses are considered to be within the same range as those transmitted in many community-acquired HCV infections because the infectivity titre of most carriers is known to be low50. These pre-clinical data (and supporting data from other small studies exploring various gpE1/gpE2 vaccine formulations51,52,53) support the initiation of a clinical prophylactic programme using adjuvanted gpE1/gpE2 that is currently in phase 1 testing.
Many studies correlate recovery from acute HCV infection with cellular immune responses to the virus, and so other relevant strat-egies for developing a vaccine will involve eliciting a broad cellular immune response to the virus or, preferably, both a humoral (anti-gpE1/gpE2) and a cellular immune response. One small study using the chimpanzee model investigated the use of a vaccination regimen employing multiple immunizations with plasmid DNA encoding the nucleocapsid (C), gpE1, gpE2 and nonstructural protein 3 (NS3) domains followed by multiple boosting with an adjuvanted mixture of recombinant C, gpE1, gpE2 and NS3 proteins51. Following challenge with a heterologous strain (which causes chronic, persistent infection in the large majority of control animals), one vaccinee experienced an ameliorated and abortive acute infection that did not progress to the carrier state, whereas the other vaccinee developed chronic infection, albeit ameliorated in terms of viral load and level of hepatitis. These data provide additional support for the feasibility of successful vaccination against HCV but also suggest that further optimization of vaccine immunogenicity is required.
Surprisingly, eliciting broad CD4+ and CD8+ T-cell responses to the virus in the absence of any antibody responses to the envelope glycoproteins (using an ISCOMATRIX®-adjuvanted13,14 NS3-4-5-Core polyprotein derived from strain HCV-1) failed to prevent chronic, persistent infection following challenge with the heterologous HCV-H strain in five out of five chimpanzee vaccinees tested, despite observing a substantial amelioration in acute viraemia and hepatitis (M.H., unpublished data). This result may be caused by insufficient priming of cellular immune responses by the vaccine regimen or protocol because recovery from acute infection has been linked with cellular immune responses to the virus in the absence of anti-envelope antibody responses19. This result also suggests that vaccine formulations capable of priming both anti-envelope neutralizing antibody and broad cellular immune responses to the virus may be more effective. Considering that this is the only vaccine formulation of several tested by us that failed to result in prevention of HCV chronicity in at least some animals, this result also emphasizes the importance of using the chimpanzee challenge model before proceeding to clinical testing.
Other approaches to HCV vaccination (summarized in Table 2), in common with those used in vaccine research for other persistent pathogens like HIV and malaria, include the use of various defective or attenuated viral vectors to enhance priming of humoral and cellular immune responses to multiple HCV gene products expressed by the vector. The use of adenoviral54, avipox55, alphaviral56,57 and vaccinia58 viral vectors, among others, are all being explored in various animal models including the chimpanzee challenge model. These approaches offer the potential of improved immunogenicity as a result of enhanced gene delivery and expression. Some of these vectors also infect and/or activate antigen-presenting dendritic cells, thus enhancing antigen presentation and stimulating innate immune responses that, in turn, lead to enhancement of adaptive immune responses to the encoded vaccine antigens. However, challenges for these approaches include the problem of pre-existing immunity to some of these vectors in the human population, thus limiting potency. Repeated vaccination to boost initial immune responses can also be limited by vector-elicited immunity. To overcome this obstacle, priming of the immune response with DNA vaccines followed by boosting with recombinant viral vectors is being employed as well as prime/boost regimens using different recombinant vectors for each immunization. One promising approach is the use of defective alphaviral delivery vectors that infect professional antigen-presenting dendritic cells, activate innate immunity as well as adaptive cellular and humoral immune responses to encoded vaccine antigens and which can be used repeatedly to boost immune responses in mice57. A defective ovine atadenovirus vector59 may also be useful in this regard. Other promising approaches being explored include the use of DNA microparticles60, which can significantly enhance the potency of DNA vaccines and HCV viral-like particles produced in insect cells that have an inherently strong immunogenicity61.
Apart from optimizing vaccine formulations to maximize humoral and cellular immune responses, future issues include expanding the range and level of cross-protection afforded by the vaccine. This will require more extensive analyses into the nature and range of cross-neutralizing antibody and cross-protective cellular immune responses and will probably involve the definition of cocktails of immunogens derived from various HCV genotypes to obtain an effective global vaccine formulation.
Proving the efficacy of the vaccine in humans is a significant challenge because accessing groups at high risk of HCV infection is no longer a simple task. With the near elimination of post-transfusion hepatitis C by donor screening, other high-risk groups suitable for efficacy testing have inherent difficulties such as lack of compliance (for example, intravenous drug users), low incidence of infection (for example, in health-care workers and paramedics), lack of supporting infrastructure (for example, in many developing countries where incidence of infection is high) and ethical issues (for example, in prisoner populations where prevalence and incidence of infection are both high). However, some of these cohorts have been used successfully in the past (for testing hepatitis B vaccines) and so these obstacles should not be insurmountable. If a vaccine is successfully developed, an important cause of global morbidity and mortality will be controlled and, even in countries with a relatively low incidence of infection, the vaccine will be reasonably cost-effective when used in the general population62.
Potential for therapeutic HCV vaccination
The current standard-of-care therapy for chronically infected HCV patients is a combination of pegylated IFN-α and ribavirin, which is costly, lengthy (6–12 months), associated with significant side effects and results in sustained viral response in only ∼50% of patients. In patients infected with genotype 1, the most common form, response rates are even lower63. With an estimated 170 million HCV carriers worldwide, it is clearly important to develop better therapeutic options. With our increasing knowledge of the virus-encoded enzymes and genetic elements vital to the life-cycle of HCV, much attention is now being focused on the development of HCV protease, replicase, helicase, antisense, silencing RNA and other specific inhibitors. However, preliminary data have directly linked responses to IFN-α and ribavirin with pretreatment titres of viral antibodies64 (presumed to be against the envelope glycoproteins), peripheral TH cell responses to the HCV core and other antigens65, as well as to intrahepatic CD8+ CTL responses to the virus66. Total pretreatment CD8+ T-cell counts in the liver have also been correlated with sustained responses to standard-of-care therapy67. Therefore, it may be possible to boost such immune responses in patients by appropriate vaccination and thereby improve the response rate to the standard-of-care therapy. Such immunotherapy may also help control the emergence of escape mutants that would be predicted to arise from any future use of HCV protease or replicase inhibitors, for example, given the extreme fluidity and heterogeneity of the HCV genome3.
Many therapeutic vaccine trials are planned or are already in progress and use diverse delivery methods and formulations (summarized in Table 3) but little information is available about their efficacy at present. What is known, however, is that use of an alum-adjuvanted recombinant gpE1 antigen was able to boost humoral and cellular immune responses to gpE1 in viraemic patients, providing encouragement that vaccination can increase immune responses in pre-existing carriers68. It remains to be seen whether boosting viral-neutralizing antibody titres or broad CD4+ TH responses or broad CD8+ T-cell responses will have the greatest impact on reducing viral load and in the response to antiviral therapy. But, as may be the case for optimal prophylaxis, boosting all of these immune responses may be ideal for immunotherapy.
HCV tries to counter innate immunity by inhibiting the induction of type-1 interferons (IFN-α/β)30,31 and downregulating NK-cell activity32,33. Therefore, therapeutic vaccine formulations could benefit by inclusion of molecules capable of triggering innate immune responses. Such molecules include oligonucleotides containing CpG motifs that trigger Toll-like receptor 9 within dendritic cells and that also enhance adaptive immune responses to vaccine antigens69. If successful, vaccination for the treatment of chronic hepatitis C would be one of the first demonstrations of immunotherapeutic intervention in chronic viral infections, although, very recently, such an approach has been used successfully to inhibit the age-related emergence of herpes zoster infections and disease in carriers70.
Future directions
In the future, it will be important to use the chimpanzee model to further define correlates of protection, duration of vaccine-mediated protection, the extent of cross-protection against diverse genotypes and mechanisms of chronicity and to determine optimal vaccine formulations for prophylactic and immunotherapeutic efficacy. In addition, human cohorts at high risk of infection need to be identified and characterized for efficacy trials. The huge burden of chronically infected HCV patients facilitates the testing of various immunotherapeutic vaccine formulations that, most probably, will be especially useful when used as adjunct therapy with antiviral drugs, including pegylated IFN-α and ribavirin as well as the new class of HCV drugs currently under development that inhibit viral enzymes and other elements crucial to the viral life-cycle. It will also be important to understand the mechanisms involved in immune dysfunction and evasion during chronic HCV infections so as to facilitate the design of further immunotherapies.
References
Alter, H. J. & Seeff, L. B. Recovery, persistence, and sequelae in hepatitis C virus infection: a perspective on long-term outcome. Semin. Liver Dis. 20, 17–35 (2000).
Lai, M. E. et al. Hepatitis C virus in multiple episodes of acute hepatitis in polytransfused thalassaemic children. Lancet 343, 388–390 (1994).
Simmonds, P. Genetic diversity and evolution of hepatitis C virus — 15 years on. J. Gen. Virol. 85, 3173–3188 (2004).
Seeff, L. B. Natural history of chronic hepatitis C. Hepatology 36, S35–S46 (2002).
Mehta, S. H. et al. Protection against persistence of hepatitis C. Lancet 359, 1478–1483 (2002).
Weiner, A. J. et al. Intrahepatic genetic inoculation of hepatitis C virus RNA confers cross-protective immunity. J. Virol. 75, 7142–7148 (2001).
Bassett, S. E. et al. Protective immune response to hepatitis C virus in chimpanzees rechallenged following clearance of primary infection. Hepatology 33, 1479–1487 (2001).
Lanford, R. E. et al. Cross-genotype immunity to hepatitis C virus. J. Virol. 78, 1575–1581 (2004).
Farci, P. et al. Lack of protective immunity against reinfection with hepatitis C virus. Science 258, 135–140 (1992).
Wakita, T. et al. Production of infectious hepatitis C virus in tissue culture from a cloned viral genome. Nature Med. 11, 791–796 (2005).
Zhong, J. et al. Robust hepatitis C virus infection in vitro. Proc. Natl Acad. Sci. USA 102, 9294–9299 (2005).
Lindenbach, B. D. et al. Complete replication of hepatitis C virus in cell culture. Science 309, 623–626 (2005).
Polakos, N. K. et al. Characterization of hepatitis C virus core-specific immune responses primed in rhesus macaques by a non-classical ISCOM vaccine. J. Immunol. 166, 3589–3598 (2001).
Pearse, M. J. & Drane, D. ISCOMATRIX adjuvant for antigen delivery. Adv. Drug Deliv. Rev. 57, 465–474 (2005).
Diepolder, H. M. et al. Possible mechanism involving T-lymphocyte response to non-structural protein 3 in viral clearance in acute hepatitis C virus infection. Lancet 346, 1006–1007 (1995).
Missale, G. et al. Different clinical behaviors of acute hepatitis C virus infection are associated with different vigor of the anti-viral cell-mediated immune response. J. Clin. Invest. 98, 706–714 (1996).
Tsai, S. L., Liaw, Y. F., Chen, M. H., Huang, C. Y. & Kuo, G. C. Detection of type 2-like T-helper cells in hepatitis C virus infection: implications for hepatitis C virus chronicity. Hepatology 25, 449–458 (1997).
Gerlach, J. T. et al. Recurrence of hepatitis C virus after loss of virus-specific CD4+ T-cell response in acute hepatitis C. Gastroenterology 117, 933–941 (1999).
Cooper, S. et al. Analysis of a successful immune response against hepatitis C virus. Immunity 10, 439–449 (1999).
Thimme, R., Oldach, D., Chang, K. M., Steiger, C., Ray, S. C. & Chisari, F. V. Determinants of viral clearance and persistence during acute hepatitis C virus infection. J. Exp. Med. 194, 1395–1406 (2001).
Grakoui, A. et al. HCV persistence and immune evasion in the absence of memory T cell help. Science 302, 659–662 (2003).
Shoukry, N. H. et al. Memory CD8+ T cells are required for protection from persistent hepatitis C virus infection. J. Exp. Med. 197, 1645–1655 (2003).
Lechner, F. et al. Analysis of successful immune responses in persons infected with hepatitis C virus. J. Exp. Med. 191, 1499–1512 (2000).
Lechner, F. et al. CD8+ T lymphocyte responses are induced during acute hepatitis C virus infection but are not sustained. Eur. J. Immunol. 30, 2479–2487 (2000).
Frese, M. et al. Interferon-gamma inhibits replication of subgenomic and genomic hepatitis C virus RNAs. Hepatology 35, 694–703 (2002)
Bartosch, B. et al. In vitro assay for neutralizing antibody to hepatitis C virus: evidence for broadly conserved neutralization epitopes. Proc. Natl Acad. Sci. USA 100, 14199–14204 (2003).
Logvinoff, C. et al. Neutralizing antibody response during acute and chronic hepatitis C virus infection. Proc. Natl Acad. Sci. USA 101, 10149–10154 (2004).
Meunier, J. C. et al. Evidence for cross-genotype neutralization of hepatitis C virus pseudo-particles and enhancement of infectivity by apolipoprotein C1. Proc. Natl Acad. Sci. USA 102, 4560–4565 (2005).
Lavillette, D. et al. Human serum facilitates hepatitis C virus infection, and neutralizing responses inversely correlate with viral replication kinetics at the acute phase of hepatitis C virus infection. J. Virol. 79, 6023–6034 (2005).
Foy, E. et al. Control of antiviral defenses through hepatitis C virus disruption of retinoic acid-inducible gene-I signaling. Proc. Natl Acad. Sci. USA 102, 2986–2991 (2005).
Li, K. et al. Immune evasion by hepatitis C virus NS3/4A protease-mediated cleavage of the Toll-like receptor 3 adaptor protein TRIF. Proc. Natl Acad. Sci. USA 102, 2992–2997 (2005).
Crotta, S. et al. Inhibition of natural killer cells through engagement of CD81 by the major hepatitis C virus envelope protein. J. Exp. Med. 195, 35–41 (2002).
Tseng, C. T. & Klimpel, G. R. Binding of the hepatitis C virus envelope protein E2 to CD81 inhibits natural killer cell functions. J. Exp. Med. 195, 43–49 (2002).
Erickson, A. L. et al. The outcome of hepatitis C virus infection is predicted by escape mutations in epitopes targeted by cytotoxic T lymphocytes. Immunity 15, 883–895 (2001).
Weiner, A. J. et al. Evidence for immune selection of hepatitis C virus (HCV) putative envelope glycoprotein variants: potential role in chronic HCV infections. Proc. Natl Acad. Sci. USA 89, 3468–3472 (1992).
Kato, N. et al. Humoral immune response to hypervariable region 1 of the putative envelope glycoprotein (gp70) of hepatitis C virus. J. Virol. 67, 3923–3930 (1993).
Farci, P. et al. The outcome of acute hepatitis C predicted by the evolution of the viral quasispecies. Science 288, 339–344 (2000).
Farci, P. et al. Prevention of hepatitis C virus infection in chimpanzees by hyperimmune serum against the hypervariable region 1 of the envelope 2 protein. Proc. Natl Acad. Sci. USA 93, 15394–15399 (1996).
Thomssen, R., Bonk, S., Propfe, C., Heermann, K. H., Kochel, H. G. & Uy, A. Association of hepatitis C virus in human sera with beta-lipoprotein. Med. Microbiol. Immunol. 181, 293–300 (1992).
Wedemeyer, H. et al. Impaired effector function of hepatitis C virus-specific CD8+ T cells in chronic hepatitis C virus infection. J. Immunol. 169, 3447–3458 (2002).
Semmo, N. et al. Preferential loss of IL-2-secreting CD4+ T helper cells in chronic HCV infection. Hepatology 41, 1019–1028 (2005).
Khakoo, S. I. et al. HLA and NK cell inhibitory receptor genes in resolving hepatitis C virus infection. Science 305, 872–874 (2004).
Ralston, R. et al. Characterization of hepatitis C virus envelope glycoprotein complexes expressed by recombinant vaccinia viruses. J. Virol. 67, 6753–6761 (1993).
Choo, Q. L. et al. Vaccination of chimpanzees against infection by the hepatitis C virus. Proc. Natl Acad. Sci. USA 91, 1294–1298 (1994).
Rosa, D. et al. A quantitative test to estimate neutralizing antibodies to the hepatitis C virus: cytofluorimetric assessment of envelope glycoprotein 2 binding to target cells. Proc. Natl Acad. Sci. USA 93, 1759–1763 (1996).
Pileri, P. et al. Binding of hepatitis C virus to CD81. Science 282, 938–941 (1998).
McKeating, J. A. et al. Diverse hepatitis C virus glycoproteins mediate viral infection in a CD81-dependent manner. J. Virol. 78, 8496–8505 (2004).
Houghton, M. et al. in Viral Hepatitis and Liver Disease (eds Rizzetto, M, Purcell, R. H., Gerin, J. L., Verme, G) pp 656–659 (Edizioni Minerva Medica, 1997).
Coates, S. et al. in Proceedings of the 11th International Symposium on Viral Hepatitis and Liver Disease (eds Jilbert, A. R., Grgacic, E. V. L., Vickery, K., Burrell, C. J., Cossart, Y. E.) pp 118–123 (Australian Center for Hepatitis Virology, 2005).
Prince, A. M. Reliability of chimpanzee model for non-A, non-B hepatitis. Lancet ii, 1134 (1985).
Rollier, C. et al. Control of heterologous hepatitis C virus infection in chimpanzees is associated with the quality of vaccine-induced peripheral T-helper immune response. J. Virol. 78, 187–196 (2004).
Puig, M., Major, M. E., Mihalik, K. & Feinstone, S. M. Immunization of chimpanzees with an envelope protein-based vaccine enhances specific humoral and cellular immune responses that delay hepatitis C virus infection. Vaccine 22, 991–1000 (2004).
Forns, X. et al. Vaccination of chimpanzees with plasmid DNA encoding the hepatitis C virus (HCV) envelope E2 protein modified the infection after challenge with homologous monoclonal HCV. Hepatology 32, 618–625 (2000).
Catalucci, D., Sporeno, E., Cirillo, A., Ciliberto, G., Nicosia, A. & Colloca, S. An adenovirus type 5 (Ad5) amplicon-based packaging cell line for production of high-capacity helper-independent deltaE1-E2-E3-E4 Ad5 vectors. J. Virol. 79, 6400–6409 (2005).
Pancholi, P., Perkus, M., Tricoche, N., Liu, Q. & Prince, A. M. DNA immunization with hepatitis C virus (HCV) polycistronic genes or immunization by HCV DNA priming-recombinant canarypox virus boosting induces immune responses and protection from recombinant HCV-vaccinia virus infection in HLA-A2.1-transgenic mice. J. Virol. 77, 382–390 (2003).
Brinster, C. et al. Hepatitis C virus non-structural protein 3-specific cellular immune responses following single or combined immunization with DNA or recombinant Semliki Forest virus particles. J. Gen. Virol. 83, 369–381 (2002).
Perri, S. et al. An alphavirus replicon particle chimera derived from venezuelan equine encephalitis and sindbis viruses is a potent gene-based vaccine delivery vector. J. Virol. 77, 10394–10403 (2003).
Abraham, J. D. et al. Comparative immunogenicity analysis of modified vaccinia Ankara vectors expressing native or modified forms of hepatitis C virus E1 and E2 glycoproteins. Vaccine 22, 3917–3928 (2004).
Wuest, T., Both, G. W., Prince, A. M., Hofmann, C. & Loser, P. Recombinant ovine atadenovirus induces a strong and sustained T cell response against the hepatitis C virus NS3 antigen in mice. Vaccine 22, 2717–2721 (2004).
O'Hagan, D. T. et al. Cationic microparticles are a potent delivery system for a HCV DNA vaccine. Vaccine 23, 672–680 (2004).
Jeong, S. H. et al. Immunization with hepatitis C virus-like particles induces humoral and cellular immune responses in nonhuman primates. J. Virol. 78, 6995–7003 (2004).
Krahn, M. D. et al. Potential cost-effectiveness of a preventive hepatitis C vaccine in high risk and average risk populations in Canada. Vaccine 23, 1549–1558 (2005).
Saadeh, S. & Davis, G. L. The evolving treatment of chronic hepatitis C: where we stand a decade out. Cleveland Clin. J. Med. 71 (Suppl. 3), S3–S7 (2004).
Baumert, T. F. et al. Antibodies against hepatitis C virus-like particles and viral clearance in acute and chronic hepatitis C. Hepatology 32, 610–617 (2000).
Cramp, M. E., Rossol, S., Chokshi, S., Carucci, P., Williams, R. & Naoumov, N. V. Hepatitis C virus-specific T-cell reactivity during interferon and ribavirin treatment in chronic hepatitis C. Gastroenterology 118, 346–355 (2000).
Nelson, D. R., Marousis, C. G., Ohno, T., Davis, G. L. & Lau, J. Y. Intrahepatic hepatitis C virus-specific cytotoxic T lymphocyte activity and response to interferon alfa therapy in chronic hepatitis C. Hepatology 28, 225–230 (1998).
Vrolijk, J. M. et al. Pretreatment intrahepatic CD8+ cell count correlates with virological response to antiviral therapy in chronic hepatitis C virus infection. J. Infect. Dis. 188, 1528–1532 (2003).
Nevens, F. et al. A pilot study of therapeutic vaccination with envelope protein E1 in 35 patients with chronic hepatitis C. Hepatology 38, 1289–1296 (2003).
Abel, K. et al. Deoxycytidyl-deoxyguanosine oligonucleotide classes A, B, and C induce distinct cytokine gene expression patterns in Rhesus monkey peripheral blood mononuclear cells and distinct alpha interferon responses in TLR9-expressing Rhesus monkey plasmacytoid dendritic cells. Clin. Diagn. Lab. Immunol. 12, 606–621 (2005).
Oxman, M. N. et al. A vaccine to prevent herpes zoster and postherpetic neuralgia in older adults. N. Engl. J. Med. 352, 2271–2284 (2005).
Leroux-Roels, G. et al. A candidate vaccine based on the hepatitis C E1 protein: tolerability and immunogenicity in healthy volunteers. Vaccine 22, 3080–3086 (2004).
Franzusoff, A., Duke, R. C., King, T. H., Lu, Y., & Rodell, T. C. Yeasts encoding tumour antigens in cancer immunotherapy. Exp. Opin. Biol. Ther. 5, 565–575 (2005).
Author information
Authors and Affiliations
Ethics declarations
Competing interests
Both authors are employed by Chiron Corp., which is developing a HCV vaccine. At least one of the authors (M.H.) has a substantial equity investment in Chiron Corp.
Additional information
Author information Reprints and permissions information is available at npg.nature.com/preprintsandpermissions.
Rights and permissions
About this article
Cite this article
Houghton, M., Abrignani, S. Prospects for a vaccine against the hepatitis C virus. Nature 436, 961–966 (2005). https://doi.org/10.1038/nature04081
Published:
Issue Date:
DOI: https://doi.org/10.1038/nature04081
This article is cited by
-
Exploring NS3/4A, NS5A and NS5B proteins to design conserved subunit multi-epitope vaccine against HCV utilizing immunoinformatics approaches
Scientific Reports (2018)
-
Nonhuman primate models of human viral infections
Nature Reviews Immunology (2018)
-
Virus-Specific Cellular Response in Hepatitis C Virus Infection
Archivum Immunologiae et Therapiae Experimentalis (2016)
-
Design, Synthesis and Structure-Activity Relationship Optimization of Lycorine Derivatives for HCV Inhibition
Scientific Reports (2015)
-
Approaching rational epitope vaccine design for hepatitis C virus with meta-server and multivalent scaffolding
Scientific Reports (2015)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
