
Abstract
Of the many materials and methodologies aimed at producing low-cost, efficient photovoltaic cells, inorganic–organic lead halide perovskite materials1,2,3,4,5,6,7,8,9,10,11,12,13,14,15,16,17 appear particularly promising for next-generation solar devices owing to their high power conversion efficiency. The highest efficiencies reported for perovskite solar cells so far have been obtained mainly with methylammonium lead halide materials1,2,3,4,5,6,7,8,9,10. Here we combine the promising—owing to its comparatively narrow bandgap—but relatively unstable formamidinium lead iodide (FAPbI3) with methylammonium lead bromide (MAPbBr3) as the light-harvesting unit in a bilayer solar-cell architecture13. We investigated phase stability, morphology of the perovskite layer, hysteresis in current–voltage characteristics, and overall performance as a function of chemical composition. Our results show that incorporation of MAPbBr3 into FAPbI3 stabilizes the perovskite phase of FAPbI3 and improves the power conversion efficiency of the solar cell to more than 18 per cent under a standard illumination of 100 milliwatts per square centimetre. These findings further emphasize the versatility and performance potential of inorganic–organic lead halide perovskite materials for photovoltaic applications.
Similar content being viewed by others


Main
An inorganic–organic lead halide perovskite is any material that crystallizes into an AMX3 (where A is an organic ammonium cation, M is Pb or Sn, and X is a halide anion) structure. The size of cation A is critical for the formation of a close-packed perovskite structure; in particular, cation A must fit into the space composed of four adjacent corner-sharing MX6 octahedra. Of the various inorganic–organic lead halide perovskite materials, methylammonium lead iodide (MAPbI3), with a bandgap of about 1.5–1.6 eV and a light absorption spectrum up to a wavelength of 800 nm, has been extensively used as a light harvester in solar cells. Several methods, including a one-step spin-coating method1,2,3,6, a two-step sequential method4, and vapour deposition in a high-vacuum chamber5, have been used to prepare MAPbI3 materials. Additionally, different device architectures such as planar5,10 and mesostructured cells1,7 have also been proposed. The highest power conversion efficiency8,9,11 (PCE) so far, of 16% to 17%, was achieved for solution-processed MAPbI3, although 19.3% in a planar device architecture was recently obtained from a reverse-bias current–voltage (I–V) curve12. We note that a PCE measured13,14,15 via a reverse-bias scan with the solar cells exhibiting a large-hysteresis I–V curve can be highly overestimated.
We have already showed that chemical modification of the X site anions (for example, substitution of I with Br) of MAPbI3 can tune the bandgaps to range between 1.5 eV and 2.3 eV by incorporating MAPbBr3 (2.3 eV bandgap); this resulted in colour variation and PCE modulation6. In contrast, HC(NH2)2PbI3, which contains formamidinium (FA) cations instead of methylammonium (MA) cations at the ‘A’ site of the AMX3 perovskite structure, has a bandgap of 1.48 eV, with an absorption edge of 840 nm (refs 16, 17, 18). We expect that this reduced bandgap may allow absorption of photons over a broader solar spectrum. The structural and opto-electrical differences of MAPbI3 and FAPbI3 are likely to originate from the difference in ionic radius of the MA (1.8 Å) and FA ions (1.9–2.2 Å). In fact, the relative ionic radii of A, M and X in the AMX3 perovskite structure have been widely used as a method of establishing the distortion of the MX6 octahedron; in particular, a relatively smaller ion radius for X favours the formation of cubic structures18. However, the photovoltaic performance of FAPbI3 has been reported to be lower than that of MAPbI3 (refs 16, 17). In addition, the black perovskite-type polymorph (α-phase), which is stable at relatively high temperatures (above 160 °C), was observed to turn into the yellow FAPbI3 polymorph (δ-phase) in an ambient humid atmosphere17. However, considering their suitable bandgap (which is lower than that in MAPbI3), the performance of FAPbI3 solar cells can be considerably improved by stabilizing the FAPbI3 phase, improving the crystallinity, and optimizing the cell architecture. In this regard, Pellet et al.19 demonstrated an improved PCE using mixed cation lead iodide perovskites by gradually substituting MA with FA cations—which increases the absorption range by shifting it redwards, allowing for a higher current density—but the performance was still dominated by MAPbI3 rather than FAPbI3. We have recently reported a 16.2% certified PCE obtained from the combination of MAPbI3 and MAPbBr3 with a bilayer architecture consisting of perovskite-infiltrated mesoporous-TiO2 electrodes, and an extremely uniform and dense upper perovskite layer obtained by solvent engineering techniques13; the absorption edge was below 770 nm. A strategy for extending the absorption range of solar light is thus to replace MAPbI3 with FAPbI3 in the combined composition of MAPbI3 and MAPbBr3.
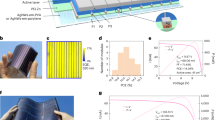
Here we report the overall PCE and structural stability of (FAPbI3)1 − x(MAPbBr3)x, with the mole ratio x ranging between 0 and 0.3. We investigated the performance of systems containing different amounts of MAPbBr3 incorporated into FAPbI3—that is, (FAPbI3)1 − x(MAPbBr3)x—by measuring their current density versus voltage (J−V). Figure 1a shows the PCEs obtained from the J−V curves for different (FAPbI3)1 − x(MAPbBr3)x systems under standard air-mass 1.5 global (AM1.5G) illumination. The PCEs were averaged from the reverse (from the open-circuit voltage Voc to the short-circuit current Isc) and forward (from Isc to Voc) sweeps, because perovskite solar cells may experience a dependence of the measured efficiency on the scan direction. However, the hysteresis of the cells fabricated from FAPbI3 and FAPbI3 incorporating 15 mol% MAPbBr3 was not relevant, even after collecting J−V curves with the relatively short scanning delay time of 40 ms (Fig. 1b). In addition, the small discrepancies related to the scan direction for the FAPbI3/MAPbBr3 systems disappeared at longer delay times (above 100 ms). That the hysteresis observed in FAPbI3-based systems is negligible compared to the hysteresis in MAPbI3 (Fig. 1b inset) may be related to the balance between electron and hole transport within the perovskite layer.

a, PCE values for cells using (FAPbI3)1 − x(MAPbBr3)x materials, which were annealed at 100 °C for 10 min (black line); the red diamond indicates the PCE value for the cell fabricated using pure FAPbI3 (x = 0); annealing was performed at 150 °C for 10 min to form the black perovskite phase. b, J–V curves for cells constructed from FAPbI3, (FAPbI3)0.85(MAPbBr3)0.15 and MAPbI3 (inset) films measured with a 40 ms scanning delay in reverse (from Voc to Isc) and forward (from Isc to Voc) modes under standard AM 1.5 G illumination. Pure FAPbI3 (x = 0) was then annealed at 150 °C. c, Ultraviolet–visible absorption spectra of (FAPbI3)1 − x(MAPbBr3)x films (with x = 0, dark yellow; x = 0.05, red; x = 0.15, green; x = 0.25, blue) annealed at 100 °C on fused silica substrates; the black line indicates absorption spectra for the pure FAPbI3 film (perovskite phase) annealed at 150 °C; the dark yellow line indicates a non-perovskite FAPbI3 form. d, EQE spectra for cells using (FAPbI3)1 − x(MAPbBr3)x materials (with x = 0, black; x = 0.05, red; x = 0.15, green; x = 0.25, blue). Pure FAPbI3 (x = 0) was annealed at 150 °C.
As reported, the electron-diffusion length in MAPbI3 (∼130 nm) is 1.4 times larger than the hole-diffusion length (∼90 nm)20, while FAPbI3 has a hole-diffusion length (∼813 nm) that is 4.6 times longer than the electron-diffusion length (∼177 nm)16. In addition, Kanatzidis et al.18 showed, by measuring the Seebeck coefficient, that MAPbI3 and FAPbI3 display an n-type and a p-type character, respectively. Since the incident light of the FTO/TiO2/perovskite/PTAA/Au (where FTO is fluorine-doped tin oxide and PTAA is poly(triarylamine)) cell configuration reaches the perovskite through the FTO side, more carriers are generated on the perovskite near TiO2 than on the perovskite near PTAA. Therefore, many more holes than electrons should travel a long distance through the perovskite layer to the PTAA layer. This indicates that the balance between electron and hole transport in cells should be considered in compositional engineering for highly efficient perovskite solar cells. Data in Table 1 show that the solar cell annealed at 100 °C with x = 0 exhibits considerably low Jsc, Voc and fill factor (FF) values. In contrast, the overall PCE, including Jsc, Voc and FF, noticeably increased when annealing was performed at 150 °C. The trend of PCE as a function of x in the cells fabricated with FAPbI3–MAPbBr3 showed that Jsc increases from 19.0 mA cm−2 (at x = 0.05) to a maximum value of 22 mA cm−2 (at x = 0.15); after this point, Jsc decreases to 20 mA cm−2 (at x = 0.30). In this range, Voc increases from 1.0 V to 1.12 V. Interestingly, the FF showed exactly the same Jsc behaviour with a maximum value of 73% at x = 0.15.
The ultraviolet–visible absorption spectra displayed in Fig. 1c show a systematic shift of the absorption band edge to shorter wavelengths when the MAPbBr3 content is increased. The reduction of Jsc observed with x larger than 0.15 is directly related to the blue-shift of absorption onset, which in turn is responsible for the reduced light-harvesting efficiency. As expected, the external quantum efficiency (EQE) spectrum shown in Fig. 1d is blue-shifted when x is increased, resulting in the reduction of Jsc. However, a relatively lower Jsc, below x = 0.15, indicates that the charge-collection efficiency is also low, because Jsc is proportional to the product of the charge-collection efficiency and the light-harvesting efficiency. The fact that the FF has a trend similar to that of Jsc supports this, as confirmed by the considerably high series resistance shown below x = 0.15 (with annealing at 100 °C). The increase of Voc over the entire range studied in this work may be attributed to the widening of the bandgap, which occurs when x increases. Most importantly, the PCE of the solar cells fabricated in this study exhibits a maximum value of 17.3% at x = 0.15, owing to the simultaneous enhancement of Jsc and FF, while Voc continues to increase (because the PCE is determined from the products of Jsc, Voc and FF).
To further elucidate possible reasons for such low values of Jsc and FF below x = 0.15, we examined the phase stability of FAPbI3. It is known that FAPbI3 is characterized by a reversible phase transition between two polymorphs, that is, a black perovskite with trigonal symmetry (space group P3m1) and a yellow non-perovskite with hexagonal symmetry (P63mc)17,18. The latter contains linear chains of [PbI6] octahedrons with face-sharing, while the former consists of a three-dimensional network of corner-sharing octahedrons.
Figure 2a shows the results of the differential scanning calorimetry (DSC) and thermogravimetric analysis of a yellow FAPbI3 powder heated at 300 °C. An endothermic peak around 160 °C was found with the DSC analysis, which appears without any weight loss up to 250 °C in the thermogravimetric analysis. The X-ray diffraction (XRD) spectra of the yellow powder measured in situ (Extended Data Fig. 1) suggest that the endothermic peak can be assigned to the phase transition from yellow non-perovskite to black perovskite, in agreement with previous studies16,21. This indicates that the yellow non-perovskite phase for the pure FAPbI3 material is likely to be thermodynamically stable. This phase transition was also found to be reversible in air, that is, the prepared yellow non-perovskite phase changed to the black perovskite phase when annealing was performed at 170 °C; the black powder turned yellow again after being stored in air for 10 days (Extended Data Fig. 2).

a, DSC and thermogravimetric curves of the as-prepared yellow FAPbI3 powder under an Ar atmosphere with a heating rate of 2 °C min−1 from room temperature to 300 °C. The green line indicates the DSC results for (FAPbI3)0.85(MAPbBr3)0.15 powder, which was prepared at room temperature by precipitation from the same compositional solution. b, XRD spectra of solvent-engineering-processed FAPbI3, (FAPbI3)1 − x(MAPbI3)x, (FAPbI3)1 − x(FAPbBr3)x, and (FAPbI3)1 − x(MAPbBr3)x films with x = 0.15, on mesoporous-TiO2/blocking-TiO2/FTO glass substrates after annealing at 100 °C for 10 min. α, δ and # denote the identified diffraction peaks corresponding to the perovskite and non-perovskite polymorphs of FAPbI3 and FTO, respectively. c, FWHM of the (−111) peak for (FAPbI3)1 − x(MAPbBr3)x films as a function of x from XRD spectra of solvent-engineering-processed (FAPbI3)1 − x(MAPbBr3)x films (see Extended Data Fig. 4 for further details). d, SEM plane view images of (FAPbI3)1 − x(MAPbBr3)x films with x = 0, 0.05 and 0.15.
This unintentional phase transition in FAPbI3 solar cells may reduce the photovoltaic performance, because the yellow phase is characterized by a large optical bandgap (Fig. 1c) and an inferior charge-transporting ability due to the linear chain-like [PbI6] octahedron structure. Notably, AMX3 (where A is Rb, Cs, MA or FA, M is Pb or Sn, and X is Cl, Br or I) metal trihalide materials exist as either two polymorphs (perovskite and non-perovskite) or only one of the two, depending on the atomic size of the components22. For example, in contrast to CsSnI3, which has two polymorphs, MASnI3, with larger MA+ cations, shows only a perovskite phase and no phase transition near room temperature (∼25 °C). RbSnI3, with smaller Rb+ cations, shows only a non-perovskite phase22,23,24. Therefore, the atomic combination of the A and X sites may lead to the stabilization of the FAPbI3 perovskite structure.
Figure 2b shows the XRD spectra of the prepared FAPbI3, (FAPbI3)1 − x(MAPbI3)x, (FAPbI3)1 − x(FAPbBr3)x, and (FAPbI3)1 − x(MAPbBr3)x films (with x = 0.15) on mesoporous-TiO2/blocking-TiO2/FTO glass substrates, after annealing at 100 °C for 10 min; these were prepared using the solvent-engineering process with precursor solutions of the desired compositions as previously reported13. The XRD spectrum of the pure FAPbI3 thin film shows the typical diffraction pattern of a hexagonal non-perovskite polymorph of FAPbI3 (P63mc)18; this can be detected from DSC (Fig. 2a), because the temperature of the annealing (100 °C) is much lower than the temperature (160 °C) at which the phase transition occurs. However, when FA+ cations in FAPbI3 are substituted by 15 mol% of MA+ cations, a strong (111) diffraction peak at 13.9° for the trigonal perovskite phase (P3m1) appears in spite of the annealing at 100 °C. The same diffraction peaks are also observed in systems containing Br− ions (15 mol%), although the secondary phase coexists in the film.
Surprisingly, a simultaneous introduction of 15 mol% of both MA+ cations and Br− anions in FAPbI3 to obtain (FAPbI3)0.85(MAPbBr3)0.15 leads to a synergetic effect that stabilizes the perovskite phase. Interestingly, this is sufficient to form a FAPbI3 perovskite phase even after incorporating 5 mol% of MAPbBr3 (Extended Data Fig. 3), although the single MA+ or Br− can only partially form a perovskite phase. We also found that a highly crystalline perovskite layer is formed with values of x larger than 0.15, according to the full width of half maximum (FWHM) of the (−111) diffraction peak (Fig. 2c). Therefore, we surmise that the enhancement of the phase stability and crystallinity results in an improvement of PCE in the x range of 0 to 0.15. The perovskite phase stabilization caused by the introduction of MAPbBr3 to FAPbI3 was also confirmed by an analysis carried out on the synthesized powders, which were prepared at room temperature by precipitation from solutions of FAPbI3, (FAPbI3)1 − x(MAPbI3)x, (FAPbI3)1 − x(FAPbBr3)x, and (FAPbI3)1 − x(MAPbBr3)x with x = 0.15. Photographs of the as-prepared powders shown in Extended Data Fig. 4 indicate that a black powder like as-prepared MAPbI3 is obtained only for (FAPbI3)0.85(MAPbBr3)0.15. In addition, the XRD spectra of the powders revealed that (FAPbI3)0.85(MAPbBr3)0.15, in contrast to FAPbI3, shows a pure perovskite phase (Extended Data Fig. 5), with no endothermic DSC peaks (Fig. 2a). This finding confirms that the co-substitution of MA to FA and Br to I can efficiently stabilize the perovskite phase. However, further investigation is required to determine the energetics of the perovskite and non-perovskite formation and to establish the composition of the stable form in perovskite halide materials.
We recently successfully fabricated a complete bilayer on a mesoporous-TiO2 using the solvent-engineering technology; our findings confirmed that surface coverage and morphology of perovskite materials is critical13. For this reason, here we analysed the surfaces deposited with different x values for MAPbBr3/FAPbI3 using the solvent-engineering process. Figure 2d shows the surface scanning electron microscope (SEM) images of (FAPbI3)1 − x(MAPbBr3)x, with x = 0, 0.05 and 0.15, on a 200-nm-thick mesoporous-TiO2/blocking-TiO2/FTO annealed for 10 min at 150 °C (x = 0) and 100 °C, respectively. Our results showed that the surface of FAPbI3 exhibits an irregular morphology with bumpy roughness. Incorporating MAPbBr3 into FAPbI3 (with x = 0.15) considerably smoothed the surface; however, systems characterized by x = 0.05 still presented large voids between crystal boundaries. The rough surface of FAPbI3 may be due to the transition from non-perovskite to perovskite phases and to the high temperature required for the formation of the perovskite phase. Indeed, manipulating the composition of FAPbI3 by adding MAPbBr3 led to the stabilization of the perovskite phase with a uniform and dense morphology as well as well-developed crystallites, which are responsible for the highly improved cell performance.
On the basis of these results, we repeated the fabrication procedure, fixing x to a value of 0.15 to enhance further the performance of the FAPbI3-MAPbBr3 system with an architecture as follows: FTO/blocking-TiO2 (70 nm)/mesoporous-TiO2:perovskite composite layer (200 nm)/perovskite upper layer (300 nm)/PTAA (50 nm)/Au (100 nm). Of the devices obtained, Fig. 3a shows the J–V curves measured via reverse and forward bias sweep for one of the best-performing solar cells. The Jsc, Voc and FF values averaged from the J–V curves of this device are 22.5 mA cm−2, 1,105 mV and 73.2%, respectively; these correspond to a PCE of 18.4% under standard AM1.5G conditions. The PCE value is in agreement with that obtained from the stabilized power output near the maximum power point, reflecting the device performance in the working condition more closely (Extended Data Fig. 6).

a, J–V curves of forward and reverse bias sweep and their averaged curve for the solar cell using the (FAPbI3)0.85(MAPbBr3)0.15 perovskite active layer. b, EQE spectrum and integrated Jsc.
In the case of the cell using thinner mesoporous-TiO2 layer (80 nm), as shown in Extended Data Fig. 7, although an unprecedented PCE of 20.3% was measured via reverse bias scan, the PCE of around 17.3% obtained from an average J–V curve and steady-state current measurement is far lower than that from the best cell using a 200-nm-thick mesoporous-TiO2 layer, owing to the low PCE of 15.5% with forward bias scan. This result is similar to those of ref. 12 and implies that substantial PCE values should be obtained from J–V curves averaged with reverse and forward bias sweep. Figure 3b shows the EQE spectrum for one of the best-performing solar cells. A very broad EQE plateau of over 80% between 400 nm and 750 nm was observed. The Jsc value integrated from EQE was found to be in good agreement with that measured by J–V. The highly performing devices exhibiting PCEs of 18.0% with very small hysteresis was certified by the standardized method in the photovoltaic calibration laboratory, confirming a PCE of 17.9% under AM1.5G full sun (Extended Data Fig. 8). To the best of our knowledge, such a PCE value is the highest ever reported for a perovskite-based solar cell, excluding the value overestimated by reverse bias scan.
We have described the compositional engineering of (FAPbI3)1 − x(MAPbBr3)x for efficient perovskite solar cells. (FAPbI3)0.85(MAPbBr3)0.15 has many advantages over other systems such MAPbI3, FAPbI3, and MAPb(I0.85Br0.15)3. This strategy may lead to more efficient and cost-effective inorganic–organic hybrid perovskite solar cells.
Methods
Materials
Unless stated otherwise, all materials were purchased from Sigma-Aldrich or Junsei Organics and used as received. PTAA was purchased from EM Index.
Synthesis of the TiO2 paste
The TiO2 nanoparticles used in the paste were prepared by hydrothermal treatment at 250 °C for 12 h from aqueous solutions of the peroxotitanium complex, as described elsewhere25,26. The peroxotitanium complex solutions were synthesized via a reaction between hydrogen peroxide and the TiO(OH)2 wet cake. The wet cake was obtained by the dropwise addition of a NH4OH solution into 0.33M TiOCl2 aqueous solution obtained by the hydrolysis of TiCl4. The resulting cake was purified by repeated washing with deionized water until no Cl− ions were detected. Then, 200 ml of 35 wt% hydrogen peroxide was added into 300 ml of a 2 wt% TiO(OH)2-dispersed aqueous solution such that the H2O2/Ti4+ ratio was ∼40; the solution was constantly stirred in an ice bath. Once the addition of H2O2 was completed, the colour of the precipitate changed from white to turbid yellowish, and finally light orange- or reddish-coloured transparent solutions were formed within 1 h.
The TiO2-nanoparticle (average diameter 50 nm, anatase) slurry obtained by the hydrothermal treatment was stirred for 1 h, while several drops of concentrated nitric acid were added to obtain a colloidal dispersion solution. Then the TiO2 nanoparticles were collected by the centrifugal method. The TiO2 wet cake was re-dispersed and centrifuged in absolute ethanol by ultrasonic irradiation. The process with absolute ethanol was repeated for three cycles. The collected TiO2 nanoparticles were dispersed in 100 ml of absolute ethanol, 4.5 g (per 1 g of TiO2) of a 10 wt% ethanolic solution of ethyl cellulose (Fluka, 46070), and 4.4 g (per 1 g of TiO2) of terpineol. After the addition of each component, the mix was stirred for 10 min and homogenized by ultrasonic irradiation. The paste was finally produced by a three-roller-mill grinder (EXAKT) after concentrating the mixture solution in a rotary evaporator.
Synthesis of the inorganic–organic mixed perovskite
CH3NH3I and NH2CH = NH2I were first synthesized by reacting 30 ml hydroiodic acid (57% in water, Aldrich), 27.86 ml CH3NH2 (40% in methanol, Junsei Chemical), and 15 g formamidine acetate (Aldrich) in a 250 ml round-bottomed flask at 0 °C for 2 h with stirring. The precipitates were recovered by evaporating the solutions at 50 °C for 1 h. The products were dissolved in ethanol, recrystallized using diethyl ether, and finally dried at 60 °C in a vacuum oven for 24 h. Similarly, CH3NH3Br and NH2CH = NH2Br were prepared using hydrobromic acid (48 wt% in water, Aldrich) according to a reported procedure27. The desired solutions of FAPbI3, (FAPbI3)1 − x(MAPbI3)x, (FAPbI3)1 − x(FAPbBr3)x, and (FAPbI3)1 − x(MAPbBr3)x (with x = 0–0.30) were prepared by dissolution of the CH3NH3I, CH3NH3Br, NH2CH = NH2I, and NH2CH = NH2Br powders with PbI2 (Aldrich) and PbBr2 (Aldrich) in the γ-butyrolactone:DMSO mixed solvent (7:3, volume ratio) at 60 °C for 10 min.
Solar cell fabrication
A dense blocking layer of TiO2 (60 nm, blocking-TiO2) was deposited onto an F-doped SnO2 (FTO, Pilkington, TEC8) substrate by spray pyrolysis using a 20 mM titanium diisopropoxide bis(acetylacetonate) solution (Aldrich) at 450 °C; this was done to prevent a direct contact between FTO and the hole-conducting layer. A 200-nm-thick mesoporous-TiO2 was spin-coated onto the blocking-TiO2/FTO substrate using TiO2 pastes diluted in 2-methoxyethanol (1 g in 5 ml) and calcinated at 500 °C for 1 h in air to remove the organic components.
The inorganic–organic lead halide perovskite solutions were then coated onto the mesoporous-TiO2/blocking-TiO2/FTO substrate by two consecutive spin-coating steps, at 1,000 rpm and 5,000 rpm for 40 s and 20 s, respectively. During the second spin-coating step, 1 ml toluene was poured onto the substrate, according to the procedure reported in ref. 13. To obtain a uniform and flat intermediate-phase film, toluene should be quickly cast in one-shot mode on a rapidly rotating (at 5,000 rpm) substrate to wash out the surplus of DMSO molecules that did not participate in the formation of the PbI2–NH2CH = NH2I–DMSO complex. The substrate was then dried on a hot plate at 100 °C or 150 °C for 10 min. A solution of PTAA (EM Index, [Mn] = 17,500 g mol−1)/toluene (10 mg ml−1) with an additive of 7.5 µl Li-bis(trifluoromethanesulphonyl) imide/acetonitrile (170 mg ml−1) and 4 µl 4-tert-butylpyridine was spin-coated on the perovskite layer/mesoporous-TiO2/blocking-TiO2/FTO substrate at 3,000 rpm for 30 s. Finally, an Au counter electrode was deposited by thermal evaporation; the active area of this electrode was fixed at 0.16 cm2. The different inorganic–organic lead halide triiodide powders were prepared by precipitation using toluene at room temperature from the desired compositional solutions obtained by dissolving CH3NH3I, CH3NH3Br, NH2CH = NH2I and NH2CH = NH2Br powders with PbI2 and PbBr2 in γ-butyrolactone.
Characterization
The XRD spectra of the prepared films were measured using a Rigaku SmartLab X-ray diffractometer; the in situ XRD experiment of the as-prepared FAPbI3 yellow powder was performed using a Rigaku Ultima IV with an X-ray tube (Cu Kα, wavelength λ = 1.5406 Å). Ultraviolet–visible absorption spectra were recorded on a Shimadzu UV 2550 spectrophotometer in the 300–800 nm wavelength range at room temperature. The morphology of the films was observed using a field-emission SEM (MIRA3 LMU, Tescan). Thermogravimetric and DSC analyses of the as-prepared powders were performed with a heating rate of 2 °C min−1 from room temperature up to 300 °C under a nitrogen atmosphere using TA Instruments SDT 2960 and DSC 2910, respectively. EQE was measured by a power source (Newport 300W Xenon lamp, 66920) with a monochromator (Newport Cornerstone 260) and a multimeter (Keithley2001). The J–V curves were measured using a solar simulator (Newport, Oriel Class A, 91195A) with a source meter (Keithley 2420) at 100 mA cm−2 AM1.5G illumination and a calibrated Si-reference cell certified by the National Renewable Energy Laboratory, USA. The J–V curves were measured by reverse scan (forward bias (1.2 V) → short circuit (0 V)) or forward scan (short circuit (0 V) → forward bias (1.2 V)). The step voltage was fixed at 10 mV and the delay time, which is a delay set at each voltage step before measuring each current, was modulated. The J–V curves for all devices were measured by masking the active area with a metal mask (area of 0.096 cm2).
References
Lee, M. M. et al. Efficient hybrid solar cells based on meso-superstructured organometal halide perovskites. Science 338, 643–647 (2012)
Kim, H.-S. et al. Lead iodide perovskite sensitized all-solid-state submicron thin film mesoscopic solar cell with efficiency exceeding 9%. Sci. Rep. 2, 1–7 (2012)
Heo, J. H. et al. Efficient inorganic-organic hybrid heterojunction solar cells containing perovskite compound and polymeric hole conductors. Nature Photon. 7, 486–491 (2013)
Burschka, J. et al. Sequential deposition as a route to high-performance perovskite-sensitized solar cells. Nature 499, 316–319 (2013)
Liu, M., Johnston, M. B. & Snaith, H. J. Efficient planar heterojunction perovskite solar cells by vapor deposition. Nature 501, 395–398 (2013)
Noh, J. H., Im, S. H., Heo, J. H., Mandal, T. N. & Seok, S. I. Chemical management for colorful, efficient, and stable inorganic-organic hybrid nanostructured solar cells. Nano Lett. 13, 1764–1769 (2013)
Ball, J. M., Lee, M. M., Hey, A. & Snaith, H. J. Low-temperature processed meso-superstructured to thin-film solar cells. Energy Environ. Sci. 6, 1739–1743 (2013)
Jeon, N. J. et al. o-Methoxy substituents in Spiro-OMeTAD for efficient inorganic–organic hybrid perovskite solar cells. J. Am. Chem. Soc. 136, 7837–7840 (2014)
Ryu, S. et al. Voltage output of efficient perovskite solar cells with high open-circuit voltage and fill factor. Energy Environ. Sci. 7, 2614–2618 (2014)
Malinkiewicz, O. et al. Perovskite solar cells employing organic charge-transport layers. Nature Photon. 8, 128–132 (2014)
Lee, J.-W. et al. High-efficiency perovskitesolar cells based on the black polymorph of HC(NH2)2PbI3 . Adv. Mater. 26, 4991–4998 (2014)
Zhou, H. et al. Interface engineering of highly efficient perovskite solar cells. Science 345, 542–546 (2014)
Jeon, N. J. et al. Solvent engineering for high-performance inorganic-organic hybrid perovskite solar cells. Nature Mater. 13, 897–903 (2014)
Snaith, H. J. et al. Anomalous hysteresis in perovskite solar cells. J. Phys. Chem. Lett. 5, 1511–1515 (2014)
Kim, H. S. & Park, N.-G. Parameters affecting I–V hysteresis of CH3NH3PbI3 perovskite solar cells: effect of perovskite crystal size and mesoporous TiO2 layer. J. Phys. Chem. Lett. 5, 2927–2934 (2014)
Eperon, G. E. et al. Formamidinium lead halide: a broad tunable perovskite for efficient planar heterojunction solar cells. Energy Environ. Sci. 7, 982–988 (2014)
Koh, T. M. et al. Formamidinium-containing metal-halide: an alternative material for near-IR absorption perovskite solar cells. J. Phys. Chem. C 118, 16458–16462 (2014)
Stoumpos, C. C. et al. Semiconducting tin and lead iodide perovskites with organic cations: phase transition, high mobilities, and near-infrared photoluminescent properties. Inorg. Chem. 52, 9019–9038 (2013)
Pellet, N. et al. Mixed-organic-cation perovskite photovoltaics for enhanced solar-light harvesting. Angew. Chem. Int. Ed. 53, 3151–3157 (2014)
Xing, G. et al. Long-range balanced electron and hole-transport lengths in organic-inorganic CH3NH3PbI3 . Science 342, 344–347 (2013)
Scaife, D. E., Weller, P. F. & Fisher, W. G. Crystal preparation and properties of cesium tin(II) trihalides. J. Solid State Chem. 9, 308–314 (1974)
Chung, I. et al. CsSnI3: semiconductor or metal? High electrical conductivity and strong near-infrared photoluminescence from a single material. High hole mobility and phase-transitions. J. Am. Chem. Soc. 134, 8579–8587 (2012)
Takahashi, Y. et al. Charge-transport in tin-iodide perovskite CH3NH3SnI3: origin of high conductivity. Dalton Trans. 40, 5563–5568 (2011)
Amat, A. et al. Cation-induced band-gap tuning in organohalide perovskites: interplay of spin-orbit coupling and octahetra tilting. Nano Lett. 14, 3608–3616 (2014)
Baek, I. C. et al. Facile preparation of large aspect ratio ellipsoidal anatase TiO2 nanoparticles and their application to dye-sensitized solar cell. Electrochem. Commun. 11, 909–912 (2009)
Seok, S. I. et al. Colloidal TiO2 nanocrystals prepared from peroxotitanium complex solutions: phase evolution from different precursors. J. Colloid Interf. Sci. 346, 66–71 (2010)
Pang, S. et al. NH2CH = NH2PbI3: an alternative organolead iodide perovskite sensitizer for mesoscopic solar cells. Chem. Mater. 26, 1485–1491 (2014)
Acknowledgements
This work was supported by the Global Research Laboratory (GRL) Program, the Global Frontier R&D Program of the Center for Multiscale Energy System, funded by the National Research Foundation in Korea, and by a grant from the Korea Research Institute of Chemical Technology (KRICT) 2020 Program for Future Technology in South Korea.
Author information
Authors and Affiliations
Contributions
N.J.J., J.H.N. and S.I.S. conceived the experiments and analysed and interpreted the data. N.J.J., Y.C.K., J.H.N. and J.S. performed the fabrication of devices, device performance measurements and characterization. N.J.J., W.S.Y. and S.R. carried out the synthesis of materials for perovskites, and S.I.S. prepared TiO2 particles and pastes. The manuscript was mainly written and revised by S.I.S. and J.H.N. The project was planned, directed and supervised by S.I.S. All authors discussed the results and commented on the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Extended data figures and tables
Extended Data Figure 1 In situ XRD spectra (heating from 100 °C to 170 °C) for FAPbI3 yellow powders prepared at room temperature.
Hexagonal non-perovskite FAPbI3 (P63mc) converted into a trigonal perovskite phase (P3m1) near 150 °C. The (−111) diffraction peak for perovskite FAPbI3 at 2θ = 14.3° appeared at a temperature of 150 °C; simultaneously the main peak of non-perovskite FAPbI3 at 11.6° disappeared.
Extended Data Figure 2 XRD spectra of FAPbI3 powders.
The as-prepared yellow FAPbI3 powder shows a non-perovskite phase and is converted to perovskite phase by annealing at 170 °C. The perovskite FAPbI3 black powder returned to the yellow non-perovskite powder after being stored in air for 10 h; the yellow powder reversibly changed to black perovskite phase by re-annealing at 170 °C.
Extended Data Figure 3 XRD spectra of (FAPbI3)1 − x(MAPbBr3)x cells as a function of x.
XRD spectra of solvent-engineering processed (FA1 − xMAx)Pb(I1 − xBrx)3 films on the mesoporous-TiO2/blocking-TiO2/FTO glass substrate after annealing at 100 °C for 10 min. α, α-phase of FAPbI3; #, peaks diffracted from FTO.
Extended Data Figure 4 Photographs of inorganic–organic hybrid halide powders.
Photographs show the colour of the as-prepared MAPbI3, annealed FAPbI3 at 170 °C, FAPbI3, (FAPbI3)1 − x(MAPbI3)x, (FAPbI3)1 − x(FAPbBr3)x, and (FAPbI3)1 − x(MAPbBr3)x powders with x = 0.15 (from left to right). The (FAPbI3)1 − x(MAPbBr3)x powder is the only black powder among the as-prepared FAPbI3-based materials.
Extended Data Figure 5 XRD spectra of the as-prepared powders at room temperature.
XRD spectra of the as-prepared FAPbI3, (FAPbI3)1 − x(MAPbI3)x, (FAPbI3)1 − x(FAPbBr3)x, and (FAPbI3)1 − x(MAPbBr3)x powders with x = 0.15 (from left to right). Only the (FAPbI3)1 − x(MAPbBr3)x powder shows a pure perovskite phase. α, black perovskite-type polymorph; δ, yellow non-perovskite polymorph.
Extended Data Figure 6 Steady-state current measurement.
Steady-state current measured at a maximum power point (0.89 V) and stabilized power output.
Extended Data Figure 7 Photovoltaic performance.
a, J–V curves measured by forward and reverse bias sweep and their averaged curve for cell using the (FAPbI3)0.85(MAPbBr3)0.15 perovskite active layer and 80-nm-thick mesoporous-TiO2 layer. η, PCE. b, Steady-state current measured at a maximum power point (0.92 V)and stabilized power output.
Rights and permissions
About this article

Cite this article
Jeon, N., Noh, J., Yang, W. et al. Compositional engineering of perovskite materials for high-performance solar cells. Nature 517, 476–480 (2015). https://doi.org/10.1038/nature14133
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/nature14133
This article is cited by
-
All-perovskite-based unassisted photoelectrochemical water splitting system for efficient, stable and scalable solar hydrogen production
Nature Energy (2024)
-
Phase dimensions resolving of efficient and stable perovskite light-emitting diodes at high brightness
Nature Photonics (2024)
-
Manipulating solvent fluidic dynamics for large-area perovskite film-formation and white light-emitting diodes
Nature Communications (2024)
-
Design of a high-efficiency perovskite solar cell based on photonic crystal in the absorption layer
Optical and Quantum Electronics (2024)
-
Optimizing lead-free Cs3Bi2I9 perovskite solar cells: exploring absorber and charge transport layers parameters for improved efficiency
Journal of Optics (2024)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
