
Abstract
Lateral DNA transfer—the movement of genetic traits between bacteria—has a profound impact on genomic evolution and speciation. The efficiency with which bacteria incorporate genetic information reflects their capacity to adapt to changing environmental conditions. Integron integrases are proteins that mediate site-specific DNA recombination between a proximal primary site (attI) and a secondary target site (attC) found within mobile gene cassettes encoding resistance or virulence factors. The lack of sequence conservation among attC sites has led to the hypothesis that a sequence-independent structural recognition determinant must exist within attC. Here we report the crystal structure of an integron integrase bound to an attC substrate. The structure shows that DNA target site recognition and high-order synaptic assembly are not dependent on canonical DNA but on the position of two flipped-out bases that interact in cis and in trans with the integrase. These extrahelical bases, one of which is required for recombination in vivo, originate from folding of the bottom strand of attC owing to its imperfect internal dyad symmetry. The mechanism reported here supports a new paradigm for how sequence-degenerate single-stranded genetic material is recognized and exchanged between bacteria.
Similar content being viewed by others

Main
Nature has evolved several classes of enzymes that mediate the capture and spread of genetic information between bacteria1. The efficiency by which these genetic traits are used is dependent on the rate of donor DNA delivery, their genomic incorporation, and the level of gene expression. Novel traits are incorporated by several methods; among these, recombination by site-specific recombinases has an important role2. These enzymes require short matching DNA sequences between the donor and genomic DNA, which undoubtedly reduces the rate of DNA assimilation among highly diverse genetic populations.
Integron integrase-mediated recombination
Integron integrases (IntIs) are site-specific recombinases that form a subclass within the tyrosine recombinase family owing to the presence of a unique insertion that is required for activity3. Unlike other members of the family, such as the bacteriophage P1 Cre and yeast Flp proteins4,5, IntIs can mediate the exchange of DNA between two architecturally distinct sites even though homology predicts only one DNA binding domain6. The mechanism by which IntIs achieve this dual site-specificity is unknown. IntIs catalyse an insertion between a primary recombination site (attI), located within a DNA element called an integron, and secondary target sites (attC), located within mobile gene cassettes7. These insertions are balanced by the excision of gene cassettes that occurs between two attC sites (Fig. 1a).

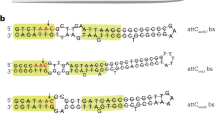
a, Integrons contain a gene, IntI, encoding a tyrosine recombinase, and an adjacent recombination site, attI. Gene cassettes (open reading frames, ORFs) are flanked by secondary sites, attC sites. IntI recombines attI and attC during integration and two attC sites during excision. Pi and Pc are promoters for IntI and gene cassettes, respectively; DR1 and DR2 are directly repeated accessory binding sites; L and R are binding sites within the core region of attI; L′ and L″ are inner repeats; R′ and R″ are flanking repeats. b, Excision by the classic tyrosine recombinase model. Each duplex attC site (step 1) is bound by two IntI molecules to form an antiparallel recombination synapse (step 2). Tyr 302 cleavage forms covalent 3′-phosphotyrosine intermediates (step 3). The free 5′-hydroxyl groups attack their partner substrates yielding a Holliday junction (HJ) intermediate (step 4), which isomerizes (step 5) before undergoing a second round of cleavage and strand-exchange reactions to yield the recombinant products5,6 (step 6). c, Proposed IntI excision through a single-stranded DNA substrate pathway. The bottom strand of the integron element, produced by conjugation or transformation, folds upon itself to yield an active stem-loop substrate (step 1). Two IntI molecules bind each folded attC site to form an antiparallel recombination synapse (step 2). The attack and strand exchange steps proceed in a similar fashion to steps 3–4 in panel b; however, the HJ intermediate requires cellular components in order to be resolved12 (steps 5–6). The reaction intermediate shown in step 2 represents the VchIntIA–VCRbs structure described here. IntI molecules coloured green and magenta are potentially active or non-active for cleavage, respectively. d, DNA sequence of VCRbs used to form VchIntIA–DNA co-crystals. Yellow boxes highlight the inner (L′ and L″) and flanking (R′ and R″) repeats. The nucleotides T12″ (red) and G20″ (blue) have an extrahelical geometry upon folding of attC bottom strands (see also Supplementary Fig. 1).
The attI site, like other site-specific recombinase binding sites, contains a core of short symmetrical dyad sequences at its recombination crossover point, as well as two upstream secondary sites that are potentially either regulatory or repressive towards illegitimate recombination8,9. In contrast, the attC sites show poor sequence conservation and vary in length (57 to 141 base pairs (bp)), containing only short regions of sequence similarity at their boundaries. These conserved regions are separated by a stretch of imperfect internal dyad symmetry7.
It might have been expected that excision of a gene cassette would occur through the classic model of Holliday junction (HJ) formation and resolution using two duplex attC sites, as observed with Cre-mediated loxP recombination10 (Fig. 1b). However, we have proposed an alternative pathway for IntI recombination that involves hairpin substrates formed from the bottom strand of the attC site, in which the HJ intermediate is resolved by as yet unknown cellular factors or perhaps DNA replication7,11,12,13,14,15,16 (Fig. 1c). The transition to these complex substrates requires single-stranded DNA stages that can potentially be generated through either transformation or conjugation17.
To address the structural attributes of our model, we have determined the crystal structure of IntI from Vibrio cholerae (VchIntIA) bound to a complex substrate (attC) that was derived from the bottom strand of a V. cholerae repeat (VCR) sequence. VCRs are the attC sites of superintegrons17. The structure shows that the site of recombination along the DNA backbone is determined by the position of two pre-existing extrahelical bases that not only act as molecular markers, positioning the integrase along the DNA, but also mediate the high-order assembly of the synaptic complex. The remainder of the protein–DNA interface is composed almost entirely of non-specific protein-to-DNA phosphate interactions. This structural mechanism of recognition and assembly allows a greater diversity of genetic traits to be captured and exchanged during lateral DNA transfer, thus increasing the rate of bacterial speciation.
A folded single-stranded recombination substrate
The DNA construct for co-crystallization was based on the conserved features of the predicted secondary structures of numerous attC bottom strands (Fig. 1d, Supplementary Fig. 1a). The resulting bulged duplex, VCRbs, formed discrete complexes with VchIntIA in electrophoretic mobility shift assays (EMSAs, see below). In addition, SDS–polyacrylamide gel electrophoresis (SDS–PAGE) analysis of equilibrium mixtures of VchIntIA–VCRbs complexes revealed a band with reduced mobility representing ∼5% of the total protein. Mass spectrometry confirmed this slower migrating species to be covalently linked VchIntIA–VCRbs molecules. This suggested the nucleophilic tyrosine (Tyr 302) within the integrase had formed the characteristic phosphoprotein intermediate.
The structure of the non-covalent VchIntIA–VCRbs complex was determined to 2.8 Å using phases obtained from a single-wavelength anomalous diffraction (SAD) experiment on selenomethionine (SeMet)-labelled protein (see Methods). These phases contributed to generating a readily interpretable electron density map. Model building and refinement allowed for rapid convergence to Rcryst = 0.234 and Rfree = 0.262 for 48–2.80 Å resolution data and good geometry. Data collection, phasing statistics and refinement results are summarized in Supplementary Table S1.
Architecture of the VchIntIA–VCRbs excision complex
The VchIntIA–VCRbs complex contains four VchIntIA molecules bound to two antiparallel VCRbs duplexes (Fig. 2a). This constitutes a recombination synapse representing the step preceding first-strand cleavage in an attC × attC cassette excision reaction (Fig. 1c, step 2). Two of the VchIntIA molecules (subunits A and C) have Tyr 302 adjacent (∼3 Å) to the DNA backbone and thus attacking the scissile phosphate between nucleotides A14′ and C15′ on strand 1 (Supplementary Fig. 2a). The equivalent tyrosines in the B and D subunits are ∼7 Å away (Supplementary Fig. 2b). The amino acids in each active site are all derived from the same subunit: that is, provided in cis. The extrahelical base T12″ is stabilized by cis-interactions within subunits B/D, and is important for DNA site recognition (see below). The extrahelical base G20″ is buried in a deep hydrophobic pocket, located in subunits A/C bound to the other VCRbs duplex, thus forming a set of trans-interactions that hold the synaptic complex together. The protein–protein interfaces between subunits bound to the same VCRbs differ from those bound in trans across the synapse. This arrangement yields an overall synapse that is only two-fold symmetric.

a, N-terminal view of the complex. Four VchIntIA molecules bind two antiparallel VCRbs duplexes to form the active synapse. The extrahelical base T12″ (red) is stabilized by cis interactions and is involved with DNA site recognition (Fig. 4a, b). The extrahelical base G20″ (blue) is buried in subunits that are bound to the other VCRbs duplex forming a set of trans interactions (Fig. 4c, d). The non-symmetric interfaces between VchIntIA molecules yield a two-fold symmetric synapse. b, Orthogonal view with respect to a. The C-terminal helices (N) bury one face in a hydrophobic pocket of the adjacent subunit in a cyclic manner (NA → B, NB → C, and so on).
Structure of VchIntIA recombinase
VchIntIA folds into two distinct domains (Supplementary Fig. 3a). The amino-terminal domain (residues 1–85) contains four α-helices (αA–αD) organized as helix-turn-helix motifs nearly orthogonal to each other pair-wise (αA–αB, αC–αD), thus resembling the corresponding λ integrase and XerD18,19 folds. Helices αB and αD contact the major groove of the VCRbs, whereas helix αA is involved in intersubunit contacts across the synaptic complex (Figs 2 and 3). The four separate N-terminal domains have a low root-mean-square deviation (∼0.5 Å) between them.

The attacking VchIntIA subunit is shown in green (right) and the non-attacking VchIntIA subunit in magenta (left). Although both subunits use their N- and C-terminal domains to encompass the DNA, forming a clamp, they do not share equivalent protein–DNA contacts (Supplementary Fig. 4). In the non-attacking subunit, the β-4,5 hairpin contacts the DNA via the extrahelical base T12″. Tyr 302 from both subunits is shown in pink.
The carboxy-terminal domain (residues 105–320) contains the characteristic insertion (residues 192–210, Supplementary Fig. 3b) found only in IntIs. The αI2 helix within this essential region3, as discussed below, has an important role in synapse formation. The rest of the C-terminal domain is structurally similar to other tyrosine recombinase family members10,18,20,21,22.
Substrate recognition: an adaptive molecular switch
VchIntIA binds its attC substrate as a dimer. However, unlike other tyrosine recombinases, it does not form symmetrical protein–DNA contacts (Supplementary Fig. 4). The attacking subunit (green, Fig. 3) positions Tyr 302 to engage the scissile phosphate, and to trigger first-strand cleavage and transfer. The N- and C-terminal domains of this subunit wrap around the DNA, forming a clamp burying 4,000 Å2 of accessible surface area. Two base contacts are made at this half-site, both through Lys 160. The positioning of this invariant catalytic residue is important, as it may have a role in the protonation of the 5′-hydroxyl-leaving group after strand cleavage23. The remainder of this interface is characterized by protein–DNA backbone phosphate contacts.
The non-attacking integrase subunit (magenta, Fig. 3) forms two distinct protein–DNA contact points, burying 5,000 Å2 of accessible surface area. One of these contacts contains the β-4,5 hairpin interacting with the flipped-out nucleotide T12″, which dictates the position of the integrase dimer along the DNA (Fig. 4a, b). This extrahelical base is inserted between two stacked histidines (His 240 and His 241; invariant among IntIs) at one end and a highly conserved proline (Pro 232) at the other end, to form a tight non-polar nucleotide–protein interface. Interruption of this interface alters the ability of VchIntIA to bind the VCRbs duplex in vitro, as discussed below.

a, Interaction with β-4,5 from a non-attacking subunit and the extrahelical base T12″. b, Detailed view of the cis interaction depicted in a. Base T12″ (red) forms a protein–nucleic acid interface by stacking between His 240 and Pro 232. Experimental electron densities after phase modification are shown (1.5σ contour level). c, Ribbon diagram showing G20″ (blue) binding in trans between Trp 157 and Trp 219. The β-4,5 hairpin from the attacking subunit does not contact the DNA. d, Detailed view of the trans interactions shown in c. The NH2 group of G20″ forms hydrogen bonds with the Oγ of Glu 145 and N1 of Trp 157 within the attacking subunits. Non-specific contacts are made with the adjoining non-attacking subunit (magenta). A 2.8-Å 2Fo - Fc simulated annealed omit electron density map (1.6σ contour level) is shown. G20″ and all protein residues labelled in d were omitted during map calculation.
The larger protein–DNA contact of this non-attacking subunit forms a DNA footprint that is roughly a mirror image of the attacking subunit interface, covering half a helical turn of DNA and sharing many of the equivalent non-specific protein–DNA phosphate contacts (Supplementary Fig. 4). Notably, this subunit—although centred on G20″—does not make direct contacts with this extrahelical base. Instead, this flipped-out base binds within a deep hydrophobic pocket located in the attacking subunit (C) across the synaptic interface (Fig. 2b).
IntIs must recognize two architecturally distinct sites during cassette integration (attI × attC). To achieve this dual site-specificity, we propose that IntIs have developed a molecular switch that allows sequence-degenerate binding (attC), as seen in the VchIntIA–VCRbs complex reported here, as well as sequence-dependent binding during attI site recognition. The molecular switch that adapts to these two different modes of binding is the β-4,5 hairpin. During VCRbs (attC) binding, the β-4,5 hairpin is found in two environments corresponding to each unique half-site along the DNA. In the non-attacking subunit, the β-4,5 hairpin recognizes the molecular marker T12″ (Fig. 4a, b), whereas in the attacking one it does not contact the DNA (Fig. 4c). We suggest that in the attacking interface, the position of the β-4,5 hairpin may have rotated away from the minor groove relative to its position when IntIs are in complex with an attI site, possibly owing to the trans-interaction mediated by G20″ (Supplementary Fig. 5). During attI binding, the β-4,5 hairpin would make contacts with the minor groove analogous to the equivalent disposition of this element in the Cre–lox system10. The increased DNA footprint for attI site binding (Supplementary Fig. 4) relative to the attC interface reported here supports such a hypothesis8.
Synaptic assembly: a multiple structure problem
Assembly of a tyrosine recombinase synapse is mediated by a highly specific set of intersubunit protein–protein interactions, induced by DNA target capture. This stepwise assembly ensures that strand exchange takes place only between appropriate DNA substrates. How can IntIs, which bind a vast array of attC sequences, guarantee competent assembly of their DNA excision synapse? The strategy they have adopted is to use an invariant flipped-out DNA base, acting as a linchpin, to position the two dimers in the synapse (Fig. 4c, d). This flipped-out base (G20″) guarantees proper geometrical assembly by inserting itself within the hydrophobic pocket, created by Trp 157 and Trp 219, across the synaptic interface. This interaction allows helix αI2 from the attacking subunit(s) to form several important DNA contacts in trans, holding the synapse together.
These trans interactions mediated by G20″ result in only a two-fold symmetric synapse, which may inhibit HJ isomerization as discussed below. Subunits bound to the same VCRbs bury only 1,600 Å2 of solvent-accessible protein surface, by docking the C-terminal helix (αN) of the attacking subunit in the neighbouring non-attacking subunit10,24 (Fig. 2b, Fig. 3). The spacing and geometry of the bases in the central region of the DNA duplex preclude intersubunit interaction between the N termini on the same VCRbs (Fig. 2a, Fig. 3). The more extensive intersubunit interface across the synapse buries 4,000 Å2 of accessible surface area. These interfacial contacts are mostly due to C-terminal helix (αN) exchange, helix αA in the attacking subunits contacting the helix-turn-helix region of the αC–αD helices within the non-attacking subunits across the synapse, and the unusual trans interactions mediated by the extrahelical base G20″. A Supplementary Movie showing the final model of the VchIntIA–VCRbs synaptic complex is available in Supplementary Information.
Integron site-specific recombination
Recently, exploiting conjugation as a medium to exclusively deliver single-stranded attC substrates, we demonstrated that the bottom strand of attC recombined with a resident attI site at a rate that was 1,000-fold higher than the comparable top strand of attC12. Disruption of the postulated13,14,15,16 secondary structure of attC affected recombination. On the basis of these results we proposed a recombination model (Fig. 1c). The structure of the VchIntIA–VCRbs complex presented here provides a structural basis for IntI-mediated site-specific recombination using the bottom strand of attC as a substrate.
The biological relevance of the observed structural aspects of our model—that is, the cis and trans interactions observed in the VchIntIA–VCRbs complex—were investigated using a combination of EMSAs and in vivo excision assays13. Four discrete bands are observed with wild-type VchIntIA–VCRbs (Supplementary Fig. 6a), potentially corresponding to the occupancy of the four available half-sites. Deletion of T12″ from VCRbs markedly reduced the overall amount of complex formation as judged by EMSA (Supplementary Fig. 6a), with, however, only a fivefold drop in excision frequency (Supplementary Fig. 6c). Mutagenesis of His 240 (H240V), which interacts with T12″ in the crystal, did not allow us to further investigate this interaction as the mutant protein formed aggregates with VCRbs (Supplementary Fig. 6b, lane 5). Notably, deletion of G20″ from VCRbs affected both the band intensity and the migration pattern, with a quantitative increase of the fastest migrating band (Supplementary Fig. 6a). This suggests a higher occupancy of only one half-site and a change in the effective radius/molecular weight or nature of higher-order complexes. This same mutated sequence tested in vivo resulted in a 10,000-fold drop in excision activity (Supplementary Fig. 6c). Preliminary in vivo mutagenesis experiments of key residues (W157I and W219I) also produced a large reduction (1,000-fold) in excision frequency. These results suggest that G20″ has a central role in maintaining an active VchIntIA–VCRbs synapse, but that other factors—namely C-terminal helix exchange, and B/C and A/D interfaces—also contribute to the assembly of the tetrameric synapse.
A folded single-strand attC substrate during integron recombination necessitates that second-strand cleavage and transfer is downregulated relative to most members of the tyrosine recombinase family. A second round of cleavage and transfer does not lead to the excision/insertion of the gene cassettes but only to rearrangements within the attC sites (Supplementary Fig. 7). This regulation probably occurs at the HJ isomerization step, and hence perturbs the next stage of the reaction in IntI recombination25. Studies based on λ, XerC/D, Flp and Cre systems support models of HJ isomerization that involve only limited branch migration resulting in subtle movements of the quaternary structure10,20,26,27,28. The two isomeric HJ intermediates have structures that are similar except for an exchange in the roles of their DNA strands29. In this model, the protein–protein interface between subunits is maintained. Small changes between subunits, which result in the loss of interfacial binding energy, are regained by reciprocal changes in symmetrically related interfaces. These HJ intermediates thus have similar free energies, which explain the lack of bias observed in their resolution in the Cre and Flp systems30. These reciprocal intersubunit changes require a pseudo four-fold symmetric synapse. We suggest that as the VchIntIA–VCRbs synapse is only two-fold symmetric—and thus contains non-equivalent subunit interfaces (Fig. 2a)—the reciprocal changes needed to maintain the iso-energetic intermediates are lost. This may lead to an increased energy barrier for isomerization and thus produce a larger population of HJ intermediates that have not isomerized—that is, with a bias to revert to the original substrates. However, this group of stalled HJ intermediates then needs to be resolved via other cellular processes (Fig. 1c).
In addition, the rotation (∼15°) of the C-terminal domains within the non-attacking subunits could also reduce the rate of second-strand cleavage (Supplementary Fig. 8). This movement is a consequence of binding the extrahelical base T12″. It results in the translation of helix αM, and repositions nucleophilic Tyr 302 away from the DNA backbone to a distance of ∼7 Å in the non-attacking subunits.
Increased genomic diversity through broad DNA-specificity
Several mechanisms mediate genetic exchange between prokaryotes, but the introduction of foreign DNA does not guarantee its assimilation. Processes such as DNA recombination are needed if persistence as an episome is not established. These recombination systems, however, place restrictions on the type of DNA that can be transferred. Homologous recombination primarily rearranges sequences among closely related taxa and is unlikely to allow for the introduction of novel traits31. Site-specific recombination, by contrast, can mediate the exchange of more diverse traits, but normally these systems are still constrained by the need for the necessary core sites for recognition and catalysis2. However, IntIs have evolved an ingenious way of allowing highly diverse genetic traits, containing little sequence homology at their DNA target sites, to be recognized and assimilated into the integron element. Although the positioning of DNA bases in an extrahelical geometry for genomic repair is well documented32, the use of extrahelical bases for DNA recognition of single-stranded DNA, as well as assembly of higher-order protein–DNA complexes, is gathering more support12,33,34. We suspect that as most of the DNA exchanged through lateral transfer is in the single-stranded form, be it through natural transformation or conjugation35, this may have contributed to the development of the mechanism reported here.
Methods
Structure determination and refinement
The preparation of VchIntIA and the DNA used to form co-crystals is described in Supplementary Information. X-ray diffraction data to 2.8 Å was collected at beamline ID14-4 of the European Synchrotron Radiation Facility (ESRF). A wavelength of 0.9793 Å, corresponding to the SeMet peak, was used for data collection at 100 K on a single co-crystal. X-ray data were processed with XDS36 and CCP437 program suites. Phases for the VchIntIA–VCRbs complex were determined by using the single-wavelength anomalous dispersion (SAD) method with SeMet-labelled protein. The position of substructure atoms, initial phases and solvent flattening were determined using BnP38. The atomic model was fitted into electron density using the program O39. The asymmetric unit contained four VchIntIA molecules and two antiparallel VCRbs duplexes. Residues 1 and 305–310 in subunit A and 305–307 in subunit C were not fit into the density. For the DNA helices, nucleotides 1–4 and 36–40 of chain E, nucleotides 1–5 and 40–43 of chain F, nucleotides 1–5 and 35–40 of chain G, and nucleotides 1–6 and 39–43 of chain H were not fit owing to poor electron density. DNA chains E and G correspond to strand 1, and DNA chains F and H correspond to strand 2 in Fig. 1d. The four protein subunits and DNA duplexes were all independently modelled and refined with CNS40 using maximum-likelihood target using amplitudes and phase probability distribution with alternate cycles of manual density fitting. The structural similarity of subunits A/C and B/D warranted the use of non-crystallographic symmetry restraints in the final rounds of refinement. This resulted in an Rcryst = 0.234 and Rfree = 0.262 at 2.8 Å, with good geometry for bond lengths and angles. All amino acids have (ϕ, ψ) backbone torsion angles in allowed regions of Ramachandran space. Statistics for the final model are given in Supplementary Table S1. The figures were produced with PYMOL (http://www.pymol.org).
VCRbs and VchIntIA mutagenic analysis
Details of EMSAs and preparation of mutants is described in Supplementary Information. The in vivo excision assay has been previously described13. Briefly, the assay measures the frequency at which a synthetic integron gene cassette—the reporter gene (lacIq)—is excised from its two flanking recombination sites, attCaadA7 and the VCR2/1. In all experiments, symmetrical mutations were introduced to both the VCR2/1 and attCaadA7 sites when possible. The equivalent base to T16″ is not present within attCaadA7 (see Supplementary Fig. 1a). This protocol was followed as previous experiments showed mutations introduced in either site independently led to a slight decrease in deletion frequency, whereas mutations introduced at identical positions in tandem resulted in considerably larger reductions in recombination frequency. This suggests that the presence of a wild-type attC site assists in integrase complex formation between a mutated site, probably in a cooperative fashion. This observation may be similar to the formation of the attC × attI synapse, where the extrahelical base G20″ within the attC site drives formation of the active synapse (D. Mazel, unpublished observations). Mutations in the attCaadA7 and VCR2/1 sites were introduced as described previously13.
References
Kazazian, H. H. Jr. Mobile elements: drivers of genome evolution. Science 303, 1626–1632 (2004)
Craig, N. L., Craigie, R., Gellert, M. & Lambowitz, A. M. (eds) Mobile DNA II (ASM Press, Washington DC, 2002)
Messier, N. & Roy, P. H. Integron integrases possess a unique additional domain necessary for activity. J. Bacteriol. 183, 6699–6706 (2001)
Chen, Y. & Rice, P. A. New insight into site-specific recombination from Flp recombinase–DNA structures. Annu. Rev. Biophys. Biomol. Struct. 32, 135–159 (2003)
Van Duyne, G. A structural view of tyrosine recombinase site-specific recombination. In Mobile DNA II (eds Craig, N. L., Craigie, R., Gellert, M. & Lambowitz, A. M.) (ASM Press, Washington DC, 2002)
Azaro, M. A. & Landy, A. Lambda integrase and the lambda Int family. In Mobile DNA II (eds Craig, N. L., Craigie, R., Gellert, M. & Lambowitz, A. M.) (ASM Press, Washington DC, 2002)
Stokes, H. W., O'Gorman, D. B., Recchia, G. D., Parsekhian, M. & Hall, R. M. Structure and function of 59-base element recombination sites associated with mobile gene cassettes. Mol. Microbiol. 26, 731–745 (1997)
Gravel, A., Fournier, B. & Roy, P. H. DNA complexes obtained with the integron integrase IntI1 at the attI1 site. Nucleic Acids Res. 26, 4347–4355 (1998)
Stark, W. M., Boocock, M. R. & Sherratt, D. J. Site-specific recombination by Tn3 resolvase. Trends Genet. 5, 304–309 (1989)
Guo, F., Gopaul, D. N. & van Duyne, G. D. Structure of Cre recombinase complexed with DNA in a site-specific recombination synapse. Nature 389, 40–46 (1997)
Hansson, K., Skold, O. & Sundstrom, L. Non-palindromic attI sites of integrons are capable of site-specific recombination with one another and with secondary targets. Mol. Microbiol. 26, 441–453 (1997)
Bouvier, M., Demarre, G. & Mazel, D. Integron cassette insertion: a recombination process involving a folded single strand substrate. EMBO J. 24, 4356–4367 (2005)
Biskri, L., Bouvier, M., Guerout, A. M., Boisnard, S. & Mazel, D. Comparative study of class 1 integron and Vibrio cholerae superintegron integrase activities. J. Bacteriol. 187, 1740–1750 (2005)
Rowe-Magnus, D. A., Guerout, A. M., Biskri, L., Bouige, P. & Mazel, D. Comparative analysis of superintegrons: engineering extensive genetic diversity in the Vibrionaceae. Genome Res. 13, 428–442 (2003)
Johansson, C., Kamali-Moghaddam, M. & Sundstrom, L. Integron integrase binds to bulged hairpin DNA. Nucleic Acids Res. 32, 4033–4043 (2004)
Francia, M. V., Zabala, J. C., de la Cruz, F. & Garcia Lobo, J. M. The IntI1 integron integrase preferentially binds single-stranded DNA of the attC site. J. Bacteriol. 181, 6844–6849 (1999)
Mazel, D., Dychinco, B., Webb, V. A. & Davies, J. A distinctive class of integron in the Vibrio cholerae genome. Science 280, 605–608 (1998)
Subramanya, H. S. et al. Crystal structure of the site-specific recombinase, XerD. EMBO J. 16, 5178–5187 (1997)
Aihara, H., Kwon, H. J., Nunes-Duby, S. E., Landy, A. & Ellenberger, T. A conformational switch controls the DNA cleavage activity of λ integrase. Mol. Cell 12, 187–198 (2003)
Chen, Y., Narendra, U., Iype, L. E., Cox, M. M. & Rice, P. A. Crystal structure of a Flp recombinase–Holliday junction complex: assembly of an active oligomer by helix swapping. Mol. Cell 6, 885–897 (2000)
Kwon, H. J., Tirumalai, R., Landy, A. & Ellenberger, T. Flexibility in DNA recombination: structure of the lambda integrase catalytic core. Science 276, 126–131 (1997)
Hickman, A. B., Waninger, S., Scocca, J. J. & Dyda, F. Molecular organization in site-specific recombination: the catalytic domain of bacteriophage HP1 integrase at 2.7 Å resolution. Cell 89, 227–237 (1997)
Krogh, B. O. & Shuman, S. Catalytic mechanism of DNA topoisomerase IB. Mol. Cell 5, 1035–1041 (2000)
Rafferty, J. B. et al. Crystal structure of DNA recombination protein RuvA and a model for its binding to the Holliday junction. Science 274, 415–421 (1996)
Gopaul, D. N. & Van Duyne, G. D. Structure and mechanism in site-specific recombination. Curr. Opin. Struct. Biol. 9, 14–20 (1999)
Arciszewska, L. K., Grainge, I. & Sherratt, D. J. Action of site-specific recombinases XerC and XerD on tethered Holliday junctions. EMBO J. 16, 3731–3743 (1997)
Biswas, T. et al. A structural basis for allosteric control of DNA recombination by λ integrase. Nature 435, 1059–1066 (2005)
Nunes-Duby, S. E., Azaro, M. A. & Landy, A. Swapping DNA strands and sensing homology without branch migration in λ site-specific recombination. Curr. Biol. 5, 139–148 (1995)
Gopaul, D. N., Guo, F. & Van Duyne, G. D. Structure of the Holliday junction intermediate in Cre–loxP site-specific recombination. EMBO J. 17, 4175–4187 (1998)
Meyer-Leon, L., Inman, R. B. & Cox, M. M. Characterization of Holliday structures in FLP protein-promoted site-specific recombination. Mol. Cell. Biol. 10, 235–242 (1990)
Ochman, H., Lawrence, J. G. & Groisman, E. A. Lateral gene transfer and the nature of bacterial innovation. Nature 405, 299–304 (2000)
Klimasauskas, S., Kumar, S., Roberts, R. J. & Cheng, X. HhaI methyltransferase flips its target base out of the DNA helix. Cell 76, 357–369 (1994)
Val, M. E. et al. The single-stranded genome of phage CTX is the form used for integration into the genome of Vibrio cholerae. Mol. Cell 19, 559–566 (2005)
Ronning, D. R. et al. Active site sharing and subterminal hairpin recognition in a new class of DNA transposases. Mol. Cell 20, 143–154 (2005)
Llosa, M., Gomis-Ruth, F. X., Coll, M. & de la Cruz, F. Bacterial conjugation: a two-step mechanism for DNA transport. Mol. Microbiol. 45, 1–8 (2002)
Kabsch, W. Automatic processing of rotation diffraction data from crystals of initially unknown symmetry and cell constants. J. Appl. Cryst. 26, 795–800 (1993)
Collaborative Computational Project, Number 4. The CCP4 suite: programs for protein crystallography. Acta Crystallogr. D 50, 760–763 (1994)
Weeks, C. M. et al. Towards automated protein structure determination: BnP, the SnB–PHASES interface. Z. Kristallogr. 217, 686–693 (2002)
Jones, T. A., Zou, J. Y., Cowan, S. W. & Kjeldgaard, M. Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr. A 47, 110–119 (1991)
Brunger, A. T. et al. Crystallography & NMR system: a new software suite for macromolecular structure determination. Acta Crystallogr. D 54, 905–921 (1998)
Acknowledgements
This work was supported by laboratory start-up funds from the Pasteur Institute to D.N.G., research grants from the CNRS to D.N.G., research grants from the Institut Pasteur and from the CNRS on transposable elements to D. Mazel, and a French Ministry of Research (MENESR) grant to D. Mazel and D.N.G. D. MacDonald is a Florence Gould Scholar funded by the Pasteur Foundation of New York. G.D. and M.B. are doctoral fellows from the Fondation pour la Recherche Médicale and MENESR, respectively. We also thank T. Ellenberger and G. Liou for critical review of this manuscript. Author Contributions D. MacDonald carried out the in vitro assays, structural determination and wrote the paper with editing from D.N.G. D. Mazel provided the conceptual framework for the substrate and invaluable background. G.D. and M.B. performed the in vivo excision frequency assays. All authors discussed the results and commented on the manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
Coordinates and structure factors are deposited in the Protein Data Bank under accession code 2A3V. Reprints and permissions information is available at npg.nature.com/reprintsandpermissions. The authors declare no competing financial interests.
Supplementary information
Supplementary Figure 1
This figure contains the predicted secondary structures of attC bottom strands. (PDF 94 kb)
Supplementary Figure 2
Supplementary Figure 2 nature04643-s02.pdf This figure contains stereo views of the VchIntIA–VCRbs active sites. (PDF 941 kb)
Supplementary Figure 3
This figure contains a stereo model of a VchIntIA attacking subunit and the sequence alignment of VchIntIA and Cre recombinase with their corresponding secondary structure elements. (PDF 259 kb)
Supplementary Figure 4
This figure contains a schematic representation of the protein-DNA contacts between VchIntIA and VCRbs. (PDF 225 kb)
Supplementary Figure 5
This figure contains a model of the proposed movement of the β 4,5 hairpin. (PDF 841 kb)
Supplementary Figure 6
This figure contains data from EMSA and in vivo excision assays. (PDF 124 kb)
Supplementary Figure 7
This figure contains proposed IntI double strand exchange pathway via a single-stranded DNA substrate. (PDF 141 kb)
Supplementary Figure 8
This figure contains the rotation of the C-terminal domain within the non-attacking subunits. (PDF 633 kb)
Supplementary Table 1
Data collection and refinement statistics (PDF 125 kb)
Supplementary Movie 1
This movie shows the final model of the VchIntIA–VCRbs synaptic complex. (WMV 4614 kb)
Supplementary Notes
This file contains Supplementary Methods, Supplementary Figure Legends and additional references. (DOC 56 kb)
Rights and permissions
About this article
Cite this article
MacDonald, D., Demarre, G., Bouvier, M. et al. Structural basis for broad DNA-specificity in integron recombination. Nature 440, 1157–1162 (2006). https://doi.org/10.1038/nature04643
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1038/nature04643
This article is cited by
-
Influence of class 2 integron integrase concentration on gene cassette insertion and excision in vivo
Brazilian Journal of Microbiology (2023)
-
The association of group IIB intron with integrons in hypersaline environments
Mobile DNA (2021)
-
Comment on “Conserved phylogenetic distribution and limited antibiotic resistance of class 1 integrons revealed by assessing the bacterial genome and plasmid collection” by A.N. Zhang et al.
Microbiome (2021)
-
Comparison of Class 2 Integron Integrase Activities
Current Microbiology (2021)
-
A comprehensive survey of integron-associated genes present in metagenomes
BMC Genomics (2020)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
