
Abstract
Our knowledge of species and functional composition of the human gut microbiome is rapidly increasing, but it is still based on very few cohorts and little is known about variation across the world. By combining 22 newly sequenced faecal metagenomes of individuals from four countries with previously published data sets, here we identify three robust clusters (referred to as enterotypes hereafter) that are not nation or continent specific. We also confirmed the enterotypes in two published, larger cohorts, indicating that intestinal microbiota variation is generally stratified, not continuous. This indicates further the existence of a limited number of well-balanced host–microbial symbiotic states that might respond differently to diet and drug intake. The enterotypes are mostly driven by species composition, but abundant molecular functions are not necessarily provided by abundant species, highlighting the importance of a functional analysis to understand microbial communities. Although individual host properties such as body mass index, age, or gender cannot explain the observed enterotypes, data-driven marker genes or functional modules can be identified for each of these host properties. For example, twelve genes significantly correlate with age and three functional modules with the body mass index, hinting at a diagnostic potential of microbial markers.
Similar content being viewed by others


Main
Various studies of the human intestinal tract microbiome based on the 16S ribosomal-RNA-encoding gene reported species diversity within and between individuals1,2,3, and the first metagenomics studies characterized the functional repertoire of the microbiomes of several American4,5 and Japanese6 individuals. Although a general consensus about the phylum level composition in the human gut is emerging1,3,7, the variation in species composition1,2 and gene pools5,8 within the human population is less clear. Furthermore, it is unknown whether inter-individual variation manifests itself as a continuum of different community compositions or whether individual gut microbiota congregate around preferred, balanced and stable community compositions that can be classified. Studying such questions is complicated by the complexity of sampling, DNA preparation, processing, sequencing and analysis protocols9 as well as by varying physiological, nutritional and environmental conditions. To analyse the feasibility of comparative metagenomics of the human gut across cohorts and protocols and to obtain first insights into commonalities and differences between gut microbiomes across different populations, we Sanger-sequenced 22 European metagenomes from Danish, French, Italian and Spanish individuals that were selected for diversity (Supplementary Notes section 1), and combined them with existing Sanger (13 Japanese6, 2 American4) and pyrosequencing (2 American5) gut data sets—totalling 39 individuals.
Global variation of human gut metagenomes
The vast majority of sequences in the newly sequenced 22 European samples belong to bacteria—only 0.14% of the reads could be classified as human contamination, all other eukaryotes together only comprised 0.5%, archaea 0.8% and viruses up to 5.8% (see Supplementary Notes section 2.1 for details).
To investigate the phylogenetic composition of the 39 samples from 6 nationalities, we mapped metagenomic reads, using DNA sequence homology, to 1,511 reference genomes (Supplementary Table 3) including 379 publicly available human microbiome genomes generated through the National Institutes of Health (NIH) Human Microbiome Project10 and the European MetaHIT consortium11 (Supplementary Methods section 4.1). To consistently estimate the functional composition of the samples, we annotated the predicted genes from the metagenomes using eggNOG12 orthologous groups (Supplementary Methods section 6.2). We ensured that comparative analysis using these procedures was not biased by data-set origin, sample preparation, sequencing technology and quality filtering (see Supplementary Notes section 1). We also investigated whether the relatively low and somewhat arbitrary amounts of sequence per sample (between 53–295 Mb) bias our results: we assigned habitat information to 1,368 of the 1,511 reference genomes, distinguished between orthologous groups from gut and ‘non-gut’ species and conclude that our data set captures most of the functions from gut species even though functions from non-gut species accumulated with each additional sample (Fig. 1a; see Supplementary Notes section 1.3).

a, Simulation of the detection of distinct orthologous groups when increasing the number of individuals (samples). Complete genomes were classified by habitat information and the orthologous groups divided into those that occur in known gut species (red) and those that have not yet been associated with gut (blue). The former are close to saturation when sampling 35 individuals (excluding infants) whereas functions from non-gut (probably rare and transient) species are not. b, Genus abundance variation box plot for the 30 most abundant genera as determined by read abundance. Genera are coloured by their respective phylum (see inset for colour key). Inset shows phylum abundance box plot. Genus and phylum level abundances were measured using reference-genome-based mapping with 85% and 65% sequence similarity cutoffs. Unclassified genera under a higher rank are marked by asterisks. c, Orthologous group abundance variation box plot for the 30 most abundant orthologous gruops as determined by assignment to eggNOG12. Orthologous groups are coloured by their respective functional category (see inset for colour key). Inset shows abundance box plot of 24 functional categories. Boxes represent the interquartile range (IQR) between first and third quartiles and the line inside represents the median. Whiskers denote the lowest and highest values within 1.5 × IQR from the first and third quartiles, respectively. Circles represent outliers beyond the whiskers.
We then characterized the phylogenetic variation across samples at the genus and phylum levels, and functional variation at gene and functional class levels. As infants are known to have very heterogeneous, unstable and distinctive microbiota6,13, we excluded the four respective Japanese samples from the analysis. Using calibrated similarity cutoffs (Supplementary Fig. 1), on average, 52.8% of the fragments in each sample could be robustly assigned to a genus in our reference genome set (ranging from 22% to 80.5%), and 80% could be assigned to a phylum (ranging from 64.9% to 91%) implying that the trends observed (Fig. 1b) represent a large fraction of the metagenome.
The phylogenetic composition of the newly sequenced samples confirms that the Firmicutes and Bacteroidetes phyla constitute the vast majority of the dominant human gut microbiota7 (Fig. 1b, inset). Bacteroides was the most abundant but also most variable genus across samples (Fig. 1b and Supplementary Notes section 2.2), agreeing with previous observations6,14. Our function identification protocol led to a high functional assignment rate: 63.5% of all predicted genes in the Sanger-sequenced samples analysed (41% of all predicted genes in two samples obtained by pyrosequencing; Supplementary Table 5) can be assigned to orthologous groups, and orthologous group abundance patterns agree with previous observations6,15 (for example, histidine kinases make up the largest group; Fig. 1c and Supplementary Notes section 2.3).
Abundant functions from low-abundance microbes
Microbes in the human gut undergo selective pressure from the host as well as from microbial competitors. This typically leads to a homeostasis of the ecosystem in which some species occur in high and many in low abundance16 (the ‘long-tail’ effect, as seen in Fig. 1b), with some low-abundance species, like methanogens17, performing specialized functions beneficial to the host. Metagenomics enables us to study the presence of abundant functions shared by several low-abundance species, which could shed light on their survival strategies in the human gut. In the samples analysed here, the most abundant molecular functions generally trace back to the most dominant species. However, we identified some abundant orthologous groups that are contributed to primarily by low-abundance genera (see Supplementary Fig. 2, Supplementary Table 6 and Supplementary Notes section 3). For example, low-abundance Escherichia contribute over 90% of two abundant proteins associated with bacterial pilus assembly, FimA (COG3539) and PapC (COG3188), found in one individual (IT-AD-5). Pili enable the microbes to colonize the epithelium of specific host organs; they help microbes to stay longer in the human intestinal tract by binding to human mucus or mannose sugars present on intestinal surface structures18. They are also key components in the transfer of plasmids between bacteria through conjugation, often leading to exchange of protective functions such as antibiotic resistance18. Pili can thus provide multiple benefits to these low-abundance microbes in their efforts to survive and persist in the human gut. This example illustrates that abundant species or genera cannot reveal the entire functional complexity of the gut microbiota. More reference genomes will facilitate better taxonomic assignment from samples and thus the detection of more low-abundance species. However, there is not much room for as yet undetected, abundant genera. Even with our limited genus assignment rate of 52.8% of all reads, we estimate that we miss another 30.7% of the already classified genera owing to our strict assignment criteria (Supplementary Fig. 1); that is, only 16.5% of all reads are likely to belong to hitherto unknown genera.
Detection of enterotypes, cross-national clusters
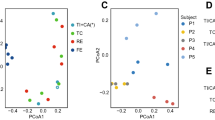
To get an overview of species variation we used phylogenetic profile similarities obtained by mapping metagenomic reads to the 1,511 reference genomes (Fig. 2a; see Supplementary Methods section 4.1). We excluded the two American Sanger-sequenced samples4 from further analysis because of an unusual, very low fraction of Bacteroidetes and suspected technical artefacts19. Multidimensional cluster analysis and principal component analysis (PCA) revealed that the remaining 33 samples formed three distinct clusters that we designate as enterotypes (see Supplementary Notes section 4.1, Supplementary Fig. 3a and Supplementary Table 8). Each of these three enterotypes are identifiable by the variation in the levels of one of three genera: Bacteroides (enterotype 1), Prevotella (enterotype 2) and Ruminococcus (enterotype 3) (Fig. 2a, d), which was reproduced using independent array-based HITChip20 data in a subset of 22 European samples (Supplementary Fig. 4 and Supplementary Notes section 4.5). The same analysis on two larger published gut microbiome data sets of different origins (16S pyrosequencing data from 154 American individuals5 and Illumina-based metagenomics data from 85 Danish individuals8; Supplementary Methods section 5) shows that these data sets could also be represented best by three clusters (Supplementary Fig. 3b, c and Supplementary Tables 9, 10). Two of these are also driven by Bacteroides and Prevotella, whereas the third cluster is mostly driven by related groups of the order Clostridiales, Blautia and unclassified Lachnospiraceae in the 16S rDNA and Illumina data, respectively (Fig. 2b, c). This can be explained by a different reference data set in the instance of the 16S rDNA data, different mapping behaviour of short reads in the case of the Illumina data or current taxonomic uncertainties in the Lachnospiraceae and Ruminococcaceae clades (see Supplementary Notes section 4.2). The differences might also hint at community subpopulations within this enterotype, which might only be detectable with substantially more samples. Correlation analysis of the Sanger data revealed that abundances of each of the three discriminating genera strongly correlate (that is, they co-occur or avoid each other) with those of other genera (Fig. 2d; see Supplementary Methods section 11), indicating that the enterotypes are in fact driven by groups of species that together contribute to the preferred community compositions.

a–c, Between-class analysis, which visualizes results from PCA and clustering, of the genus compositions of 33 Sanger metagenomes estimated by mapping the metagenome reads to 1,511 reference genome sequences using an 85% similarity threshold (a), Danish subset containing 85 metagenomes from a published Illumina data set8 (b) and 154 pyrosequencing-based 16S sequences5 (c) reveal three robust clusters that we call enterotypes. IBD, inflammatory bowel disease. Two principal components are plotted using the ade4 package in R with each sample represented by a filled circle. The centre of gravity for each cluster is marked by a rectangle and the coloured ellipse covers 67% of the samples belonging to the cluster. IBD, inflammatory bowel disease. d, Abundances of the main contributors of each enterotype from the Sanger metagenomes. See Fig. 1 for definition of box plot. e, Co-occurrence networks of the three enterotypes from the Sanger metagenomes. Unclassified genera under a higher rank are marked by asterisks in b and e.
We demonstrate further the robustness of the enterotypes using two distinct statistical concepts. First, we used the silhouette coefficient21 to validate that the three clusters are superior to clusterings obtained from various randomizations of the genus profile data, indicating a potential role for the interactions between co-occurring genera (see Supplementary Fig. 5 and Supplementary Notes section 4.3). Second, we used supervised learning and cross-validation to establish that these clusters have non-random characteristics that can be modelled and subsequently used to classify new samples (learning on clusters from randomized genus profiles led to considerably worse classification performance; see Supplementary Fig. 6 and Supplementary Notes section 4.4). These consistent results indicate that enterotypes will be identifiable in human gut metagenomes also from larger cohorts.
We then clustered the 33 samples using a purely functional metric: the abundance of the assigned orthologous groups (Fig. 3a). Remarkably, this clustering also showed a similar grouping of the samples with only minor differences (five samples placed in different clusters compared to Fig. 2a), indicating that function and species composition roughly coincide with some exceptions such as Spanish sample ES-AD-3, whose genus composition belongs to enterotype 2 whereas its functional composition is similar to members of enterotype 1. This individual has high levels of phage-related genes compared to the other samples (see Supplementary Fig. 7), hinting at partial temporal variability and dynamics of the microbiota, and perhaps indicating phage or virus bursts.

a, Between-class analysis (see Fig. 2) of orthologous group abundances showing only minor disagreements with enterotypes (unfilled circles indicate the differing samples). The blue cloud represents the local density estimated from the coordinates of orthologous groups; positions of selected orthologous groups are highlighted. b, Four enzymes in the biotin biosynthesis pathway (COG0132, COG0156, COG0161 and COG0502) are overrepresented in enterotype 1. c, Four enzymes in the thiamine biosynthesis pathway (COG0422, COG0351, COG0352 and COG0611) are overrepresented in enterotype 2. d, Six enzymes in the haem biosynthesis pathway (COG0007, COG0276, COG407, COG0408, COG0716 and COG1648) are overrepresented in enterotype 3.
The robustness and predictability of the enterotypes in different cohorts and at multiple phylogenetic and functional levels indicates that they are the result of well-balanced, defined microbial community compositions of which only a limited number exist across individuals. These enterotypes are not as sharply delimited as, for example, human blood groups; they are, in contrast, densely populated areas in a multidimensional space of community composition. They are nevertheless likely to characterize individuals, in line with previous reports that gut microbiota are quite stable in individuals and can even be restored after perturbation22,23,24,25.
Variation between enterotypes
To determine the phylogenetic and functional basis of the enterotypes, we investigated in detail their differences in composition at the phylum, genus, gene and pathway level as well as correlations in abundance of co-occurring genera (Figs 2, 3; also see Supplementary Methods sections 10, 11 and 12). Enterotype 1, containing eight samples, is enriched in Bacteroides (P < 0.01; Supplementary Fig. 8), which co-occurs, for example, with Parabacteroides (see Supplementary Table 11 for enriched genera and Fig. 2e for correlation networks of co-occurring genera in each enterotype). The drivers of this enterotype seem to derive energy primarily from carbohydrates and proteins through fermentation, as these closely related genera have a very broad saccharolytic potential26 and because genes encoding enzymes involved in the degradation of these substrates (galactosidases, hexosaminidases, proteases) along with glycolysis and pentose phosphate pathways are enriched in this enterotype (see Supplementary Tables 12, 13). Enterotype 2 contains six samples and is enriched in Prevotella (P < 0.01; Supplementary Fig. 9) and the co-occurring Desulfovibrio, which can act in synergy to degrade mucin glycoproteins present in the mucosal layer of the gut: Prevotella is a known mucin-degrader and Desulfovibrio may enhance the rate-limiting mucin desulphation step by removing the sulphate27. Enterotype 3 is the most frequent and is enriched in Ruminococcus (P < 0.01; Supplementary Fig. 10) as well as co-occurring Akkermansia, both known to comprise species able to degrade mucins28. It is also enriched in membrane transporters, mostly of sugars, indicating the efficient binding of mucin and its subsequent hydrolysis as well as uptake of the resulting simple sugars by these genera. The enriched genera indicate that enterotypes use different routes to generate energy from fermentable substrates available in the colon, reminiscent of a potential specialization in ecological niches or guilds. In addition to the conversion of complex carbohydrates into absorbable substrates, the gut microbiota is also beneficial to the human host by producing vitamins. Although all the vitamin metabolism pathways are represented in all samples, enterotypes 1 and 2 were enriched in biosynthesis of different vitamins: biotin (Fig. 3b), riboflavin, pantothenate and ascorbate in the former, and thiamine (Fig. 3c) and folate in the latter. These phylogenetic and functional differences among enterotypes thus reflect different combinations of microbial trophic chains with a probable impact on synergistic interrelations with the human hosts.
Functional biomarkers for host properties
Enterotypes do not seem to differ in functional richness (Supplementary Fig. 11), and virtually none of several measured host properties, namely nationality, gender, age or body mass index (BMI), significantly correlates with the enterotypes (with the exception of enterotype 1, which is enriched in Japanese individuals). However, some strong correlations do occur between host properties and particular functions, at the genes or module level (a module is a part of a pathway that is functionally tightly interconnected; see Supplementary Methods sections 6, 13 and Supplementary Notes section 6). The only significant correlation between a host property and a taxonomic group is a negative one between age and the abundance of an unknown Clostridiales genus (P < 0.02) containing three obligate anaerobes (Supplementary Fig. 12a; see Supplementary Notes section 6.2). It should be noted that age is not constant across the nationalities (in our data set, Italians are relatively old and Japanese young), but that individuals did not stratify by nationality, indicating that this is not a confounding factor. Our data did not reveal any correlation between BMI and the Firmicutes/Bacteroidetes ratio and we thus cannot contribute to the ongoing debate on the relationship between this ratio and obesity29,30.
In contrast to the minor phylogenetic signal, we found several significant functional correlations with each of the host properties studied (after correcting for multiple testing to avoid artefacts; see Supplementary Methods section 13), indicating that metagenomics-derived functional biomarkers might be more robust than phylogenetic ones. For example, the abundance of ten orthologous groups varies more between than within nationalities (Supplementary Table 14), although overall, the functional composition in total was remarkably similar among the nations (also with respect to the functional core; see Supplementary Fig. 13). For gender, we find five functional modules and one orthologous group that significantly correlate (P < 0.05; for example, enriched aspartate biosynthesis modules in males; see Supplementary Table 16). In addition, twelve orthologous groups significantly correlate with age (Supplementary Table 17). For instance, starch degradation enzymes such as glycosidases and glucan phosphorylases increase with age (which could be a reaction to decreased efficiency of host breakdown of dietary carbohydrates with age31) and so does the secA preprotein translocase (Supplementary Fig. 14). Conversely, an orthologous group coding for the facultative σ24 subunit of RNA polymerase, which drives expression under various stress responses and is linked to intestinal survival32, decreases with age (Fig. 4a). One explanation for this could be the reduced need for stress response in the gut due to the age-associated decline in host immune response33 (immunosenescence). Our analyses also identified three marker modules that correlate strongly with the hosts’ BMI (Supplementary Table 19 and Supplementary Fig. 14), two of which are ATPase complexes, supporting the link found between the gut microbiota’s capacity for energy harvest and obesity in the host34. Interestingly, functional markers found by a data-driven approach (derived from the metagenomes without previous knowledge) gave much stronger correlations than genes for which a link would be expected (for example, susC/susD, involved in starch utilization26; Fig. 4b). Linear models combining the abundance of only a few functional modules correlate even better with host properties (Fig. 4c, d). It should be noted that given the possibility of many confounding variables owing to the heterogeneity and size of our cohort, these observations will need to be substantiated using larger, independent cohorts in the future. Furthermore, patterns in metagenomics data can (partly) reflect indirect factors9 such as genome size35 (the smaller the average genome size of a sample, the higher the relative fraction of single copy genes therein), which, however, does not matter for diagnostics.

a, Pairwise correlation of RNA polymerase facultative σ24 subunit (COG1595) with age (P = 0.03, rho = −0.59). b, Pairwise correlation of SusD, a family of proteins that bind glycan molecules before they are transported into the cell, and BMI (P = 0.27, rho = −0.29, weak correlation). c, Multiple orthologous groups (OGs) (COG0085, COG0086, COG0438 and COG0739; see Supplementary Table 18) significantly correlating with age when combined into a linear model (see Supplementary Methods section 13 and ref. 40 for details; P = 2.75 × 10−5, adjusted R2 = 0.57). d, Two modules, ATPase complex and ectoine biosynthesis (M00051), significantly correlating with BMI when combined into a linear model (P = 6.786 × 10−6, adjusted R2 = 0.82).
Although individual host properties do not explain the enterotypes, the latter might be driven by a complex mixture of functional properties, by host immune modulation or by hitherto unexplored physiological conditions such as transit time or pH of luminal contents. Furthermore, the three major enterotypes could be triggered by the three distinct pathways for hydrogen disposal36 (Supplementary Notes section 6.4). Indeed, despite their low abundance, Methanobrevibacter (a methanogen) and Desulfovibrio (a known sulphate-reducer) are enriched in enterotypes 3 and 1, respectively.
Taken together, we have demonstrated the existence of enterotypes in the human gut microbiome and have identified three of them that vary in species and functional composition using data that spans several nations and continents. As our current data do not reveal which environmental or even genetic factors are causing the clustering, and as faecal samples are not representative of the entire intestine, we anticipate that the enterotypes introduced here will be refined with deeper and broader analysis of individuals’ microbiomes. Presumably, enterotypes are not limited to humans but also occur in animals. Their future investigation might well reveal novel facets of human and animal symbiotic biology and lead to the discovery of those microbial properties correlated with the health status of individuals. We anticipate that they might allow classification of human groups that respond differently to diet or drug intake. Enterotypes appear complex, are probably not driven by nutritional habits and cannot simply be explained by host properties such as age or BMI, although there are functional markers such as genes or modules that correlate remarkably well with individual features. The latter might be utilizable for diagnostic and perhaps even prognostic tools for numerous human disorders, for instance colorectal cancer and obesity-linked co-morbidities such as metabolic syndrome, diabetes and cardiovascular pathologies.
Methods Summary
Sample collection
Human faecal samples from European individuals were collected and frozen immediately, and DNA was purified as described previously37. Sequencing was carried out by Sanger-sequencing random shotgun DNA libraries of 3 kb using standard protocols established at Genoscope. For sequence processing, cloning vector, sequencing primers and low-quality bases were end-trimmed from raw Sanger reads, and possible human DNA sequences were removed. Reads were processed by the SMASH comparative metagenomics pipeline38 for assembly and gene prediction.
Informed consent was obtained from the 22 European subjects. Sample collection and experiments were approved by the following ethics committees: MetaHIT (Danish), ethical committee of the Capital Region of Denmark; MetaHIT (Spanish), CEIC, Hospital Vall d’Hebron; MicroObes, Ethical Committee for Studies with Human Subjects of Cochin Hospital in Paris, France; MicroAge, Joint Ethical Committee of the University of Camerino.
Phylogenetic annotation
Phylogenetic annotation of samples was performed by (1) aligning reads (Sanger/Illumina) against a database of 1,511 reference genomes (listed in Supplementary Table 3); or (2) classifying 16S rDNA reads using RDP classifier39. Genus and phylum abundance was estimated after normalizing for genome size for the former, and for 16S gene copy number for the latter.
Functional annotation
Genes were functionally annotated using BLASTP against eggNOG (v2) and KEGG (v50) databases. Protein abundances were estimated after normalizing for protein length. Functional abundance profiles at eggNOG, KEGG orthologous group, functional module and pathway level were created.
Clustering and classification
Samples were clustered using Jensen–Shannon distance and partitioning around medoid (PAM) clustering. Optimal number of clusters was estimated using the Calinski–Harabasz (CH) index. We used the silhouette validation technique for assessing the robustness of clusters. Additionally, within a cross-validation scheme, we trained predictive decision tree models on clusters obtained using the same clustering method and evaluated the classification of hold-out samples by accuracy, average precision and average precision gain.
Statistics
Correlations between metadata and feature abundances were computed as described previously40, based on multiple-testing corrected pairwise Spearman correlation analysis and stepwise regression for multi-feature model building. For categorical metadata and enterotype comparisons, samples were pooled into bins (male/female, obese/lean, one enterotype/rest, specific nationality/rest etc) and significant features were identified using Fisher’s exact test with multiple testing correction of P values.
Change history
08 June 2011
An author was omitted. His name has been added to the HTML and PDF and described in the accompanying Corrigendum.
References
Eckburg, P. B. et al. Diversity of the human intestinal microbial flora. Science 308, 1635–1638 (2005)
Hayashi, H., Sakamoto, M. & Benno, Y. Phylogenetic analysis of the human gut microbiota using 16S rDNA clone libraries and strictly anaerobic culture-based methods. Microbiol. Immunol. 46, 535–548 (2002)
Lay, C. et al. Colonic microbiota signatures across five northern European countries. Appl. Environ. Microbiol. 71, 4153–4155 (2005)
Gill, S. R. et al. Metagenomic analysis of the human distal gut microbiome. Science 312, 1355–1359 (2006)
Turnbaugh, P. J. et al. A core gut microbiome in obese and lean twins. Nature 457, 480–484 (2009)
Kurokawa, K. et al. Comparative metagenomics revealed commonly enriched gene sets in human gut microbiomes. DNA Res. 14, 169–181 (2007)
Zoetendal, E. G., Rajilic-Stojanovic, M. & de Vos, W. M. High-throughput diversity and functionality analysis of the gastrointestinal tract microbiota. Gut 57, 1605–1615 (2008)
Qin, J. et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature 464, 59–65 (2010)
Raes, J. & Bork, P. Molecular eco-systems biology: towards an understanding of community function. Nature Rev. Microbiol. 6, 693–699 (2008)
Nelson, K. E. et al. A catalog of reference genomes from the human microbiome. Science 328, 994–999 (2010)
MetaHIT Consortium . MetaHIT Draft Bacterial Genomes at the Sanger Institute. 〈http://www.sanger.ac.uk/resources/downloads/bacteria/metahit/〉 (9 July 2010)
Muller, J. et al. eggNOG v2.0: extending the evolutionary genealogy of genes with enhanced non-supervised orthologous groups, species and functional annotations. Nucleic Acids Res. 38, D190–D195 (2010)
Palmer, C., Bik, E. M., Digiulio, D. B., Relman, D. A. & Brown, P. O. Development of the human infant intestinal microbiota. PLoS Biol. 5, e177 (2007)
Tap, J. et al. Towards the human intestinal microbiota phylogenetic core. Environ. Microbiol. 11, 2574–2584 (2009)
Jensen, L. J. et al. STRING 8—a global view on proteins and their functional interactions in 630 organisms. Nucleic Acids Res. 37, D412–D416 (2009)
Dethlefsen, L., Huse, S., Sogin, M. L. & Relman, D. A. The pervasive effects of an antibiotic on the human gut microbiota, as revealed by deep 16S rRNA sequencing. PLoS Biol. 6, e280 (2008)
Walker, A. Say hello to our little friends. Nature Rev. Microbiol. 5, 572–573 (2007)
Krogfelt, K. A. Bacterial adhesion: genetics, biogenesis, and role in pathogenesis of fimbrial adhesins of Escherichia coli . Rev. Infect. Dis. 13, 721–735 (1991)
Salonen, A. et al. Comparative analysis of fecal DNA extraction methods with phylogenetic microarray: effective recovery of bacterial and archaeal DNA using mechanical cell lysis. J. Microbiol. Methods 81, 127–134 (2010)
Rajilic-Stojanovic, M. et al. Development and application of the human intestinal tract chip, a phylogenetic microarray: analysis of universally conserved phylotypes in the abundant microbiota of young and elderly adults. Environ. Microbiol. 11, 1736–1751 (2009)
Rousseeuw, P. J. Silhouettes: a graphical aid to the interpretation and validation of cluster analysis. J. Comput. Appl. Math. 20, 53–65 (1987)
Vanhoutte, T., Huys, G., Brandt, E., d & Swings, J. Temporal stability analysis of the microbiota in human feces by denaturing gradient gel electrophoresis using universal and group-specific 16S rRNA gene primers. FEMS Microbiol. Ecol. 48, 437–446 (2004)
Tannock, G. W. et al. Analysis of the fecal microflora of human subjects consuming a probiotic product containing Lactobacillus rhamnosus DR20. Appl. Environ. Microbiol. 66, 2578–2588 (2000)
Seksik, P. et al. Alterations of the dominant faecal bacterial groups in patients with Crohn’s disease of the colon. Gut 52, 237–242 (2003)
Costello, E. K. et al. Bacterial community variation in human body habitats across space and time. Science 326, 1694–1697 (2009)
Martens, E. C., Koropatkin, N. M., Smith, T. J. & Gordon, J. I. Complex glycan catabolism by the human gut microbiota: the Bacteroidetes Sus-like paradigm. J. Biol. Chem. 284, 24673–24677 (2009)
Wright, D. P., Rosendale, D. I. & Roberton, A. M. Prevotella enzymes involved in mucin oligosaccharide degradation and evidence for a small operon of genes expressed during growth on mucin. FEMS Microbiol. Lett. 190, 73–79 (2000)
Derrien, M., Vaughan, E. E., Plugge, C. M. & de Vos, W. M. Akkermansia muciniphila gen. nov., sp. nov., a human intestinal mucin-degrading bacterium. Int. J. Syst. Evol. Microbiol. 54, 1469–1476 (2004)
Ley, R. E., Turnbaugh, P. J., Klein, S. & Gordon, J. I. Microbial ecology: human gut microbes associated with obesity. Nature 444, 1022–1023 (2006)
Schwiertz, A. et al. Microbiota and SCFA in lean and overweight healthy subjects. Obesity 18, 190–195 (2009)
Woodmansey, E. J. Intestinal bacteria and ageing. J. Appl. Microbiol. 102, 1178–1186 (2007)
Kovacikova, G. & Skorupski, K. The alternative sigma factor σE plays an important role in intestinal survival and virulence in Vibrio cholerae . Infect. Immun. 70, 5355–5362 (2002)
Fujihashi, K. & Kiyono, H. Mucosal immunosenescence: new developments and vaccines to control infectious diseases. Trends Immunol. 30, 334–343 (2009)
Turnbaugh, P. J. et al. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature 444, 1027–1031 (2006)
Raes, J., Korbel, J. O., Lercher, M. J., von Mering, C. & Bork, P. Prediction of effective genome size in metagenomic samples. Genome Biol. 8, R10 (2007)
Gibson, G. R. et al. Alternative pathways for hydrogen disposal during fermentation in the human colon. Gut 31, 679–683 (1990)
Godon, J. J., Zumstein, E., Dabert, P., Habouzit, F. & Moletta, R. Molecular microbial diversity of an anaerobic digestor as determined by small-subunit rDNA sequence analysis. Appl. Environ. Microbiol. 63, 2802–2813 (1997)
Arumugam, M., Harrington, E. D., Foerstner, K. U., Raes, J. & Bork, P. Smash Community: a metagenomic annotation and analysis tool. Bioinformatics 26, 2977–2978 (2010)
Wang, Q., Garrity, G. M., Tiedje, J. M. & Cole, J. R. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 73, 5261–5267 (2007)
Gianoulis, T. A. et al. Quantifying environmental adaptation of metabolic pathways in metagenomics. Proc. Natl Acad. Sci. USA 106, 1374–1379 (2009)
Acknowledgements
The authors are grateful to C. Creevey, G. Falony and members of the Bork group at EMBL for discussions and assistance. We thank the EMBL IT core facility and Y. Yuan for managing the high-performance computing resources. The research leading to these results has received funding from the European Community’s Seventh Framework Programme (FP7/2007-2013): MetaHIT, grant agreement HEALTH-F4-2007-201052, EMBL, the Lundbeck Foundation Centre for Applied Medical Genomics in Personalized Disease Prediction, Prevention and Care (LuCAMP), Novo Nordisk Foundation and the International Science and Technology Cooperation Project in China (0806). Obese/non-obese volunteers for the MicroObes study were recruited from the SU.VI.MAX cohort study coordinated by P. Galan and S. Hercberg, and metagenome sequencing was funded by Agence Nationale de la Recherche (ANR); volunteers for MicroAge study were recruited from the CROWNALIFE cohort study coordinated by S. Silvi and A. Cresci, and metagenome sequencing was funded by GenoScope. Ciberehd is funded by the Instituto de Salud Carlos III (Spain). J.R. is supported by the Institute for the encouragement of Scientific Research and Innovation of Brussels (ISRIB) and the Odysseus programme of the Fund for Scientific Research Flanders (FWO). We are thankful to the Human Microbiome Project for generating the reference genomes from human gut microbes and the International Human Microbiome Consortium for discussions and exchange of data.
Author information
Authors and Affiliations
Consortia
Contributions
All authors are members of the Metagenomics of the Human Intestinal Tract (MetaHIT) Consortium. Jun W., F.G., O.P., W.M.d.V., S.B., J.D., Jean W., S.D.E. and P.B. managed the project. N.B., F.C., T.H., C.M. and T. N. performed clinical analyses. M.L. and F.L. performed DNA extraction. E.P., D.L.P., T.B., J.P. and E.U. performed DNA sequencing. M.A., J.R., S.D.E. and P.B. designed the analyses. M.A., J.R., T.Y., D.R.M., G.R.F., J.T., J.-M.B., M.B., L.F., L.G., M.K., H.B.N., N.P., J.Q., T.S.-P., S.T., D.T., E.G.Z., S.D.E. and P.B. performed the analyses. M.A., J.R., P.B. and S.D.E. wrote the manuscript. M.H., T.H., K.K. and the MetaHIT Consortium members contributed to the design and execution of the study.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Additional information
Raw Sanger read data from the European faecal metagenomes have been deposited in the NCBI Trace Archive with the following project identifiers: MH6 (33049), MH13 (33053), MH12 (33055), MH30 (33057), CD1 (33059), CD2 (33061), UC4 (33113), UC6 (33063), NO1 (33305), NO3 (33307), NO4 (33309), NO8 (33311), OB2 (33313), OB1 (38231), OB6 (38233), OB8 (45929), A (63073), B (63075), C (63077), D (63079), E (63081), G (63083). Contigs, genes and annotations are available to download from http://www.bork.embl.de/Docu/Arumugam_et_al_2011/.
Supplementary information
Supplementary Information
The file contains Supplementary Methods, Supplementary Notes and Supplementary References. A minor error in Supplementary Information section 2.2 was corrected on 02 June 2011. (PDF 769 kb)
Supplementary Figures
This file contains Supplementary Figures 1-27 with legends. (PDF 3115 kb)
Supplementary Tables
The file contains Supplementary Tables 1 - 2 and 4 - 24 (see separate file for Supplementary Table 3). (PDF 520 kb)
Supplementary Table 3
The file contains Supplementary Table 3. (PDF 1175 kb)
Rights and permissions
About this article
Cite this article
Arumugam, M., Raes, J., Pelletier, E. et al. Enterotypes of the human gut microbiome. Nature 473, 174–180 (2011). https://doi.org/10.1038/nature09944
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/nature09944
This article is cited by
-
The role of gut microbiota in human metabolism and inflammatory diseases: a focus on elderly individuals
Annals of Microbiology (2024)
-
Deciphering the gut microbiome of grass carp through multi-omics approach
Microbiome (2024)
-
Faecalibacterium prausnitzii as a potential Antiatherosclerotic microbe
Cell Communication and Signaling (2024)
-
Identification of enterotype and its predictive value for patients with colorectal cancer
Gut Pathogens (2024)
-
Unlocking the secrets: exploring the influence of the aryl hydrocarbon receptor and microbiome on cancer development
Cellular & Molecular Biology Letters (2024)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
