
Abstract
Voltage changes across the cell membrane control the gating of many cation-selective ion channels. Conserved from bacteria to humans1, the voltage-gated-ligand superfamily of ion channels are encoded as polypeptide chains of six transmembrane-spanning segments (S1–S6). S1–S4 functions as a self-contained voltage-sensing domain (VSD), in essence a positively charged lever that moves in response to voltage changes. The VSD ‘ligand’ transmits force via a linker to the S5–S6 pore domain ‘receptor’2, thereby opening or closing the channel. The ascidian VSD protein Ci-VSP gates a phosphatase activity rather than a channel pore, indicating that VSDs function independently of ion channels3. Here we describe a mammalian VSD protein (HV1) that lacks a discernible pore domain but is sufficient for expression of a voltage-sensitive proton-selective ion channel activity. Hv1 currents are activated at depolarizing voltages, sensitive to the transmembrane pH gradient, H+-selective, and Zn2+-sensitive. Mutagenesis of Hv1 identified three arginine residues in S4 that regulate channel gating and two histidine residues that are required for extracellular inhibition of Hv1 by Zn2+. Hv1 is expressed in immune tissues and manifests the characteristic properties of native proton conductances (). In phagocytic leukocytes4, are required to support the oxidative burst that underlies microbial killing by the innate immune system4,5. The data presented here identify Hv1 as a long-sought voltage-gated H+ channel and establish Hv1 as the founding member of a family of mammalian VSD proteins.
Similar content being viewed by others

Main
Hv1 was identified as a novel conserved human gene using bioinformatic searches based on known cation channels. The Hv1 gene is located on chromosome 12 at q24.11 and is encoded by seven exons, the first of which is predicted to be non-coding. Hv1 messenger RNA is encoded by multiple independent human expressed sequence tags that contain in-frame stop codons preceding the initiator ATG and 3′ poly-A+ tails. The predicted Hv1 protein is 273 amino acids in length (31.7 kDa, pI = 6.62) and forms four transmembrane segments (S1–S4), according to hydropathy analyses (Supplementary Fig. S1a, b). The overall transmembrane (TM) structure and placement of charged residues in hydrophobic domains is conserved among the vertebrate Hv1 orthologues (Supplementary Fig. S1a) and similar to both Ci-VSP (ref. 3) and the VSD of Kv1.2 (ref. 2). The amino-terminal ∼100 amino acids in Hv1 are homologous to protein and lipid phosphatases, but unlike the catalytically active Ci-VSP, core active site residues required for phosphatase activity6 are not conserved in Hv1.
Both the distribution of expressed sequence tags (UniGene, see http://www.ncbi.nlm.nih.gov/UniGene/ESTProfileViewer.cgi?uglist=Hs.334637) and high-throughput expression studies (GNF SymAtlas, see http://symatlas.gnf.org/SymAtlas/; search on MGC15619 protein) indicate that Hv1 mRNA is enriched in immune tissues such as lymph node, B-lymphocytes, monocytes and spleen. We found marked lymph node expression of Hv1 mRNA on a human RNA dot blot (Supplementary Fig. S1d, spot F7). Hv1 protein expression was detected with an affinity-purified rabbit polyclonal anti-peptide antibody that recognizes Hv1, green flurorescent protein (GFP)-Hv1 and Hv1-haemagglutinin (HA) at their appropriate molecular weights in western blots of transfected HEK cells, but not mock-transfected controls (Supplementary Fig. S2b). The antibody recognized Hv1 in human immune tissues including lymph node (Supplementary Fig. S2a), and the human leukocyte cell lines HL-60 and Jurkat (Supplementary Fig. S2b, c).
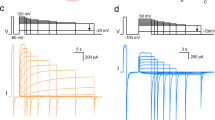
In preliminary experiments, we observed large (> 1 nA at +100 mV) voltage-dependent, outwardly rectifying whole-cell currents in transfected HEK cells bathed in standard Ringer's, low-Cl- (where all but 8 mM Cl- was substituted with  , SCN- or Br-), or NMDG+(N-methyl-d-glucamine)·Cl (Na+, K+, Ca2+ and Mg2+-free) solutions, suggesting that Hv1 currents were primarily permeable to H+. To study H+ currents under better pH control, we used solutions with high H+ buffering capacity (100 mM buffer used near its pKa: MES, pH 5.5; Bis-Tris, pH 6.5; HEPES, pH 7.5) similar to those used previously to study native (refs 7, 8). In response to depolarizing voltage steps, cells transfected with Hv1 generated time-, voltage-, and pH-dependent currents with relatively slow activation kinetics and a rapidly decaying inward tail current (Fig. 1a–c). The kinetics of Hv1 activation (τact = 715 ± 124 ms) and deactivation (τdeact = 65.0 ± 12.1 ms) were estimated from single exponential fits to the currents at +80 mV or -80 mV, respectively. Both τact and τdeact are strongly voltage-dependent (Fig. 1e) and τact was noticeably variable between experiments, perhaps owing to small fluctuations in room temperature and the highly temperature-sensitive gating of Hv1 (Supplementary Fig. S3c, d).
, SCN- or Br-), or NMDG+(N-methyl-d-glucamine)·Cl (Na+, K+, Ca2+ and Mg2+-free) solutions, suggesting that Hv1 currents were primarily permeable to H+. To study H+ currents under better pH control, we used solutions with high H+ buffering capacity (100 mM buffer used near its pKa: MES, pH 5.5; Bis-Tris, pH 6.5; HEPES, pH 7.5) similar to those used previously to study native (refs 7, 8). In response to depolarizing voltage steps, cells transfected with Hv1 generated time-, voltage-, and pH-dependent currents with relatively slow activation kinetics and a rapidly decaying inward tail current (Fig. 1a–c). The kinetics of Hv1 activation (τact = 715 ± 124 ms) and deactivation (τdeact = 65.0 ± 12.1 ms) were estimated from single exponential fits to the currents at +80 mV or -80 mV, respectively. Both τact and τdeact are strongly voltage-dependent (Fig. 1e) and τact was noticeably variable between experiments, perhaps owing to small fluctuations in room temperature and the highly temperature-sensitive gating of Hv1 (Supplementary Fig. S3c, d).

Depolarizing voltage steps (5 mV increments) were applied to an Hv1-transfected HM1 cell (a–c) to elicit outward H+ currents and deactivating inward tail currents (- 80 mV). TMA5.5o, TMA6.5o or TMA7.5o (TMA6.5i for all) were used to impose pH gradients indicated in the diagrams. a,  = 0.1, Vh = -40 mV (Vstep = +30 mV to +80 mV, scale bar 0.2 nA, 1 s; b, = 1, Vh = -40 mV (Vstep = -30 mV to +20 mV), scale bar 0.1 nA, 1 s; c, = 10, Vh = -70 mV (Vstep = -60 mV to -10 mV), scale bar 0.2 nA, 1 s. d, Hv1 currents at the end of the depolarizing step (Istep, open symbols) and the absolute value of Itail (- 80 mV, filled symbols) are plotted as a function of the step voltage. Data shown are from the cell shown in a–c. Circles, = 0.1 (Vthr = 70 ± 5.8 mV, n = 3); triangles, = 1.0 (Vthr = 11.7 ± 2.1 mV, n = 6); squares, = 10 (Vthr = -31.7 ± 3.3 mV, n = 3). e, Representative currents for Erev measurement (Vh = -40 mV, Vstep = +40 mV, Vtail = -100 mV to +40 mV). Small Istep (< 200 pA) was chosen to minimize H+i depletion (note constant outward current level). f, Monoexponential fits of Itail were extrapolated to t = 0 and Erev was estimated from the zero-current intercept of linear fits to the data. Open circles, TMA6.5i, TMA5.5o ( = 0.1); filled triangles, TMA6.5i, TMA6.5o ( = 1); open squares, TMA6.5i, TMA7.5o ( = 10). Average Erev values: Erev = 53.3 ± 1.4 mV, = 0.1, n = 10; Erev = 0.9 ± 0.7 mV, = 1.0, n = 14; Erev = -56.5 ± 2.6 mV, = 10, n = 5. Data represent mean ± s.e.m. from n experiments. g, Vthr is plotted against the Nernst potential for H+ at 24 °C. The data are fitted to Vthr = 0.82Erev + 13.8 mV (solid line). Data represent mean ± s.e.m. from n = 3–7 experiments. The Vthr versus Erev relationship for native (Vthr = 0.79 Erev + 23 mV4, dotted line) is shown for comparison.
= 0.1, Vh = -40 mV (Vstep = +30 mV to +80 mV, scale bar 0.2 nA, 1 s; b, = 1, Vh = -40 mV (Vstep = -30 mV to +20 mV), scale bar 0.1 nA, 1 s; c, = 10, Vh = -70 mV (Vstep = -60 mV to -10 mV), scale bar 0.2 nA, 1 s. d, Hv1 currents at the end of the depolarizing step (Istep, open symbols) and the absolute value of Itail (- 80 mV, filled symbols) are plotted as a function of the step voltage. Data shown are from the cell shown in a–c. Circles, = 0.1 (Vthr = 70 ± 5.8 mV, n = 3); triangles, = 1.0 (Vthr = 11.7 ± 2.1 mV, n = 6); squares, = 10 (Vthr = -31.7 ± 3.3 mV, n = 3). e, Representative currents for Erev measurement (Vh = -40 mV, Vstep = +40 mV, Vtail = -100 mV to +40 mV). Small Istep (< 200 pA) was chosen to minimize H+i depletion (note constant outward current level). f, Monoexponential fits of Itail were extrapolated to t = 0 and Erev was estimated from the zero-current intercept of linear fits to the data. Open circles, TMA6.5i, TMA5.5o ( = 0.1); filled triangles, TMA6.5i, TMA6.5o ( = 1); open squares, TMA6.5i, TMA7.5o ( = 10). Average Erev values: Erev = 53.3 ± 1.4 mV, = 0.1, n = 10; Erev = 0.9 ± 0.7 mV, = 1.0, n = 14; Erev = -56.5 ± 2.6 mV, = 10, n = 5. Data represent mean ± s.e.m. from n experiments. g, Vthr is plotted against the Nernst potential for H+ at 24 °C. The data are fitted to Vthr = 0.82Erev + 13.8 mV (solid line). Data represent mean ± s.e.m. from n = 3–7 experiments. The Vthr versus Erev relationship for native (Vthr = 0.79 Erev + 23 mV4, dotted line) is shown for comparison.
In the absence of a H+ gradient (the ratio of internal to external H+ concentration = 1.0), the voltage threshold (Vthr) for activation of outward currents (Istep) was 11 mV (Fig. 1d). An outwardly directed H+ gradient, equivalent to net intracellular acidification ( = 10), caused Vthr to shift 40 mV negative (Fig. 1d). The reversal potential (Erev) of open Hv1 channels determined from Itail (Fig. 1e) was ∼0 mV under symmetrical recording conditions ( = 1) and shifted >50 mV negative or positive in inward ( = 0.1) or outward ( = 10) H+ gradients, respectively (Fig. 1f). A plot of Erev versus [H+] was well fitted to a line with slope = 54.7 mV/log[H+] (data not shown), indicating that the Hv1 current was highly selective for H+. A plot of Vthr versus the calculated Nernst potential for H+ was well fitted by the line Vthr = 0.82Erev + 13.8 mV (Fig. 1g).
Although the outwardly rectifying Istep–V relation was linear at voltages ∼10–70 mV positive to Erev (Fig. 1d, Fig. 2a, c), we were surprised to find that the Hv1 Istep–V deviated from linearity and appeared to saturate at voltages ∼100 mV positive to Erev (Fig. 2a, c). We infer that the apparent nonlinearity in Istep results from depletion of H+i caused by rapid channel-mediated H+ efflux that is not adequately compensated by delivery of H+i from the buffer during the voltage step. Slow Hv1 kinetics near Vthr further hinder our ability to measure accurately the channels' voltage dependence because extraordinarily long voltage pulses are required to reach true steady state. Erosion of the H+ gradient during Istep and underestimation of Istep are both expected to flatten the Itail–V relation. We were nonetheless able to fit Hv1 Itail–V relations ( = 10) to a Boltzmann function in order to approximate the voltage-dependence of the Hv1 open channel probability (Po, Fig. 2c). In an outward H+ gradient, Hv1 channels appear to be half open at ∼60 mV and to move the equivalent of about one electronic charge across the membrane during channel opening (zδ = 0.9). The midpoint of the Istep–V relation shifted >40 mV when the H+ gradient was altered, consistent with the observed shift in Vthr (Fig. 1).

a, Hv1 currents (- 60 mV to +120 mV, Vh = -40 mV, = 10, Na6.5i, Na7.5o) in a representative cell. Scale bar 2 nA, 400 ms. Under symmetrical conditions (TMA6.5, = 1), τact = 715 ± 124 ms (+ 80 mV), n = 9 and τdeact = 65.0 ± 12.1 ms (- 80 mV), n = 7. b, R205A (- 60 mV to +180 mV, Vh = -60 mV, = 10, Na6.5i, Na7.5o). Scale bar 2 nA, 10 ms. Note the 40-fold difference in timescale compared to a. c, The Itail–V relation (- 80 mV, = 10, Na6.5i, Na7.5o) was normalized to the maximum current obtained from a Boltzmann fit to the data for each cell expressing Hv1 (filled circles) or R205A (open circles) to estimate Po - V. Data were fitted to a Boltzmann function (solid lines) and normalized to the extrapolated maximum current. Points represent mean ± s.e.m. of normalized data. A comparison of curve fits from individual experiments (Hv1, V0.5 = 58.0 ± 5.6 mV, zδ = 0.90 ± 0.04, n = 6; R205A, V0.5 = 99.5 ± 18.3 mV, zδ = 0.57* ± 0.03, n = 3; *P = 0.03 by Student's non-paired t-test) indicates that significantly less effective charge is moved in R205A than in Hv1 during channel gating. Data represent mean ± s.e.m. from n experiments.
In an initial attempt to determine whether voltage-dependent gating in Hv1 involved basic residues in S4, as it does in other voltage-gated channels, we individually replaced each of three arginine residues with alanine. Representative traces from cells expressing Hv1 or the point mutant R205A are shown in Fig. 2a, b. Relative to Hv1, two arginine mutants (R205A, R208A) exhibited dramatically faster activation and deactivation kinetics, whereas R211A exhibited only faster deactivation kinetics (Supplementary Fig. S3e–g). R205A exhibited a positive shift in the half-maximal voltage for channel activation (V0.5) and a statistically significant decrease in the slope factor (zδ) derived from Boltzmann fits to Itail–V (Fig. 2c).
Like , HV1 current was rapidly and reversibly blocked by bath superfusion of ZnCl2 in the absence of EGTA (Fig. 3a, e). We therefore mutated putatively extracellular residues that might coordinate metal ions to inhibit Hv1 currents. Zn2+ potently inhibited IHv1 (half-maximal inhibitory concentration, IC50 ≈ 2 µM) and slowed τact (Fig. 3a–c, data not shown). Apparent Zn2+ potency decreased 10- or 30-fold when we mutated H193 or H140 to alanine, respectively. Mutation of both residues (H140A/H193A) practically abolished Zn2+ sensitivity (Fig. 3d). All three histidine mutants exhibited the expected H+-selectivity and -dependent shift in Vthr that are characteristic of Hv1 (data not shown).

Inhibition of Istep (+ 25 mV to +50 mV) in cells superfused with EGTA-free TMA6.5 or Na6.5 ( = 1) was typically faster than the sampling interval (10 s) and washout was always complete. Average IC50 = 2.2 ± 0.6 µM, nH = 1.0 ± 0.2, n = 4; = 1, + 40 mV, TMA6.5. Data represent mean ± s.e.m. from n experiments. The applied voltages and [Zn2+] for the records shown (Na6.5, = 1) were: a, Hv1, Vstep = +40/Vtail = -80 mV, [Zn2+] = 0, 0.1, 1, 10, 100 µM; b, H140A, +40/ - 60 mV, [Zn2+] = 0, 1, 10, 100 µM; c, H193A, +30/ - 80 mV, [Zn2+] = 0, 10, 100 µM; d, H140A/H193A, +50/ - 20 mV, [Zn2+] = 0, 1 mM. Scale bars: 100 pA, 1 s. e, Representative Zn2+ concentration-response curves for Hv1 (filled squares, IC50 = 1.9 µM, nH = 0.9), H193A (open triangles, IC50 = 17.9 µM, nH = 1.2), H140A (filled circles, IC50 = 74.3 µM, nH = 1.0), and H140A/H193A (open squares, maximum inhibition = 11.1 ± 3.4%, n = 3).
The surprising finding of this study is that expression of a voltage-sensor domain, without its normally associated pore domain, is sufficient to reconstitute voltage-dependent proton channel activity. have been recorded in a wide variety of cells from molluscs9 to mammals (reviewed in ref. 4). In mammalian phagocytes, provides necessary charge compensation for the gp91phox electron current that underlies Zn2+-sensitive superoxide anion production4,5,10,11. Loss of functional gp91phox in humans causes X-linked chronic granulomatous disease, which is characterized by decreases in microbial killing by phagocytes12,13.
The properties of expressed Hv1 are consistent with those of (refs 4, 14, 15). Hv1 is H+-selective, exhibits a -40 mV shift in Vthr when pHi is decreased one unit, is blocked by micromolar Zn2+, and is highly temperature sensitive. Hv1 kinetics fall within the range reported for in a variety of mammalian cells4. Kinetic heterogeneity among native could reflect the existence of unidentified Hv1 homologues, temperature differences during recording, post-translational modification of Hv1, or second-messenger modulation of H+ channels. The small endogenous reported in HEK16 and CHO17 (but not COS-718) cells appeared neither to facilitate nor interfere with expression of Hv1 current in this study. Slow activation kinetics and large H+ currents in Hv1-transfected cells will tend to exacerbate difficulties in measuring steady-state channel behaviour and in maintaining the imposed H+ gradient during strongly depolarizing voltage pulses, and could explain why the voltage dependence of expressed Hv1 channels appears shallow relative to native currents4.
An accepted model for voltage-gated channels is one in which positively charged residues in S4 function as the primary voltage sensors19,20. Charge-neutralizing mutagenesis of S4 arginine residues (R205A, R208A, R211A) demonstrates that these basic residues are intimately involved in Hv1 gating and may directly sense transmembrane voltage. Voltage-dependent gating in Hv1 is distinguished from other cation channels by its sensitivity to a H+ gradient, indicating that further studies are required to clarify whether the mechanism of voltage sensing in Hv1 is similar to that in other voltage-gated channels. Starace and Bezanilla21 replaced an S4 arginine in Shaker with histidine (R362H) to create a H+-permeable channel at negative membrane potentials. In Hv1, neutralizing S4 arginines or histidines in the extracellular linkers did not prevent the H+ conductance, indicating that H+ permeation does not require proton acceptors at these positions. Mutagenesis of putative H+ acceptors may be of limited use in defining the conduction pathway given that H+ conductance could be indirectly attenuated owing to allosteric effects. The mechanism of proton permeation may involve a voltage-dependent movement of S4 to align residues that create a ‘proton wire’, as in gramicidin22, or exposure of an H+ acceptor that bridges two water-filled crevices21. The latter model unites both pore and wire features in that the proton acceptor can also be thought of as the binding site for H+ within a pore that confers H+ selectivity.
Polyvalent cations, particularly Zn2+, are the only known blockers of native (refs 4, 15). Zn2+ block of native proton current was predicted to involve two extracellularly accessible amino acids; apparent Zn2+ potency is modulated by both [H+]o and membrane voltage15. Strikingly, we identified two histidine residues that appear to function coordinately to confer Zn2+ inhibition to Hv1. In addition, Zn2+ slows τact, mirroring its effect on (ref. 15) and further supporting the conclusion that Hv1 activity underlies native .
Expression of Hv1 alone is sufficient to reconstitute most properties of , making it unlikely that is mediated by gp91phox (refs 23, 24). Future studies are required to ascertain whether Hv1 physically associates with or is regulated by components of the NADPH oxidase complex, protein kinase C, or arachidonic acid25,26,27, whether Hv1 channel activity is sufficient to control O2- secretion and reactive oxygen species production28,29, and what factors account for differences between ‘phagocyte’ and ‘epithelial’ type (ref. 4). Finally, future structural and biophysical studies should help to elucidate the proton permeation pathway within this voltage sensor domain protein.
Note added in proof: Okamura and colleagues also report that the mouse Hv1 gene encodes a proton channel, which they call VSOP (voltage-sensor only protein)30.
Methods
Hv1 complementary DNA was amplified by polymerase chain reaction (PCR) from an expressed sequence tag clone (IMAGE 6424182) and subcloned into pQBI25-fC3 (QBiogene) to create GFP–Hv1, pcDNA3.1(- ) for expression of non-tagged Hv1, or pBSIISK - (Stratagene) for in vitro mRNA transcription. Site-directed mutagenesis was used to create point mutations in GFP–Hv1: H140A, H193A, R205A, R208A, R211A, and the H140A/H193A double mutant; Hv1-HA was used for biochemical studies. A ∼300 nucleotide antisense RNA probe labelled with α-33P-UTP was synthesized by in vitro transcription (Ambion) and hybridized (68 °C) to a human multiple tissue mRNA panel (MTE3, Clontech). Incorporated radioactivity was detected by a phosphorimager (Molecular Dynamics).
Currents reported here were recorded 18–36 h after transfection of GFP–Hv1 cDNA in the HEK-293 cell clone HM1. Recording solutions were similar to those described previously18 and contained 100 mM pH buffer (MES, Bis-Tris or HEPES) near its pKa (5.5, 6.5 or 7.5, respectively) in tetramethylammonium methanesulphonate or NaCl adjusted to ∼300 mOsm. H+ gradients ( = [H+]intracellular/[H+]extracellular) were imposed by gravity-fed bath superfusion of differentially buffered solutions. Currents were recorded with an Axopatch 200A (Axon Instruments; 1 kHz filter) and digitized at 2 kHz (10 kHz for Arg mutants) using Clampex9 (Axon Instruments). Data were analysed using Clampfit9 (Axon Instruments) and Origin 6 (Microcal). Recordings were performed at 22–24 °C unless otherwise stated. Data shown and stated in text and legends represent mean ± s.e.m. for the indicated number of experiments.
Affinity-purified 4234 polyclonal rabbit antibodies (Chemicon) were raised against the keyhole limpet haemocyanin (KLH)-conjugated Hv1 peptide CSEKEQEIERLNKL. Transfected cells were lysed in 1% Triton X-100 and crude membranes were subjected to SDS–polyacrylamide gel electrophoresis (PAGE) (NuPage, Invitrogen). Following transfer to nitrocellulose, blots were probed with the indicated concentration of 4234 followed by an anti-rabbit HRP-conjugated secondary antibody (Zymed). Immunoreactivity was detected by enhanced chemiluminescence (Pierce). A human immune tissue western blot (1525, ProSci) was probed with 4234 as described.
References
Yu, F. H. & Catterall, W. A. The VGL-chanome: a protein superfamily specialized for electrical signaling and ionic homeostasis. Sci. STKE 2004, re15 (2004)
Long, S. B., Campbell, E. B. & Mackinnon, R. Crystal structure of a mammalian voltage-dependent Shaker family K+ channel. Science 309, 897–903 (2005)
Murata, Y., Iwasaki, H., Sasaki, M., Inaba, K. & Okamura, Y. Phosphoinositide phosphatase activity coupled to an intrinsic voltage sensor. Nature 435, 1239–1243 (2005)
DeCoursey, T. E. Voltage-gated proton channels and other proton transfer pathways. Physiol. Rev. 83, 475–579 (2003)
DeCoursey, T. E., Morgan, D. & Cherny, V. V. The voltage dependence of NADPH oxidase reveals why phagocytes need proton channels. Nature 422, 531–534 (2003)
Goldberg, J. et al. Three-dimensional structure of the catalytic subunit of protein serine/threonine phosphatase-1. Nature 376, 745–753 (1995)
Schilling, T., Gratopp, A., DeCoursey, T. E. & Eder, C. Voltage-activated proton currents in human lymphocytes. J. Physiol. (Lond.) 545, 93–105 (2002)
DeCoursey, T. E., Cherny, V. V., DeCoursey, A. G., Xu, W. & Thomas, L. L. Interactions between NADPH oxidase-related proton and electron currents in human eosinophils. J. Physiol. (Lond.) 535, 767–781 (2001)
Thomas, R. C. & Meech, R. W. Hydrogen ion currents and intracellular pH in depolarized voltage-clamped snail neurones. Nature 299, 826–828 (1982)
Henderson, L. M., Chappell, J. B. & Jones, O. T. Superoxide generation by the electrogenic NADPH oxidase of human neutrophils is limited by the movement of a compensating charge. Biochem. J. 255, 285–290 (1988)
Rada, B. K., Geiszt, M., Kaldi, K., Timar, C. & Ligeti, E. Dual role of phagocytic NADPH oxidase in bacterial killing. Blood 104, 2947–2953 (2004)
Babior, B. M. NADPH oxidase. Curr. Opin. Immunol. 16, 42–47 (2004)
Smith, R. M. & Curnutte, J. T. Molecular basis of chronic granulomatous disease. Blood 77, 673–686 (1991)
DeCoursey, T. E. & Cherny, V. V. Temperature dependence of voltage-gated H+ currents in human neutrophils, rat alveolar epithelial cells, and mammalian phagocytes. J. Gen. Physiol. 112, 503–522 (1998)
Cherny, V. V. & DeCoursey, T. E. pH-dependent inhibition of voltage-gated H+ currents in rat alveolar epithelial cells by Zn2+ and other divalent cations. J. Gen. Physiol. 114, 819–838 (1999)
Eder, C. & DeCoursey, T. E. Voltage-gated proton channels in microglia. Prog. Neurobiol. 64, 277–305 (2001)
Cherny, V. V., Henderson, L. M. & DeCoursey, T. E. Proton and chloride currents in Chinese hamster ovary cells. Membr. Cell Biol. 11, 337–347 (1997)
Morgan, D., Cherny, V. V., Price, M. O., Dinauer, M. C. & DeCoursey, T. E. Absence of proton channels in COS-7 cells expressing functional NADPH oxidase components. J. Gen. Physiol. 119, 571–580 (2002)
Bezanilla, F. The voltage sensor in voltage-dependent ion channels. Physiol. Rev. 80, 555–592 (2000)
Tombola, F., Pathak, M. M. & Isacoff, E. Y. How far will you go to sense voltage? Neuron 48, 719–725 (2005)
Starace, D. M. & Bezanilla, F. A proton pore in a potassium channel voltage sensor reveals a focused electric field. Nature 427, 548–553 (2004)
Pomes, R. & Roux, B. Molecular mechanism of H+ conduction in the single-file water chain of the gramicidin channel. Biophys. J. 82, 2304–2316 (2002)
DeCoursey, T. E., Morgan, D. & Cherny, V. V. The gp91phox component of NADPH oxidase is not a voltage-gated proton channel. J. Gen. Physiol. 120, 773–779 (2002)
Henderson, L. M. & Meech, R. W. Evidence that the product of the human X-linked CGD gene, gp91-phox, is a voltage-gated H+ pathway. J. Gen. Physiol. 114, 771–786 (1999)
Bokoch, G. M. Regulation of innate immunity by Rho GTPases. Trends Cell Biol. 15, 163–171 (2005)
DeCoursey, T. E., Cherny, V. V., Morgan, D., Katz, B. Z. & Dinauer, M. C. The gp91phox component of NADPH oxidase is not the voltage-gated proton channel in phagocytes, but it helps. J. Biol. Chem. 276, 36063–36066 (2001)
Price, M. O. et al. Creation of a genetic system for analysis of the phagocyte respiratory burst: high-level reconstitution of the NADPH oxidase in a nonhematopoietic system. Blood 99, 2653–2661 (2002)
Bokoch, G. M. & Knaus, U. G. NADPH oxidases: not just for leukocytes anymore! Trends Biochem. Sci. 28, 502–508 (2003)
Qu, A. Y., Nanda, A., Curnutte, J. T. & Grinstein, S. Development of a H+-selective conductance during granulocytic differentiation of HL-60 cells. Am. J. Physiol. 266, C1263–C1270 (1994)
Sasaki, M., Takagi, M. & Okamura, Y. A novel protein with a voltage sensor domain is a voltage-gated proton channel. Science (in the press)
Acknowledgements
We thank T. DeCoursey, C. Miller, R. MacKinnon and P. Bezanilla for comments on the manuscript, and K.-H. Lee for technical assistance. This work was supported by the Sandler Program for Asthma Research and the Howard Hughes Medical Institute.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
Reprints and permissions information is available at npg.nature.com/reprintsandpermissions. The authors declare no competing financial interests.
Supplementary information
Supplementary Figure Legends
This file contains text to accompany the Supplementary Figures. (DOC 56 kb)
Supplementary Figure 1
Hv1 amino acid sequence and expression profile. (PDF 1523 kb)
Supplementary Figure 2
a, Human tissue Western blot probed with 4234 antibody (5 µg/ml) demonstrates expression of native Hv1 protein (~32 kDa) in immune tissues. b, Western blot of total cell lysates prepared from Jurkat (lane 1) or HEK-293T cells transfected with the indicated cDNA (lanes 3-5). c, in HL-60 cells, Hv1 protein appeared to be increased when cells were cultured in the presence of 1.3% DMSO (lane 2). (PDF 1536 kb)
Supplementary Figure 3
a, Hv1-like currents were not detectable in HM1 cells. b, Native GvH+ in a DMSO-differentiated HL-60 cell. c, Temperature dependence of Hv1 currents. d, Monoexponential fits of τACT from the same cell as shown in panel c illustrate strong temperature-dependence of Hv1 kinetics. e, R205A currents. f, R208A currents. g, R211A currents. (PDF 2674 kb)
Rights and permissions
About this article
Cite this article
Ramsey, I., Moran, M., Chong, J. et al. A voltage-gated proton-selective channel lacking the pore domain. Nature 440, 1213–1216 (2006). https://doi.org/10.1038/nature04700
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/nature04700
This article is cited by
-
Control of intracellular pH and bicarbonate by CO2 diffusion into human sperm
Nature Communications (2023)
-
Vertebrate OTOP1 is also an alkali-activated channel
Nature Communications (2023)
-
Ion currents through the voltage sensor domain of distinct families of proteins
Journal of Biological Physics (2023)
-
Role of the Voltage-Gated Proton Channel Hv1 in Nervous Systems
Neuroscience Bulletin (2023)
-
Multiple mechanisms contribute to fluorometry signals from the voltage-gated proton channel
Communications Biology (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
