
Abstract
Bacteria share their ecological niches with other microbes. The bacterial type VI secretion system is one of the key players in microbial competition, as well as being an important virulence determinant during bacterial infections. It assembles a nano-crossbow-like structure in the cytoplasm of the attacker cell that propels an arrow made of a haemolysin co-regulated protein (Hcp) tube and a valine–glycine repeat protein G (VgrG) spike and punctures the prey’s cell wall. The nano-crossbow is stably anchored to the cell envelope of the attacker by a membrane core complex. Here we show that this complex is assembled by the sequential addition of three type VI subunits (Tss)—TssJ, TssM and TssL—and present a structure of the fully assembled complex at 11.6 Å resolution, determined by negative-stain electron microscopy. With overall C5 symmetry, this 1.7-megadalton complex comprises a large base in the cytoplasm. It extends in the periplasm via ten arches to form a double-ring structure containing the carboxy-terminal domain of TssM (TssMct) and TssJ that is anchored in the outer membrane. The crystal structure of the TssMct–TssJ complex coupled to whole-cell accessibility studies suggest that large conformational changes induce transient pore formation in the outer membrane, allowing passage of the attacking Hcp tube/VgrG spike.
Similar content being viewed by others


Main
In the environment, bacteria have evolved collaborative or aggressive mechanisms to communicate, exchange information and chemicals, or compete for space and resources1,2,3. One of the main weapons of bacterial conflicts is a multi-protein device called the type VI secretion system (T6SS) that is assembled in the attacker bacterium4. The T6SS is a versatile nanomachine that can deliver toxin proteins directly not only into prey prokaryotes but also into eukaryotic cells during bacterial infections3,5,6,7,8,9. Anti-host activities have been shown to inhibit phagocytosis and therefore to disable macrophages, while the anti-bacterial activities allow the bacterium to destroy competitors and to have a privileged access to the niche, to nutrients or to new DNA3,9,10. The T6SS is composed of 13 different proteins, encoded by genes that are usually clustered11. It assembles a tubular puncturing device that is evolutionarily, structurally and functionally similar to the tail of contractile bacteriophages. Its sheath is a tubular structure, hundreds of nanometres long, that extends in the cytoplasm and is built by the polymerization of TssBC building blocks12,13,14. It is assembled on an assembly platform, the baseplate13,15,16,17, and maintained in an extended, metastable conformation16,17,18. The attacking arrow, wrapped by the sheath, comprises an inner tube that is built by stacked Hcp hexameric rings19 and tipped by a puncturing spike composed of VgrG20. Upon contact with the prey, structural rearrangements of the sheath subunits induce its contraction and propulsion of the Hcp tube/VgrG spike towards the target cell, allowing toxin delivery16,17,21. The phage-like T6SS tail is anchored to the attacker cell membrane by a trans-envelope complex22. This membrane complex not only serves as a docking station but has been proposed as a channel for the passage of the inner tube after sheath contraction, thereby preventing membrane damage in the attacker16,17. The membrane core complex of the T6SS (that is, the minimal module required to function and conserved in all T6SS) is composed of the TssL and TssM inner-membrane proteins and the TssJ outer membrane lipoprotein15,16,17,22,23,24,25,26. These proteins are connected through a network of interactions between TssM and TssL, and TssM and TssJ22,24,25. Although the localization and topology of these subunits, their interactions and the crystal structures of the soluble domains of TssJ and TssL have been described17,22,23,24,25,26,27,28,29, we still lack crucial information on the biogenesis and overall architecture of this complex and how it is used as a channel during T6SS action.
Localization, dynamics and biogenesis of the T6SS membrane core complex
We first sought to determine the assembly pathway of the entero-aggregative Escherichia coli (EAEC) T6SS membrane core complex. Strains producing fluorescently labelled T6SS membrane subunits were engineered. The sequence encoding the super-folder green fluorescent protein (sfGFP) was inserted upstream of the stop codon of the tssJ gene or downstream of the start codon of the tssL and tssM genes. In these constructs, the fusion proteins were produced from their native chromosomal loci. Hcp release and anti-bacterial assays demonstrated that the sfGFP–TssL and sfGFP–TssM fusion proteins were functional (Extended Data Fig. 1a). By contrast, strains producing TssJ–sfGFP or TssJ–mCherry had a non-functional T6SS (Extended Data Fig. 1b). Fluorescence microscopy analyses showed that sfGFP–TssL and sfGFP–TssM cluster at discrete positions at the cell periphery, in agreement with their membrane localization (Fig. 1a and Extended Data Fig. 1c). These foci are stable and static (Fig. 1a and Extended Data Fig. 1d). Statistical analyses further showed that one or two foci are observable in cells expressing the T6SS (Fig. 1b) and that these clusters are randomly distributed around the cell (Fig. 1c). Co-localization experiments with strains producing sfGFP–TssM and mCherry-tagged TssL showed that the two subunits are present in the foci, demonstrating that each focus corresponds to the position of an assembled membrane complex (Fig. 1d). To test whether these foci are used to anchor the phage-like tail tubular structure, mCherry was fused to the tssB sheath gene, at its original chromosomal locus in the strain producing sfGFP–TssL. Time-lapse recordings showed that T6SS sheathes polymerize and extend from the membrane complex (Fig. 1e). On the basis of these results, we conclude that membrane complexes comprising TssL and TssM (and probably TssJ) assemble at discrete positions in the cell and are then used to recruit the tail-complex subunits. Statistical analyses showed that the number of sfGFP–TssL or sfGFP–TssM foci per cell is higher than the number of sheathes (Extended Data Fig. 1e), suggesting that the membrane complexes exist in a pre-assembled form. Interestingly, long-term time-lapse recordings showed that these membrane complexes can be re-used for new tail polymerization events (Extended Data Fig. 1f). To gain further information on the biogenesis of this initial step, tssJ or tssM were deleted in the sfGFP–TssL-producing strain, and tssJ or tssL were deleted in the sfGFP–TssM-producing strain. The rationale behind these experiments is that if a protein assembled early is missing, the recruitment of late proteins will be affected, yielding a diffuse fluorescent signal. Figure 1f shows that the recruitment of sfGFP–TssM and sfGFP–TssL is affected in the absence of TssJ, and that of sfGFP–TssL is affected in the absence of TssM. Conversely, the absence of TssL had no effect on TssM recruitment (Fig. 1f and Extended Data Fig. 1g). On the basis of these results, we conclude that TssJ is used as a nucleation factor and that the biogenesis of the T6SS membrane core complex is pursued by the inward sequential addition of TssM and TssL (Fig. 1g).

a, Time-lapse fluorescence microscopy recordings showing localization and dynamics of the sfGFP–TssM and sfGFP–TssL fusion proteins. Individual images were taken every 30 s. The positions of the foci are indicated by arrowheads. Scale bars, 1 μm. Larger fields are presented in Extended Data Fig. 1c. b, Statistical analysis of sfGFP–TssM and sfGFP–TssL localization. Shown are box-and-whisker plots of the measured number of sfGFP–TssM and sfGFP–TssL foci per cell for each strain with the lower and upper boundaries of the boxes corresponding to the 25% and 75% percentiles respectively. The black horizontal bar represents the median values for each strain and the whiskers represent the 10% and 90% percentiles. The number of cells studied per strain is indicated above the bars. c, Spatial repartition of the sfGFP–TssM and sfGFP–TssL foci. Shown is a superposition of the different foci analysed in a single cell. d, sfGFP–TssM and mCh–TssL proteins co-localize. Fluorescence microscopy recordings showing co-localization between sfGFP–TssM and mCh–TssL fusion proteins. The positions of the foci are indicated by the arrowheads. Scale bar, 1 μm. e, The membrane complex serves as a docking site for tail sheath polymerization. Time-lapse fluorescence microscopy recordings showing co-localization between sfGFP–TssL and TssB–mCh fusion proteins. Individual images were taken every 30 s. Assembly/contraction of the sheath and TssL localization events is schematized in the bottom row of panels. Scale bars, 1 μm. f, Assembly pathway of the T6SS TssJLM membrane complex. Fluorescence microscopy recordings showing sfGFP–TssM and sfGFP–TssL localization in the absence of the TssJ or TssL and TssJ or TssM proteins respectively. The positions of the foci are indicated by the arrowheads. Scale bars, 1 μm. The quantification of the sfGFP–TssM and sfGFP–TssL clusters per cell is presented in Extended Data Fig. 1g. g, Schematic representation of the sequential biogenesis of the T6SS membrane complex. The names of the proteins, their localizations and topologies are shown.
Architecture of the T6SS membrane core complex
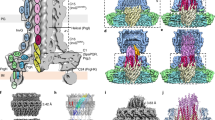
To gain further insights on the architecture of the T6SS membrane core complex, the tssJ, tssL and tssM genes were co-expressed in E. coli BL21(DE3). Constructs were designed to add StrepII, Flag and 6×His tags at the carboxy (C) terminus of TssJ, amino (N) terminus of TssL and N terminus of TssM, respectively (Extended Data Fig. 2). Total membranes were isolated and solubilized using detergents. Two-step affinity chromatography followed by gel filtration resulted in the purification of a complex containing TssJ, TssL and TssM (Fig. 2a and Extended Data Fig. 2f–h). In this complex, we determined the TssM–TssL stoichiometry as 1 to 1 (Extended Data Fig. 2h). Purified complexes were visualized by negative-stain electron microscopy (EM) (Fig. 2b and Extended Data Fig. 3a). A data set was collected, and reference-free classification and averaging revealed characteristic views of the complex (class averages) (Fig. 2b). We observed rocket-shaped and ring-shaped views corresponding to side and end views of the T6SS membrane core complex respectively (Fig. 2b). Rotational symmetry analysis of end-view class-averages revealed a clear five-fold symmetry in the whole TssJLM population (Extended Data Fig. 3b). The complex is composed of a base and a tip complex linked by arches (Fig. 2b). The negative-stain data set was used to reconstruct a 11.6-Å resolution three-dimensional (3D) volume of the complex with five-fold symmetry applied (Fig. 2c and Extended Data Fig. 3c, d). Local resolution calculations using ResMap30 indicated that the local resolution was significantly lower in the base (Extended Data Fig. 3e, f). This impaired a correct interpretation of this part of the TssJLM map. Since this could be due to flexibility between the base and the rest of the complex, we performed a local 3D refinement on the base region only, which yielded a 3D reconstruction of the base at 16.6-Å resolution. A composite map where this new reconstruction of the base replaces the equivalent densities in the reconstruction of the whole complex is shown in Fig. 2c. The T6SS membrane core complex is 300 Å in height and 205 Å in diameter (Fig. 2c). It is made of a base that is decorated at its bottom by five hooks and is pierced at its centre by a small hole of 15 Å in diameter (Fig. 2c). Ten arches connect this base to a tip complex of 160 Å in diameter covered by a small cap. Remarkably, five arches gather at the centre of the tip complex to define a 15- to 20-Å diameter channel. The five other arches form a scaffold at the periphery of this complex (Extended Data Fig. 4a). Overall, the tip complex is made of internal and external pillars arranged in concentric rings (Extended Data Fig. 4a).

a, SDS–polyacrylamide gel electrophoresis (SDS–PAGE) analysis of the purified EAEC TssJLM complex. The bands corresponding to TssM (130 kDa), TssL (24 kDa) and TssJ (18 kDa) after SDS–PAGE and Coomassie blue staining are indicated. b, Representative views (class averages) of purified TssJLM complexes. End to side views are shown from top left to bottom right. Scale bar, 10 nm. c, Structure of the TssJLM complex. Side, cut-away, bottom and top views are shown from top left to bottom right respectively. The different regions of the complex are indicated on the cut-away view.
To define how the core complex is inserted in the cell envelope, we first performed differential solubilization of the inner and outer membranes. The total membrane fraction was solubilized with N-lauryl sarkosyl, a detergent that preferentially solubilizes inner-membrane proteins. This differential solubilization resulted in the fractionation of the core complex in both inner and outer membrane fractions (Extended Data Fig. 4b), indicating that this complex resides in both membranes. To determine its orientation in the cell envelope, the purified core complex was incubated with anti-StrepII antibodies or Ni-NTA-coated gold particles targeting the TssJ C-terminal StrepII and TssM N-terminal 6×His tags respectively (Extended Data Fig. 4c), before EM analyses. Anti-StrepII antibodies labelled the tip complex/cap while the base was labelled by the Ni-NTA gold particles (Extended Data Fig. 4c). We concluded that the TssJ C terminus is located in the tip complex while the TssM N terminus is located in the base (Extended Data Fig. 4c). When the N-terminal cysteine residue of the TssJ lipoprotein was substituted by Ser (C1S) to prevent its acylation, an intact TssJC1S–L–M core complex was formed (Extended Data Fig. 4b), but differential solubilization proved the complex mis-localized to the inner membrane fraction only (Extended Data Fig. 4b). Hence, TssJ acylation tethers the apex of the complex to the outer membrane whereas the base of the complex is located in the cytoplasm.
We next analysed the EM reconstruction to assign the different regions of the core complex to its components. The volume corresponding to one arch and the corresponding pillar within the tip complex (Extended Data Fig. 4a) is comparable in size and shape to that of the isolated TssM periplasmic domain (amino acids 386–1129; TssMp) in complex with TssJ obtained by small-angle X-ray scattering (SAXS) (Extended Data Fig. 4d, e). Segmentation of this volume yielded five different sub-volumes (Fig. 3a). We propose that the sub-volume closest to the cap corresponds to TssJ, in agreement with its location close to the outer membrane. The other four sub-volumes would correspond to sub-domains of TssMp. Sub-volume 4 is in close contact with TssJ, suggesting that it corresponds to the C-terminal domain of TssM domain, which was previously shown to mediate contact with TssJ25. With sub-volume 3, it forms the tip complex while sub-volumes 1 and 2 correspond to the arches (Fig. 3a). Interestingly, the last TssM transmembrane segment crossing the inner membrane is located just upstream of TssMp. This would place the inner membrane at the bottom of the arches or at the top of the base. The volume of the base (1,450 Å3) is much bigger than the estimated volume occupied by ten copies of the cytoplasmic domains of TssM and TssL (825 Å3). The crystal structure of the TssL cytoplasmic domain dimer28,29 could be fitted in the hooks with 88% correlation (Extended Data Fig. 4f). This indicates that the remainder of the base could correspond to the cytoplasmic domain of TssM and the 40 transmembrane segments bound to detergent (Extended Data Fig. 4f).

a, Segmentation of the TssJLM complex reconstruction. Each volume encompassing one arch and the corresponding pillar within the tip complex is segmented in five different domains (shown in different colours). b, Crystal structure of the TssM26Ct–TssJsol complex represented as ribbons. TssJsol is coloured orange, while TssM26Ct is coloured cyan (α-domain) and blue (β-domain). The C-terminal α5-helix and the extended stretch are coloured magenta. The β-hairpin (β1–β2) is highlighted in the dashed circle. Two orthogonal views of the crystal structure and its surface representation are shown from left to right (coloured as in a). c, TssM26Ct–TssJ crystal structure docked into the EM volume corresponding to TssJ and the TssM periplasmic domains 3 and 4 extracted from both internal and external pillars of the tip complex. d, Energy-minimized atomic model of the tip complex structure (left panel, side view; right panel, top view).
To gain more insight into the structure of the TssMp–TssJ complex, TssMp was produced and purified as described previously25. To help crystallization, TssMp complex was subjected to controlled proteolytic digestion31. A protease-resistant fragment of an apparent size of ∼32 kDa (called hereafter TssM32Ct; residues 836–1129; Extended Data Fig. 5a) was further purified and co-crystallized with nb25, a specific camelid single-chain nanobody31,32. The structure of the TssM32Ct–nb25 complex was solved by molecular replacement using the X-ray structure of nb25 reported previously32 (Extended Data Fig. 5b and Extended Data Table 1). In the complex, the TssM32Ct amino-acid chains are defined in the electron density map between residues 868 and 1107. We therefore purified a new TssMp fragment (TssM26Ct) encompassing the crystallographic visible chain. This shorter fragment crystallized readily alone as well as in complex with the unacylated TssJ protein (Extended Data Table 1). The structure of TssM26Ct is composed of two domains. The N-terminal domain (residues 870–974) is a bundle of four α-helices, covered on one side by a β-hairpin (Fig. 3b) and on the other by the C-terminal elongated stretch of the protein. The C-terminal domain (residues 975–1085) is a nine-stranded β-sandwich that contacts nb25 or TssJ (Fig. 3b and Extended Table 2a, b). This C-terminal domain is followed by a stretch of residues (1086–1107) comprising helix α5 (Fig. 3b). TssJ binds to the apex of the C-terminal domain, and the 590-Å2 interaction area involves contacts between TssJ loops L1–2, L3–4 and L5–6 with TssM26Ct loops L3–4 and L5–6 (Extended Data Fig. 5c and Extended Data Table 2b), in agreement with a previous study demonstrating the importance of TssJ loop L1–2 for TssM–TssJ complex formation25. Superimposition of the structures of TssM32Ct–nb25 and TssM26Ct–TssJ shows that nb25 and TssJ cannot bind simultaneously to TssM (Extended Data Fig. 5d), explaining the nb25 in vivo inhibitory effect on T6SS function32. The comparison between TssM26Ct–TssJ crystal structure and the volume proposed to correspond to TssJ and domains 3 and 4 of TssMp determined by EM resulted in 95% correlation between the two structures (Fig. 3c). This confirms the location of TssM26Ct–TssJ in the tip complex (Fig. 3d).
Cell surface accessibility and transient pore formation
The orientation of the TssJ N terminus places the outer membrane above TssJ, where the cap is located (Figs 2c and 4a and Extended Data Fig. 5e). Accordingly, close inspection of the proposed oligomeric structure of the TssM26Ct–TssJ complex could not reveal any obvious transmembrane region (Extended Data Fig. 5f). To test this, we engineered functional cysteine derivatives between the β-strands of the C-terminal domain of TssM (Extended Data Fig. 6a). The extracellular accessibility of these residues was assessed by incubating whole cells with an outer membrane-impermeant cysteine-reactive maleimide. We observed that positions 989, 1005, 1035, 1075 and 1109 were labelled whereas positions 972, 1019, 1062 and 1092 were not (Extended Data Fig. 6b and Extended Data Table 2c). With the exception of position 1092, all other positions were labelled when cell lysates were used instead of intact cells (Extended Data Table 2c). The labelled cysteine substitutions are on the tip of TssM facing the outer membrane (Fig. 4a). Interestingly, residues 989 and 1005 are buried at the interface with TssJ (Extended Data Fig. 6c). Therefore, for these residues to be labelled, the TssM–TssJ complex has to dissociate. This result also suggests that the tip of TssM26Ct is exposed to the cell exterior. To test whether TssM stably crosses the outer membrane or accesses the cell exterior temporarily, similar experiments were conducted in a tssBC-deleted background. In the absence of the TssB and TssC sheath components, the TssJLM membrane complex is properly assembled but the T6SS is inactive as no sheath assembly or contraction could occur. In tssBC cells, only position 1109 was labelled (Extended Data Fig. 6b and Fig. 4a). These results suggest that the TssM α5-helix crosses the outer membrane permanently, exposing the C-terminal extension to the extracellular medium whereas part of TssM26Ct domain is exposed transiently at the cell surface during the T6SS mechanism of action.

a, Cell surface accessibility studies. Crystal structure of the TssM26Ct represented as ribbons, coloured cyan (α-domain) and blue (β-domain). The C-terminal α5-helix and the extended stretch are coloured magenta. The C terminus (lacking in the crystallized fragment) is represented as a random structure beyond the last residue in the crystallographic model. The cysteine substitutions (in sphere representation) used for labelling experiments are positioned in the TssM26Ct crystal structure. Cysteines with extracellular accessibility when the T6SS is active are coloured yellow, while the unlabelled ones are coloured grey. b, Model of action. The proposed mechanism of action involves five sequential stages. Stage 1: the assembled TssJLM complex is not integrally inserted in the outer membrane, but anchored to it by the TssJ N-terminal lipid moiety. This stage corresponds to the EM reconstruction of the purified TssJLM complex (bottom inset) and the crystal structure of the TssM26Ct–TssJ complex (top inset) presented in this study. Stage 2: upon assembly of T6SS baseplate and tail components, the C-terminal extremity of TssM inserts into the outer membrane and is therefore accessible at the cell surface (yellow star). This stage corresponds to the ‘resting’ state of the T6SS membrane complex. Stages 3 and 4: the membrane core complex opens to allow the passage of the Hcp tube/VgrG spike or the sheath contraction force induces conformational changes of the TssJLM complex. A molecular model of a C10 symmetrized TssJ/TssM26Ct ring is presented (top inset). The apical loops of TssM are exposed at the cell surface (yellow stars). Stage 5: after release of the Hcp tube/VgrG spike, the TssJLM membrane complex returns to the resting state, ready to perform another cycle of secretion.
Closing remarks and outlook
The data presented here allow an unprecedented understanding of the biogenesis, architecture and role of the T6SS TssJLM membrane core complex. This complex anchors the phage tail-like structure to the cell envelope and is thought to serve as conduit to guide the Hcp tube/VgrG spike upon sheath contraction15,16,17. Using fluorescence microscopy, we demonstrate that the three subunits are recruited in a specific order, starting from the outer membrane TssJ lipoprotein and pursued by the sequential addition of TssM and TssL, a hierarchy in agreement with previously published localization and interaction studies17,22,23,24,25,26,27. Therefore, T6SS biogenesis is initiated by an outer membrane lipoprotein nucleation factor and progresses inwards, like the assembly mechanisms of other bacterial secretion systems33,34,35,36,37,38,39. Our fluorescence microscopy analyses also showed that the T6SS membrane core complex assembles randomly in the cell envelope, without specific localization. The complex is stable and can be used for several events of sheath assembly/contraction, increasing the amount of toxin effectors delivered to the target cell.
The TssJLM complex has a five-fold symmetry and is composed of ten copies of each component that assemble a 1.7-MDa structure crossing the inner membrane, the periplasm and anchored to the outer membrane via the TssJ N-terminal lipid moiety. Its architecture is unique compared with other trans-envelope bacterial secretion systems (Extended Data Fig. 7a). On the basis of our accessibility experiments, we propose that upon assembly of other T6SS subunits with the membrane core complex, the TssM C-terminal extension (C-terminal extended stretch following helix α5 in the crystal structure and the remaining 22 non-visible amino acids) will change its conformation and will cross the outer membrane. The base of the TssJLM complex defines a small cavity and hole that cannot accommodate the VgrG protein and potential effectors bound to it (Fig. 4b, stages 1 and 2)9,20. We propose that the base changes its conformation to receive the baseplate components. This state would correspond to a ‘resting’ state of the T6SS machinery (Fig. 4b, stage 2). Ten arches cross the periplasm and are followed by ten pillars positioned in two concentric layers in the tip complex. The inner pillars define a channel of 15–20 Å in diameter that is not large enough to allow the passage of the ∼110 Å Hcp tube16,17,18 (Extended Data Fig. 7b). Interestingly, it was previously shown that TssM undergoes large conformational changes during secretion26. Therefore, we propose that the inner TssM pillars are pushed outwards to define a wider TssM ring with internal dimensions compatible with the passage of the Hcp tube/VgrG spike (Fig. 4b, stages 3 and 4, and Extended Data Fig. 7b, c). In other secretion systems, specific components are dedicated to assemble the outer membrane pore. No obvious transmembrane region could be found in the TssM C-terminal domain or in TssJ. It is unlikely that the C-terminal portion of TssM would form a pore of sufficient dimension by itself. Therefore, we propose that the stroke of the Hcp–VgrG arrow would mechanically push the C-terminal TssM domain towards the cell exterior, allowing the transient formation of a pore through the outer membrane (Fig. 4b, stage 4). To avoid deleterious effects for the bacterium, one may expect that the C-terminal domain of TssM returns to its initial ‘resting’ conformation at the periplasmic face of the outer membrane once the Hcp tube has been released, closing the outer membrane channel (Fig. 4b, stage 5). Overall, the membrane core complex appears to act like a docking station for the phage-like T6SS device. It nucleates the assembly of the rest of the secretion system and then guides the Hcp tube/VgrG spike through the bacterial cell envelope upon sheath contraction. Further studies will be necessary to fully understand the complete assembly process of the T6SS, the trigger that releases sheath contraction and how the Hcp tube/VgrG spike crosses both bacterial and host membranes.
Methods
No statistical methods were used to predetermine sample size. The experiments were not randomized. The investigators were not blinded to allocation during experiments and outcome assessment.
Strains, media and chemicals
The strains, plasmids and oligonucleotides used in this study are listed in Supplementary Table 1. The E. coli K-12 DH5α strain was used for cloning steps whereas E. coli K-12 BL21(DE3) and T7-Iq pLys strains were used for protein purification. The enteroaggregative E. coli EAEC strain 17-2 was used to engineer gene knockouts and fusions with fluorescent labels. Strains were routinely grown in lysogeny broth (LB) rich medium (or Terrific broth medium for protein purification) or in Sci-1-inducing medium (SIM; M9 minimal medium, glycerol 0.2%, vitamin B1 1 µg ml−1, casaminoacids 100 µg ml−1, LB 10%, supplemented or not with bactoagar 1.5%)40 with shaking at 37 °C. Plasmids were maintained by the addition of ampicillin (100 μg ml−1 for E. coli K-12, 200 μg ml−1 for EAEC) or kanamycin (50 μg ml−1). Expression of genes from pETG20A and pRSF vectors was induced with 1 mM of isopropyl-β-d-thio-galactopyrannoside (IPTG, Eurobio) for 16 h.
Strain construction
Gene deletion into the enteroaggregative E. coli 17-2 strain was achieved by using a modified one-step inactivation procedure41 as previously described23 using plasmid pKOBEG42. Briefly, a kanamycin cassette was amplified from plasmid pKD441 using oligonucleotide pairs carrying 5′ 50-nucleotide extensions homologous to regions adjacent to the gene to be deleted. After electroporation of 600 ng of column-purified PCR product, kanamycin-resistant clones were selected and verified by colony-PCR. The kanamycin cassette was then excised using plasmid pCP20 (ref. 41). Gene deletions were confirmed by colony-PCR. The same procedure was used to introduce the mCherry- or sfGFP-coding sequences downstream from the start codon (vector pKD4-sfGFP or pKD4-mCherry) or the mCherry-coding sequence upstream from the stop codon (vector pmCherry-KD4). This procedure yields strains producing fusion proteins from their original chromosomal loci.
Plasmid construction
PCRs were performed using the Phusion DNA polymerase (Thermo Scientific). Restriction enzymes were purchased from New England Biolabs and used according to the manufacturer’s instructions. Custom oligonucleotides were synthesized by Sigma Aldrich and are listed in Supplementary Table 1. Enteroaggregative E. coli 17-2 chromosomal DNA was used as a template for all PCRs. E. coli strain DH5α was used for cloning procedures. The pETG20A vector derivative encoding the periplasmic domain of the TssM periplasmic domain (TssMp, residues 386–1129) or the TssJ soluble domain have been previously described25. The fragment encoding TssM26Ct (residues 869–1107) was cloned into the pETG20A vector by restriction-free cloning43. The pRSF–TssJS intermediate plasmid was constructed by restriction cloning. Briefly, the sequence encoding the full-length tssJ gene (residues 1–178) was PCR-amplified using primers RSF-sJ-F and RSF-sJ-R. The PCR introduced a 5′ NdeI and a 3′ XhoI restriction site and a C-terminal streptavidin extension. The tssJ PCR product was sub-cloned into the pRSF-Duet (Novagen) MCS2 corresponding restriction sites. The pRSF–TssJS-FL-HM (encoding C-terminally StrepII-tagged TssJ, N-terminally Flag-tagged TssL and N-terminally 6×His-tagged TssM) was constructed by restriction-free cloning43 as previously described22. Briefly, the sequence encoding the full-length tssL (residues 1–217) and full-length tssM (residues 1–1129) genes were PCR-amplified using the primer pairs RSF-fL-F/RSF-fL-R and RSF-hM-F/RSF-hM-R, respectively. The two PCR products (tssL and tssM) were synthesized with 30-base-pair overhangs, from both 5′ and 3′ ends, corresponding to the designed integration sites into the pRSF–TssJS plasmid. The double-stranded product of the first PCR was then used as oligonucleotides for a second PCR using the target vector as template. The introduction of the C1S mutation in TssJ was performed by QuikChange mutagenesis of the pRSF–TssJS-FL-HM plasmid using oligonucleotides Jcs-F and Jcs-R. Plasmid pIBA37-TssM was constructed by restriction-free cloning and cysteine derivatives were obtained by QuikChange mutagenesis using pIBA37-TssM-C757S mutant as template.
Hcp release assay
Cells producing Flag- or HA-tagged Hcp from plasmids pUCHcpFlag or pOKHcpHA22,23 were grown in SIM to an absorbance A600 nm ∼ 0.8. Supernatant and cell fractions were separated as previously described23. Briefly, 2 × 109 cells were harvested and collected by centrifugation at 2,000g for 5 min. The supernatant fraction was then subjected to a second low-speed centrifugation and then at 16,000g for 15 min. The supernatant was filtered on sterile polyester membranes with a pore size of 0.2 μm (Membrex 25 PET, membraPure) before overnight precipitation with trichloroacetic acid 15% on ice. Cells and precipitated supernatants were resuspended in loading buffer and analysed by SDS–PAGE and immunoblotting with the anti-Flag or anti-HA antibody. As control for cell lysis, western blots were probed with antibodies raised against the periplasmic TolB protein. The assays were performed from three independent cultures, and a representative experiment is shown.
Interbacterial competition assay
The antibacterial growth competition assay was performed as described for the studies on the Citrobacter rodentium and EAEC Sci-2 T6SSs21,44 with modifications. The wild-type E. coli strain W3110 bearing the KanR pUA66-rrnB plasmid45 was used as prey in the competition assay. Attacker and prey cells were grown for 16 h in LB medium, then diluted in SIM to allow maximal expression of the sci-1 gene cluster40. Once the culture reached A600 nm ∼ 0.8, the cells were harvested and normalized to A600 nm = 0.5 in SIM. Attacker and prey cells were mixed to a 4:1 ratio and 20-µl drops of the mixture were spotted in triplicate onto a pre-warmed dry SIM agar plate supplemented or not with anhydrotetracyclin 0.02 µg ml−1. After overnight incubation at 37 °C, the bacterial spots were then cut off, and cells were resuspended in SIM to A600 nm = 0.5. Two hundred microlitres of serial dilutions were plated on kanamycin LB plates and the number of colonies was scored after overnight incubation at 37 °C. The assays were performed from at least three independent cultures, with technical triplicates and a representative technical triplicate shown.
Fluorescence microscopy, image treatment and statistical analyses
Fluorescence microscopy experiments were performed essentially as described21,46. Briefly, cells were grown overnight in LB medium and diluted to A600 nm ∼ 0.04 in SIM. Exponentially growing cells (A600 nm ∼ 0.8–1) were harvested, washed in phosphate buffered saline buffer (PBS), resuspended in PBS to A600 nm ∼ 50, spotted on a thin pad of 1.5% agarose in PBS, covered with a cover slip and incubated for 1 h at 37 °C before microscopy acquisition. For each experiment, ten independent fields were manually defined with a motorized stage (Prior Scientific) and stored (x, y, z, Perfect Focus System (PFS) offset) in our custom automation system designed for time-lapse experiments. Fluorescence and phase contrast micrographs were captured every 30 s using an automated and inverted epifluorescence microscope TE2000-E-PFS (Nikon) equipped with PFS. PFS automatically maintains focus so that the point of interest within a specimen is always kept in sharp focus at all times despite mechanical or thermal perturbations. Images were recorded with a CoolSNAP HQ 2 (Roper Scientific) and a ×100/1.4 DLL objective. The excitation light was emitted by a 120 W metal halide light. All fluorescence images were acquired with a minimal exposure time to reduce bleaching and phototoxicity effects. The sfGFP images were recorded by using the ET-GFP filter set (Chroma 49002) with an exposure time of 200–400 ms. The mCherry images were recorded by using the ET-mCherry filter set (Chroma 49008) using an exposure time of 100–200ms. Slight movements of the whole field during the time of the experiment were corrected by registering individual frames using StackReg and Image Stabilizer plugins for ImageJ. sfGFP and mCherry fluorescence channels were adjusted and merged using ImageJ (http://rsb.info.nih.gov/ij/). sfGFP fluorescence sets of data were treated to monitor foci detection. Noise and background were reduced using the ‘Subtract Background’ (20 pixels Rolling Ball) plugin from Fiji (Image J/National Institutes of Health). The sfGFP foci were automatically detected by simple image processing: (1) create a mask of cell surface and dilate, (2) detect the individual cells using the ‘Analyse Particle’ plugin of Fiji and (3) identify foci by the ‘Find Maxima’ process in Fiji. To avoid false positive results, each event was manually controlled in the original data. Microscopy analyses were performed at least six times, each in technical triplicate, and a representative experiment is shown. Box-and-whisker plots representing the number of detected foci for each strain were made with R software. To compare each population, t-tests were performed in R. Sub-pixel resolution tracking of fluorescent foci: Fluorescent foci were detected using a local and sub-pixel resolution maxima detection algorithm and tracked over time with a specifically developed plug-in for ImageJ. The x and y coordinates were obtained for each fluorescent focus on each frame. The mean square displacement was calculated as the distance of the foci from its location at t0 at each time using R software and plotted over time. For each strain tested, the mean square displacement of at least ten individual focus trajectories was calculated.
Inner and outer membrane separation
Cells were broken using an Emulsiflex-C5 (Avestin) and the crude membrane fraction was isolated by ultracentrifugation at 100,000g for 45 min. Outer and inner membranes were separated by differential solubilization. Inner membranes were solubilized by 0.5% sodium N-lauroyl sarcosyl in 50 mM Tris-HCl pH 8.0 for 30 min at 20 °C. The insoluble material containing the outer membrane fraction was isolated by ultracentrifugation at 100,000g for 20 min. The outer membrane pellet was then solubilized in SDS-loading buffer. The assay was performed in triplicate, from three independent cultures and a representative experiment is shown.
TssJLM complex production and purification
The pRSF–TssJS-FL-HM plasmid was transformed into the E. coli BL21(DE3) expression strain (Invitrogen). Cells were grown at 37 °C in lysogeny broth (LB) to A600 nm ∼ 0.7 and the expression of the tssJLM genes was induced with 1.0 mM IPTG for 16 h at 16 °C. Cell pellets were resuspended in ice-cold 50 mM Tris-HCl pH 8.0, 50 mM NaCl, 1 mM EDTA and 1 mM TCEP, supplemented with 100 μg ml−1 of DNase I, 100 μg ml−1 of lysozyme and EDTA-free protease inhibitor (Roche). After sonication, MgCl2 was added to the final concentration of 10 mM and the cell suspension was further broken using an Emulsiflex-C5 (Avestin). The broken cell suspension was clarified by centrifugation at 38,500g for 20 min. The membrane fraction was then collected by centrifugation at 98,000g for 45 min. Membranes were mechanically homogenized and solubilized in 50 mM Tris-HCl pH 8.0, 50 mM NaCl, 0.5% (w/v) n-dodecyl-β-d-maltopyranoside (DDM, Anatrace), 0.75% (w/v) decyl maltose neopentyl glycol (DM-NPG, Anatrace), 0.5% (w/v) digitonin (Sigma-Aldrich), 100 μM TCEP and 1 mM EDTA at 22 °C for 45 min. The suspension was clarified by centrifugation at 98,000g for 20 min. The supernatant was loaded onto a 5-ml StrepTrap HP (GE Healthcare) column and then washed with 50 mM Tris-HCl pH 8.0, 50 mM NaCl, 0.05% (w/v) DM-NPG (Affinity buffer) at 4 °C. The TssJLM core complex was eluted in the affinity buffer supplemented with 2.5 mM desthiobiotin (IBA) into a 5-ml HisTrap HP (GE Healthcare) column. The column was then washed in the affinity buffer supplemented with 20 mM imidazole and the TssJLM core complex was eluted in the same buffer supplemented with 500 mM imidazole. Peak fractions were pooled and loaded onto a Superose 6 10/300 column (GE Healthcare) equilibrated in 50 mM Tris-HCl pH 8.0, 50 mM NaCl, 0.025% (w/v) DM-NPG. The TssJLM core complex eluted as a single monodisperse peak close to the void volume of the column. The sample was used immediately for EM sample preparation.
Stoichiometry analyses
The purified TssJLM core membrane complex was diluted to a final concentration of 0.1 mg ml−1 and denaturated at 100 °C for 10 min after the addition of 1% sodium dodecyl sulfate. The denatured sample was incubated in presence of 40 µM of Alexa Fluor 532 C5-maleimide (Invitrogen) and 1 mM TCEP (Pierce) for 2 h at room temperature. The labelled proteins were separated by SDS–PAGE and protein-bound fluorescence was visualized and quantified using a Fujifilm FLA-3000 scanner. The assay was performed in triplicate, from three independent TssJLM complex preparations, and a representative experiment is shown. The quantification is expressed with the standard deviation from the three biological replicates.
EM and image processing
Determination of the TssJLM core membrane complex structure was achieved by negative-stain EM. Nine microlitres of suitably diluted (∼0.01 mg ml−1) TssJLM complex sample was spotted to glow-discharged carbon-coated copper grids (Agar Scientific). After 30 s of absorption, the sample was blotted, washed with three drops of water and then stained with 2% uranyl acetate. Images were recorded automatically using the EPU software on an FEG microscope operating at a voltage of 200 kV and a defocus range of 0.6–25 nm, using an FEI Falcon-II detector (Gatan) at a nominal magnification of 50,000, yielding a pixel size of 1.9 Å. A dose rate of 25 electrons per square ångström per second, and an exposure time of 1 s, were used. A total of 72,146 particles were automatically selected from 1,200 independent images and extracted within boxes of 280 pixels × 280 pixels using EMAN2/BOXER47. The defocus value was estimated and the contrast transfer function was corrected by phase flipping using EMAN2 (e2ctf). All 2D and 3D classifications and refinements were performed using RELION 1.3 (refs 48, 49). We used three rounds of reference-free 2D class averaging to clean up the automatically selected data set. Only highly populated classes displaying high-resolution features were conserved during this procedure and a final data set of 26,544 particles was assembled. An initial 3D model was generated in EMAN2 using 30 classes. Three-dimensional classification was then performed in Relion with five classes. The particles corresponding to the most populated class (∼16,738) were used for refinement. Relion auto-refine procedure was used to obtain a final reconstruction at 11.56-Å resolution after masking and with C5 symmetry imposed. Reported resolutions are based on the ‘gold standard’ Fourier shell correlation (FSC) 0.143 criterion, and FSC curves were corrected for the effects of a soft mask on them by using high-resolution noise substitution50. Before visualization, all density maps were corrected for the modulation transfer function of the detector and then sharpened by applying an ad hoc negative B-factor (−1,000). Local resolution variations were estimated using ResMap51.
Three-dimensional maps display and analysis
Three-dimensional reconstructions were displayed and rendered in USCF Chimera segmented using the SEGGER module implemented in UCSF Chimera52. Segments corresponding to individual structural domains are represented in Fig. 3c–e. All other maps were left un-segmented. A volume/mass conversion of 0.81 Da Å−3 was used to estimate the volume occupied by TssM and TssL cytoplasmic domains.
Protein production and purification for SAXS and X-ray analyses
The periplasmic domain of the TssM protein, TssMp (residues 386–1129), was produced and purified as described previously25. The purified recombinant TssMp was digested with trypsin (at a 1,000:1 molecular ratio) at room temperature for 24 h. The reaction was quenched by the addition of 1 mM phenyl-methane-sulfonyl fluoride (PMSF) and the insoluble TssMp fragments were discarded by centrifugation at 20,000g for 30 min. A proteolysis-resistant fragment of apparent size ∼32 kDa (called hereafter TssM32Ct) was further purified by consecutive ion-exchange (Mono Q 5/50 GL column, GE Healthcare) and size-exclusion (Superdex 75 16/600 HL column) chromatographies using an Äkta system (GE Healthcare). The purified fragment was subjected to N-terminal Edman sequencing. A PVDF membrane was rinsed three times with a water/ethanol mixture (10/90) and inserted in the A cartridge of a Procise 494A sequencer. After five cycles of Edman degradation, the sequence DYGSL was identified by mass spectrometry, indicating that cleavage after Arg834 generated a C-terminal fragment of theoretical mass 32,398 Da, in agreement with the 32-kDa band observed by SDS–PAGE analyses.
For production and purification of the TssM26Ct fragment (Thr869 to Glu1107), E. coli BL21(DE3) cells cultivated in the TB medium carrying plasmid pETG20A-TssM26Ct were grown to A600 nm ∼ 0.6 and the expression of TssM26Ct was induced by the addition of 0.5 mM IPTG for 16 h at 17 °C. Cells were collected by centrifugation at 10,000g at 4 °C for 15 min. The cell pellet was resuspended in lysis buffer and lysed by sonication. The lysate was clarified by centrifugation at 20,000g at 4 °C for 15 min, and the supernatant containing the Trx–His6–tev–TssM26ct fusion protein was purified by consecutive Ni2+-affinity and size-exclusion (Superdex 75 column) chromatographies on an Äkta purifier (GE Healthcare). The fractions containing the protein of interest were pooled and the 6×His-tagged TEV protease was added (5% w/w). The cleaved protein was purified using Ni2+ affinity, removing the Trx-His6, followed by size-exclusion chromatography (Superdex 75 column) on an Äkta purifier (GE Healthcare). Over 100 mg of TssM26ct fragment were obtained per litre of culture. The purified protein was verified by mass spectrometry, before being concentrated up to 8.7 mg ml−1 in 20 mM Tris-HCl pH 8.0, NaCl 150 mM.
The production of nanobody nb25 and the formation of its complex with TssM32Ct have been described previously31,32. The production of unacylated TssJ was previously described25. The TssM26Ct–TssJ complex was obtained by mixing TssM26Ct (8.7 mg ml−1) with purified TssJ (30 mg ml−1) in a 1:1.2 molecular ratio and the complex was then concentrated up to 15 mg ml−1 using centricon (cut-off of 10,000 Da) in 20 mM Tris-HCl pH 8.0, 150 mM NaCl.
SAXS and ab initio 3D shape reconstruction
SAXS analyses were performed at the ID29 beamline (European Synchrotron Radiation Facility, Grenoble, France) at a working energy of 12.5 keV (λ = 0.931 Å). Thirty microlitres of protein solution at 1.6 mg ml−1 in Tris-HCl 20 mM pH 8.0, NaCl 150 mM, were loaded by a robotic system into a 2-mm quartz capillary mounted in a vacuum. Ten independent 10-s exposures were collected on a Pilatus 6M-F detector placed at a distance of 2.85 m for each protein concentration. Individual frames were processed automatically and independently at the beamline by the data collection software (BsxCUBE), yielding radially averaged normalized intensities as a function of the momentum transfer q, with q = 4πsin(θ)/λ, where 2θ is the total scattering angle and λ is the X-ray wavelength. Data were collected in the range q = 0.04–6 nm–1. The ten frames were combined to give the average scattering curve for each measurement. Data points affected by aggregation, possibly induced by radiation damage, were excluded. Scattering from the buffer alone was also measured before and after each sample analysis and the average of these two buffer measures was used for background subtraction using the program PRIMUS53 from the ATSAS package54. PRIMUS was also used to perform Guinier analysis55 of the low q data, which provides an estimate of the radius of gyration (Rg). Regularized indirect transforms of the scattering data were performed with the program GNOM56 to obtain P(r) functions of interatomic distances. The P(r) function has a maximum at the most probable intermolecular distance and goes to zero at Dmax, the maximum intramolecular distance. The values of Dmax were chosen to fit with the experimental data and to have a positive P(r) function. Three-dimensional bead models that fitted with the scattering data were built with the program DAMMIF57. Twenty independent DAMMIF runs were performed using the scattering profile of the TRX–His–TssJp and TRX–His–TssMp complexes, with data extending up to 0.35 nm–1, using slow mode settings, assuming no symmetry and allowing for a maximum 500 steps to grant convergence. The models resulting from independent runs were superimposed using the DAMAVER suite58. This yielded an initial alignment of structures based on their axes of inertia followed by minimization of the normalized spatial discrepancy59. The normalized spatial discrepancy was therefore computed between a set of 20 independent reconstructions, with a range of normalized spatial discrepancies from 0.678 to 0.815. The aligned structures were then averaged, giving an effective occupancy to each voxel in the model, and filtered at half-maximal occupancy to produce models of the appropriate volume that were used for all subsequent analyses. All the models were similar in terms of agreement with the experimental data, as measured by DAMMIF χ parameter and the quality of the fit to the experimental curve. The DAMFILT average volume was used as the final model of the TRX–His–TssJ and TRX–His–TssMp complexes.
Ni-TNA-Nanogold labelling
The TssJLM complex was spotted onto a glow-discharged carbon coated grid (CF-400, Electron Microscopy Sciences). After 1 min, excess liquid was blotted, and the grid was washed on a drop of cold purification buffer (50 mM Tris pH 8, 50 mM NaCl, 0.025% (w/v) DM-NPG) containing 50 mM imidazole, quickly blotted and deposited on a second drop of the same buffer in the presence of 5 nM Ni-TNA-Nanogold beads (Nanoprobes). After 2 min, the grid was rinsed sequentially for 20 s with one drop of purification buffer, one drop of the same buffer without detergent and three drops of 2% uranyl acetate. Images were collected on an FEI Tecnai F20 FEG microscope operating at a voltage of 200 kV, equipped with a direct electron detector (Falcon II). Particles were selected manually using EMAN2. The assay was performed at least in triplicate, from independent TssJLM complex preparations, and representative particles are shown.
Anti-Strep labelling
The TssJLM complex was mixed with monoclonal anti-Strep antibodies (Sigma) at a ratio of complex:antibody of 2:1. The mixture was incubated at 4 °C for 30 min and the labelled complex was isolated by gel filtration. The sample was analysed by negative-stain EM as described above for negative-stain EM of the unlabelled TssJLM complex. The assay was performed at least in triplicate, from independent TssJLM complex preparations, and representative particles are shown.
Crystallization and structure determination
The crystallization of the TssM32Ct–nb25 complex has been described previously31. For TssM26Ct alone, several kits were used for crystallization screening, including STURA, WIZARD, MDL, INDEX and PEGs. A hit was observed in the PEGs kit, within a well containing 0.2 M zinc acetate and 20% (m/v) PEG3350. Crystal optimization was performed by varying PEG3350 amount in the 15–25% range in 0.1 M sodium acetate and 0.2 M ZnCl2 at pH varying between 3.8 and 5.5. Crystals appeared after few days in 20% PEG3350, 0.1 M sodium acetate pH 4 and 0.2 M ZnCl2. Crystals were tested at the European Synchrotron Radiation Facility (ESRF) ID23-1 beamline after cryo-cooling in the crystallization liquor supplemented with 12.5% propylene glycol.
The TssM26Ct–TssJ complex was screened for crystallization using the PEGs and PACT1 kits. Hits were observed in PACT1. All contained Zn2+: 0.01 M zinc chloride, 0.1 M sodium acetate pH 5, 20% (w/v) PEG 6000; or 0.01 M zinc chloride, 0.1 M MES pH 6, 20% (w/v) PEG 6000. Crystal optimization was performed by using PEG6000 in the 10–20% range in 0.1 M sodium acetate/MES pH 4.75–6, and ZnCl2 at 0, 0.01, 0.05 and 0.2 M. No crystals were obtained in conditions without ZnCl2 or containing 0.2 M ZnCl2. By contrast, well-shaped crystals appeared in 50 mM ZnCl2, 15% PEG6000 and 0.1 sodium acetate pH 4.75. Crystals were cryo-cooled with polypropylene glycol 12.5% but diffracted to only ∼4.0 Å at the Soleil Proxima 2 beamline (Saint Aubin, France). Further crystals were obtained in LIMBRO plates. Large crystals were obtained by mixing 6 µl of protein and 2 µl of well solution in 50 mM ZnCl2, 15% PEG6000, 94 mM sodium acetate pH 4.75 and 6 mM MES pH 6. Crystals were dipped in polypropylene glycol for ∼20 s and exposed at the ESRF ID23-1 beamline.
Data collection was performed at 100 K at the Soleil Proxima 1 beamline (Saint Aubin, France) for TssM32Ct–nb25 and at the ID23-1 beamline (ESRF synchrotron, Grenoble, France) for TssM26Ct alone and for the TssM26Ct–TssJ complex. Data were processed by the XDS60 package and scaled with XSCALE (Extended Data Table 1).
The structure of the TssM32Ct–nb25 complex was determined by molecular replacement with Molrep61 using the previously determined structure of nb25 (Protein Data Bank accession number 4QGY)32. After refining the positions of the two nb25 molecules in the asymmetric unit by rigid body refinement with AutoBuster62, an electron density map was calculated at 1.92-Å resolution. Features such as α-helices were easily identified, making it possible to trace manually the model of TssM32Ct using COOT63, alternated with cycles of refinement with AutoBuster62, with non-crystallographic symmetry restraints, and translation, rotation and screw-rotation (TLS) group refinement64, features used in all refinement procedures described below. The final structure at 1.92-Å resolution had Rwork/Rfree values of 18.4/21.0%, 96.3% of the residues in the preferred area of the Ramachandran plot and no outliers (Extended Data Table 1).
The structure of TssM26Ct alone was solved by molecular replacement with Molrep61 using the refined model of TssM32Ct from the TssM32Ct–nb25 complex. The initial structure model was improved through iterative refinement with AutoBuster62 and manual refitting with COOT63. The final structure at 1.51-Å resolution had Rwork/Rfree values of 19.2/20.2%, 97.4% of the residues in the preferred area of the Ramachandran plot and no outliers (Extended Data Table 1).
The structure of the TssM26Ct–TssJ complex was solved by molecular replacement with Molrep61 using the refined structure of TssM26ct and the previously determined TssJ structure (Protein Data Bank 3RX9)25 in which all the other conformations were removed. A first round of rigid body refinement and four cycles of Phenix65 cartesian-simulated annealing were performed. The resulting model was improved through iterative refinement with AutoBuster62 and manual refitting with COOT63. The final structure at 2.24-Å resolution had Rwork/Rfree values of 20.0/22.3%, 96.9% of the residues in the preferred area of the Ramachandran plot and four outlier residues in very poorly defined loops (Extended Data Table 1). The TssM32Ct–nb25, TssM26Ct and TssM26Ct–TssJ structures form similar homodimers in the asymmetric unit. However, as reported by PISA66, and the known topologies of TssM and TssJ23,24, these dimers are not biologically relevant. Molecular contacts were analysed by the PISA server33 and figures were prepared with Chimera52 and Pymol67.
The crystal structures of the TssM32Ct–nb25 complex, and of the TssM26Ct fragment and TssM26Ct–TssJ complexes, have been deposited in the Protein Data Bank under accession numbers 4Y7M, 4Y7L and 4Y7O respectively.
Docking TssM26ct–TssJ structure and TssLcyto
The crystal structures of the TssM26ct–TssJ complex and of the TssL cytoplasmic domain (Protein Data Bank 3U66)28 were docked automatically using Chimera52 after map segmentation.
Refinement of docked TssM26Ct-J pentamer in the EM density map
The atomic model of the docked TssM26Ct-J structure was refined in the EM density with RSRef68. First, missing side-chain and polar hydrogen atoms were added with Modeller69. The structure was minimized using 2,000 steps of least-squares conjugate gradient refinement in the presence of distance restraints for hydrogen bonds and backbone dihedral angle restraints to maintain secondary structures. The minimization was performed with the real-space objective function calculated by RSRef in CNS70. The C5 symmetry was enforced by strict non-crystallographic symmetry restraints. The total energy included internal parameters (bond length, bond angle, improper and dihedral angles) and non-bonded interactions with full Van der Waals and electrostatic potentials using a 7.5 Å cutoff. The final correlation coefficient between the EM reconstruction and the refined TssM26Ct-J atomic model was 0.929 (as calculated by RSRef), whereas it was 0.706 before minimization.
Modelling of TssM26Ct-J decamer
The atomic position of a TssM26Ct-J protomer from the outer ring of the pentamer served as the starting structure to generate a TssM26Ct-J decamer model with cyclic ten-fold symmetry using CNS70. The symmetry was enforced by strict non-crystallographic symmetry restraints (rotations of 36° around the symmetry axis). First, 5,000 steps of rigid body minimization were performed including only inter-protomer energetic contributions (full Van der Waals and electrostatic potentials). After a short all-atom minimization (300 steps), 1.5 ps of molecular dynamics simulation at 1,000 K was performed, followed by 300 steps of minimization and 10 ps of molecular dynamics simulation at 200 K. Minimizations and molecular dynamics simulations were realized with both intra-protomer and inter-protomer energetic contributions activated, and the backbone conformation of the protomer was restrained with harmonic constraints.
Substituted cysteine accessibility method
Cysteine accessibility experiments were performed on whole cells, mainly as described71,72 with modifications. A 20-ml culture of wild-type or ΔtssBC strains producing a periplasmic cysteine-less TssM (Cys727-to-Ser) or derivatives bearing cysteine substitutions were induced for tssM gene expression with 0.05 μg ml−1 anhydrotetracyclin (AHT) for 1 h. Cells were harvested and resuspended in buffer A (100 mM Hepes (pH 7.5), 150 mM NaCl, 25 mM MgCl2) to a final A600 nm of 12 in 500 μl of buffer A. Bovine serum albumin (BSA)-coupled maleimide (Sigma-Aldrich) was added to a final concentration of 100 µM (from a 20 mM stock freshly dissolved in DMSO) and the cells were incubated for 30 min at 25 °C. β-Mercaptoethanol (20 mM final concentration) was added to quench the biotinylation reaction, then cells were washed twice in buffer A and resuspended in buffer A containing N-ethyl maleimide (final concentration 5 mM) to block all free sulfhydryl residues. After incubation for 20 min at 25 °C, cells were disrupted by sonication. Membranes recovered by ultracentrifugation at 100,000g for 40 min were resuspended in Laemmli buffer before SDS–PAGE analysis and immunodetection with anti-Flag antibodies (to detect the TssM proteins). Controls were performed by labelling total membranes from the same samples instead of whole cells. The assay was performed in triplicate, from three independent cultures, and a representative experiment is shown.
SDS–PAGE, protein transfer, immunostaining and antibodies
SDS–PAGE was performed on Bio-Rad Mini-PROTEAN systems using standard protocols. For immunostaining, proteins were transferred onto 0.2-µm nitrocellulose membranes (Amersham Protran). Immunoblots were probed with primary antibodies and goat secondary antibodies coupled to alkaline phosphatase, and developed in alkaline buffer in presence of 5-bromo-4-chloro-3-indolylphosphate and nitroblue tetrazolium. The anti-TolB was from our laboratory collection, whereas the anti-HA (3F10 clone, Roche), anti-Flag (M2 clone, Sigma Aldrich), anti-StrepII (Sigma Aldrich), anti-5his (Sigma Aldrich) monoclonal antibodies and alkaline-phosphatase-conjugated goat anti-rabbit or mouse secondary antibodies (Millipore) were purchased as indicated.
Accession codes
Primary accessions
Electron Microscopy Data Bank
Protein Data Bank
Data deposits
The EM structure of the TssJLM complex has been deposited in the Electron Microscopy Data Bank (EMDB) under accession number emd-2927. The crystal structures of the TssM32Ct–nb25 complex, and of the TssM26Ct fragment and TssM26Ct–TssJ complexes, have been deposited in the Protein Data Bank under accession numbers 4Y7M, 4Y7L and 4Y7O, respectively.
References
West, S. A., Griffin, A. S. & Gardner, A. Evolutionary explanations for cooperation. Curr. Biol. 17, 661–672 (2007)
Blango, M. G. & Mulvey, M. A. Bacterial landlines: contact-dependent signaling in bacterial populations. Curr. Opin. Microbiol. 12, 177–181 (2009)
Russell, A. B., Peterson, S. B. & Mougous, J. D. Type VI secretion system effectors: poisons with a purpose. Nature Rev. Microbiol. 12, 137–148 (2014)
Silverman, J. M., Brunet, Y. R., Cascales, E. & Mougous, J. D. Structure and regulation of the type VI secretion system. Annu. Rev. Microbiol. 66, 453–472 (2012)
Pukatzki, S., Ma, A. T., Revel, A. T., Sturtevant, D. & Mekalanos, J. J. Type VI secretion system translocates a phage tail spike-like protein into target cells where it cross-links actin. Proc. Natl Acad. Sci. USA 104, 15508–15513 (2007)
Russell, A. B. et al. Type VI secretion delivers bacteriolytic effectors to target cells. Nature 475, 343–347 (2011)
Russell, A. B. et al. Diverse type VI secretion phospholipases are functionally plastic antibacterial effectors. Nature 496, 508–512 (2013)
Ma, L. S., Hachani, A., Lin, J. S., Filloux, A. & Lai, E. M. Agrobacterium tumefaciens deploys a superfamily of type VI secretion DNase effectors as weapons for interbacterial competition in planta. Cell Host Microbe 16, 94–104 (2014)
Durand, E., Cambillau, C., Cascales, E., Journet, L. & Vgr, G. Tae, Tle, and beyond: the versatile arsenal of Type VI secretion effectors. Trends Microbiol. 22, 498–507 (2014)
Borgeaud, S., Metzger, L. C., Scrignari, T. & Blokesch, M. Bacterial evolution. The type VI secretion system of Vibrio cholerae fosters horizontal gene transfer. Science 347, 63–67 (2015)
Cascales, E. The type VI secretion toolkit. EMBO Rep. 9, 735–741 (2008)
Bönemann, G., Pietrosiuk, A., Diemand, A., Zentgraf, H. & Mogk, A. Remodelling of VipA/VipB tubules by ClpV-mediated threading is crucial for type VI protein secretion. EMBO J. 28, 315–325 (2009)
Basler, M., Pilhofer, M., Henderson, G. P., Jensen, G. J. & Mekalanos, J. J. Type VI secretion requires a dynamic contractile phage tail-like structure. Nature 483, 182–186 (2012)
Kudryashev, M. et al. Structure of the type VI secretion system contractile sheath. Cell 160, 952–962 (2015)
Coulthurst, S. J. The type VI secretion system – a widespread and versatile cell targeting system. Res. Microbiol. 164, 640–654 (2013)
Ho, B. T., Dong, T. G. & Mekalanos, J. J. A view to a kill: the bacterial type VI secretion system. Cell Host Microbe 15, 9–21 (2014)
Zoued, A. et al. Architecture and assembly of the type VI secretion system. Biochim. Biophys. Acta 1843, 1664–1673 (2014)
Bönemann, G., Pietrosiuk, A. & Mogk, A. Tubules and donuts: a type VI secretion story. Mol. Microbiol. 76, 815–821 (2010)
Brunet, Y. R., Hénin, J., Celia, H. & Cascales, E. Type VI secretion and bacteriophage tail tubes share a common assembly pathway. EMBO Rep. 15, 315–321 (2014)
Shneider, M. M. et al. PAAR-repeat proteins sharpen and diversify the type VI secretion system spike. Nature 500, 350–353 (2013)
Brunet, Y. R., Espinosa, L., Harchouni, S., Mignot, T. & Cascales, E. Imaging type VI secretion-mediated bacterial killing. Cell Rep. 3, 36–41 (2013)
Aschtgen, M. S., Gavioli, M., Dessen, A., Lloubès, R. & Cascales, E. The SciZ protein anchors the enteroaggregative Escherichia coli type VI secretion system to the cell wall. Mol. Microbiol. 75, 886–899 (2010)
Aschtgen, M. S., Bernard, C. S., de Bentzmann, S., Lloubès, R. & Cascales, E. SciN is an outer membrane lipoprotein required for type VI secretion in enteroaggregative Escherichia coli. J. Bacteriol. 190, 7523–7531 (2008)
Ma, L. S., Lin, J. S. & Lai, E. M. An IcmF family protein, ImpLM, is an integral inner membrane protein interacting with ImpKL, and its walker a motif is required for type VI secretion system-mediated Hcp secretion in Agrobacterium tumefaciens. J. Bacteriol. 191, 4316–4329 (2009)
Felisberto-Rodrigues, C. et al. Towards a structural comprehension of bacterial type VI secretion systems: characterization of the TssJ-TssM complex of an Escherichia coli pathovar. PLoS Pathog. 7, e1002386 (2011)
Ma, L. S., Narberhaus, F. & Lai, E. M. IcmF family protein TssM exhibits ATPase activity and energizes type VI secretion. J. Biol. Chem. 287, 15610–15621 (2012)
Aschtgen, M. S., Zoued, A., Lloubès, R., Journet, L. & Cascales, E. The C-tail anchored TssL subunit, an essential protein of the enteroaggregative Escherichia coli Sci-1 type VI secretion system, is inserted by YidC. MicrobiologyOpen 1, 71–82 (2012)
Durand, E. et al. Structural characterization and oligomerization of the TssL protein, a component shared by bacterial type VI and type IVb secretion systems. J. Biol. Chem. 287, 14157–14168 (2012)
Chang, J. H. & Kim, Y. G. Crystal structure of the bacterial type VI secretion system component TssL from Vibrio cholerae. J. Microbiol. 53, 32–37 (2015)
Kucukelbir, A., Sigworth, F. J. & Tagare, H. D. Quantifying the local resolution of cryo-EM density maps. Nature Methods 11, 63–65 (2014)
Nguyen, V. S. et al. Production, crystallization and X-ray diffraction analysis of a complex between a fragment of the TssM T6SS protein and a camelid antibody. Acta Crystallogr F. 71, 266–271 (2015)
Nguyen, V. S. et al. Inhibition of type VI secretion by an anti-TssM llama nanobody. PLoS ONE 10, e0122187 (2015)
Diepold, A. et al. Deciphering the assembly of the Yersinia type III secretion injectisome. EMBO J. 29, 1928–1940 (2010)
Judd, P. K., Kumar, R. B. & Das, A. Spatial location and requirements for the assembly of the Agrobacterium tumefaciens type IV secretion apparatus. Proc. Natl Acad. Sci. USA 102, 11498–11503 (2005)
Hardie, K. R., Lory, S. & Pugsley, A. P. Insertion of an outer membrane protein in Escherichia coli requires a chaperone-like protein. EMBO J. 15, 978–988 (1996)
Drake, S. L., Sandstedt, S. A. & Koomey, M. PilP, a pilus biogenesis lipoprotein in Neisseria gonorrhoeae, affects expression of PilQ as a high-molecular-mass multimer. Mol. Microbiol. 23, 657–668 (1997)
Burghout, P. et al. Role of the pilot protein YscW in the biogenesis of the YscC secretin in Yersinia enterocolitica. J. Bacteriol. 186, 5366–5375 (2004)
Crago, A. M. & Koronakis, V. Salmonella InvG forms a ring-like multimer that requires the InvH lipoprotein for outer membrane localization. Mol. Microbiol. 30, 47–56 (1998)
Daefler, S. & Russel, M. The Salmonella typhimurium InvH protein is an outer membrane lipoprotein required for the proper localization of InvG. Mol. Microbiol. 28, 1367–1380 (1998)
Brunet, Y. R., Bernard, C. S., Gavioli, M., Lloubès, R. & Cascales, E. An epigenetic switch involving overlapping fur and DNA methylation optimizes expression of a type VI secretion gene cluster. PLoS Genet. 7, e1002205 (2011)
Datsenko, K. A. & Wanner, B. L. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl Acad. Sci. USA 97, 6640–6645 (2000)
Chaveroche, M. K., Ghigo, J. M. & d’Enfert, C. A rapid method for efficient gene replacement in the filamentous fungus Aspergillus nidulans. Nucleic Acids Res. 28, e97 (2000)
van den Ent, F. & Löwe, J. RF cloning: a restriction-free method for inserting target genes into plasmids. J. Biochem. Biophys. Methods 67, 67–74 (2006)
Gueguen, E. & Cascales, E. Promoter swapping unveils the role of the Citrobacter rodentium CTS1 type VI secretion system in interbacterial competition. Appl. Environ. Microbiol. 79, 32–38 (2013)
Zaslaver, A. et al. A comprehensive library of fluorescent transcriptional reporters for Escherichia coli. Nature Methods 3, 623–628 (2006)
Zoued, A. et al. TssK is a trimeric cytoplasmic protein interacting with components of both phage-like and membrane anchoring complexes of the type VI secretion system. J. Biol. Chem. 288, 27031–27041 (2013)
Tang, G. et al. EMAN2: an extensible image processing suite for electron microscopy. J. Struct. Biol. 157, 38–46 (2007)
Scheres, S. H. RELION: implementation of a Bayesian approach to cryo-EM structure determination. J. Struct. Biol. 180, 519–530 (2012)
Scheres, S. H. Semi-automated selection of cryo-EM particles in RELION-1.3. J. Struct. Biol. 189, 114–122 (2015)
Chen, S. et al. High-resolution noise substitution to measure overfitting and validate resolution in 3D structure determination by single particle electron cryomicroscopy. Ultramicroscopy 135, 24–35 (2013)
Kucukelbir, A., Sigworth, F. J. & Tagare, H. D. Quantifying the local resolution of cryo-EM density maps. Nature Methods 11, 63–65 (2014)
Pettersen, E. F. et al. UCSF Chimera – a visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 (2004)
Konarev, P. V., Volkov, V. V., Sokolova, A. V., Koch, M. H. & Svergun, D. I. PRIMUS: a Windows PC-based system for small-angle scattering data analysis. J. Appl. Crystallogr. 36, 1277–1282 (2003)
Konarev, P. V., Petoukhov, M. V., Volkov, V. V. & Svergun, D. I. ATSAS 2.1, a program package for small-angle scattering data analysis. J. Appl. Crystallogr. 39, 277–286 (2006)
Guinier, A. La diffraction des rayons X aux très petits angles; application à l’étude de phénomènes ultramicroscopiques. Ann. Phys. (Paris) 12, 161–237 (1939)
Svergun, D. I. Determination of the regularization parameter in indirect-transform methods using perceptual criteria. J. Appl. Crystallogr. 25, 495–503 (1992)
Franke, D. & Svergun, D. I. DAMMIF, a program for rapid ab-initio shape determination in small-angle scattering. J. Appl. Crystallogr. 42, 342–346 (2009)
Volkov, V. V. & Svergun, D. I. Uniqueness of ab initio shape determination in small-angle scattering. J. Appl. Crystallogr. 36, 860–864 (2003)
Kozin, M. B. & Svergun, D. I. Automated matching of high- and low-resolution structural models. J. Appl. Crystallogr. 34, 33–41 (2001)
Kabsch, W. XDS. Acta Crystallogr. D 66, 125–132 (2010)
Vagin, A. & Teplyakov, A. Molecular replacement with MOLREP. Acta Crystallogr. D 66, 22–25 (2010)
Blanc, E. et al. Refinement of severely incomplete structures with maximum likelihood in BUSTER-TNT. Acta Crystallogr. D 60, 2210–2221 (2004)
Emsley, P., Lohkamp, B., Scott, W. G. & Cowtan, K. Features and development of Coot. Acta Crystallogr. D 66, 486–501 (2010)
Winn, M. D., Murshudov, G. N. & Papiz, M. Z. Macromolecular TLS refinement in REFMAC at moderate resolutions. Methods Enzymol. 374, 300–321 (2003)
Adams, P. D. et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D 66, 213–221 (2010)
Krissinel, E. & Henrick, K. Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 372, 774–797 (2007)
The PyMOL Molecular Graphics System. v.1.5.0.4 (Schrödinger, LLC, 2014)
Chapman, M. S., Trzynka, A. & Chapman, B. K. Atomic modeling of cryo-electron microscopy reconstructions – joint refinement of model and imaging parameters. J. Struct. Biol. 182, 10–21 (2013)
Sali, A. & Blundell, T. L. Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 234, 779–815 (1993)
Brunger, A. T. Version 1.2 of the Crystallography and NMR system. Nature Protocols 2, 2728–2733 (2007)
Bogdanov, M., Zhang, W., Xie, J. & Dowhan, W. Transmembrane protein topology mapping by the substituted cysteine accessibility method (SCAMTM): application to lipid-specific membrane protein topogenesis. Methods 36, 148–171 (2005)
Goemaere, E. L., Devert, A., Lloubès, R. & Cascales, E. Movements of the TolR C-terminal domain depend on TolQR ionizable key residues and regulate activity of the Tol complex. J. Biol. Chem. 282, 17749–17757 (2007)
Du, D. et al. Structure of the AcrAB–TolC multidrug efflux pump. Nature 509, 512–515 (2014)
Hodgkinson, J. L. et al. Three-dimensional reconstruction of the Shigella T3SS transmembrane regions reveals 12-fold symmetry and novel features throughout. Nature Struct. Mol. Biol. 16, 477–485 (2009)
Low, H. H. et al. Structure of a type IV secretion system. Nature 508, 550–553 (2014)
Acknowledgements
This work was funded by Agence Nationale de la Recherche (ANR) grants ANR-10-JCJC-1303-03 to E.C., Bip:Bip to R.F., ANR-14-CE14-0006-02 to C.C. and E.C., and supported by the French Infrastructure for Integrated Structural Biology (FRISBI) ANR-10-INSB-05-01. E.D. was supported by a post-doctoral fellowship from the Fondation pour la Recherche Médicale (SPF20101221116) and ANR grants Bip:Bip and ANR-10-JCJC-1303-03. V.S.N. was supported by a PhD grant from the French Embassy in Vietnam (792803C). A.Z., L.L. and M.S.A. were recipients of doctoral fellowships from the French Ministère de la Recherche. A.Z. received a Fondation pour la Recherche Médicale fellowship (FDT20140931060). We thank O. Francetic for providing anti-DglA and anti-OmpF antibodies. We thank E. Marza, P. Violinova Krasteva and H. Remaut for comments on the manuscript, and T. Mignot, M. Guzzo and L. Espinosa for advice about the fluorescence microscopy experiments and the statistical analyses. We also thank the members of the R.F. and E.C. research groups for discussions and suggestions, and R. Lloubès, J. Sturgis and A. Galinier for encouragement. We thank the ERSF and Soleil Synchrotron radiation facilities for beamline allocation.
Author information
Authors and Affiliations
Contributions
E.D., A.Z., C.C., E.C. and R.F. designed the experiments. A.Z. constructed the EAEC mutant and fluorescent strains and performed the fluorescence microscopy experiments and statistical analyses. L.L. and M.S.A. constructed the TssM cysteine derivatives and performed the accessibility experiments. E.D. assisted by An.D. purified the TssJLM complex and performed its biochemical characterization. E.D. and G.P.A. collected the EM data. E.D. and R.F. obtained the 3D reconstruction of the TssJLM complex. V.S.N., S.S., A.R. and C.C. purified, crystallized and solved the X-ray structures. Al.D. generated the nanobody. B.B. obtained the energy-minimized models of the closed and open states of the TssM26Ct–TssJ complex.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Extended data figures and tables
Extended Data Figure 1 Functional and dynamic properties of fluorescently labelled Tss proteins.
a, GFP–TssM and GFP–TssL fusion proteins are functional. Top: Hcp release assay. Hcp release was assessed by separating whole cells (C) and supernatant (S) fractions from the indicated strains. A total of 1 × 109 cells and the TCA-precipitated material from the supernatant of 2 × 109 cells were analysed by western blot using anti-Flag monoclonal antibody (lower panel) and anti-TolB polyclonal antibodies as a lysis control (upper panel). The molecular mass markers (in kilodaltons) are indicated on the left. Bottom: anti-bacterial assay. The anti-bacterial activity was assessed by mixing kanamycin-resistant prey E. coli K-12 cells with the indicated attacker cells for 16 h at 37 °C in SIM. The number of recovered E. coli prey cells is indicated in the graph (as log of colony-forming units (c.f.u.)). The circles indicate values from three independent assays, and the average is indicated by the bar. b, TssJ–sfGFP and TssJ–mCh fusion proteins are non-functional. Hcp release was assessed by separating whole cells (C) and supernatant (S) fractions from the indicated strains. A total of 1 × 109 cells and the TCA-precipitated material from the supernatant of 2 × 109 cells were analysed by western blot using anti-Flag monoclonal antibody (lower panel) and anti-TolB polyclonal antibodies as a lysis control (upper panel). The molecular mass markers (in kilodaltons) are indicated on the left. c, sfGFP–TssM and sfGFP–TssL cluster in foci. Large fields of fluorescence microscopy recordings showing localization of the sfGFP–TssL (left) and sfGFP–TssM (right) fusion proteins. The positions of selected foci are indicated by arrowheads. Scale bars, 5 μm. d, sfGFP–TssM and sfGFP–TssL foci are stable and static. Mean square displacement (in arbitrary units (a.u.)) of sfGFP–TssM (blue line) and sfGFP–TssL (red line) clusters were measured by sub-pixel tracking of fluorescent foci and plotted over time (in minutes). e, The TssBC sheath tubular structures assemble on TssJLM membrane complexes. Statistical analyses reporting the average number of sheath per cell compared with the number of membrane complexes per cell, highlighting the observation that the number of membrane complexes is at least equal to the number of sheathes. Lower and upper boundaries of the boxes correspond to the 25% and 75% percentiles respectively. Black bold horizontal bar, median values for each strain; whiskers, 10% and 90% percentiles; n indicates the number of cells studied per strain. f, Long-term fluorescence microscopy recordings. Time-lapse fluorescence microscopy recordings showing localization and dynamics of the sfGFP–TssL and TssB–mCherry fusion proteins. Individual images were taken every 15 min. Assembly/contraction of the sheath and TssL localization events are schematized in the lowest panel. Scale bars, 1 μm. g, Statistical analysis of sfGFP–TssM and sfGFP–TssL localization in various tss backgrounds. Shown are box-and-whisker plots of the measured number of sfGFP–TssM and sfGFP–TssL foci per cell for each indicated strain with the lower and upper boundaries of the boxes corresponding to the 25% and 75% percentiles respectively (horizontal bar, the median values for each strain; whiskers, the 10% and 90% percentiles); n indicates the number of cells studied per strain.
Extended Data Figure 2 Expression and purification of the T6SS membrane core complex.
a–e, T6SS operon genomic organization and constructs used for in vitro analyses. a, Schematic representation of the T6SS sci-1 gene cluster from entero aggregative E. coli. The numbers on top refer to the gene locus tag (EC042_XXXX). Genes encoding core components (identified by their names on bottom, for example, ‘B’ refers to the tssB gene) are coloured grey. Genes of unknown function are coloured white. The three genes used to reconstitute the core membrane complex are coloured orange (tssJ), blue (tssL) and green (tssM). b, Schematic representation of the engineered constructs: the tssJ, tssL and tssM genes were amplified with an additional Shine Dalgarno (SD) sequence and 3′ StrepII, 5′ Flag and 5′ 6×His tags respectively. These three fragments were cloned into the pRSF-Duet vector (c). This construct allows the production of the C-terminally StrepII-tagged TssJ outer membrane (OM) lipoprotein and N-terminally Flag-tagged TssL and 6×His-tagged TssM inner-membrane (IM) proteins (d, e). The proteins are schematized and their boundaries and principal characteristics (TM, transmembrane segments; SP, signal peptide; CYS, acylated cysteine) are indicated (d) and their topologies are shown (e). The additional TssM constructs (TssMp, TssM32Ct and TssM26Ct) used for SAXS or X-ray analyses are shown at the bottom. f–h, Purification and biochemical characterization of the T6SS membrane core complex. f, Analytical size-exclusion chromatography analysis of the purified TssJLM complex (continuous line) on a Superose 6 column, calibrated with 75-, 158-, 440- and 660-kDa molecular mass markers (dotted lines). The molecular mass of each marker (in kilodaltons) is indicated on the top of the corresponding peak. An arrow indicates the position of the peak fraction corresponding to the TssJLM complex. g, SDS–PAGE of the purified TssJLM complex analysed by Coomassie staining (CB) or immunoblotting using anti-His (α-His), -Flag (α-Flag) and -StrepII (α-STREP) antibodies. h, Left: cysteine labelling of the purified TssJLM complex in reducing and denaturing conditions as described in Methods. The total number of cysteine residues was nine for TssM, five for TssL and none for TssJ (the N-terminal cysteine is acylated). Right: the relative amount of TssL compared with TssM (densitometry relative to the number of free cysteine residues, fixed at 1 for TssM).
Extended Data Figure 3 Architecture of the T6SS membrane core complex.
a, Negative-stain EM of the EAEC TssJLM complex. Representative micrograph of the data set used for image processing. Isolated TssJLM complexes were clearly visible (white circles). b, Plot of the rotational autocorrelation function for a representative class average of an end-view. c, FSC curve of the TssJLM reconstruction. The ‘gold standard’ FSC curve was calculated in Relion using the masked reconstruction of the TssJLM complex. The resolution at 0.143 correlation was 11.56 Å. d, Top: side and corresponding cut-away views of the 3D reconstruction for the whole TssJLM complex. Bottom: local resolution as calculated by Resmap. The TssJLM volume (left reconstruction, side view; right reconstruction, cut-away view) is coloured according to the local resolution from high resolution (∼12 Å) in blue to low resolution (>30 Å) in red. e, FSC curve of the TssJLM base. The ‘gold standard’ FSC curve was calculated in Relion using the unmasked reconstruction of the TssJLM base. The resolution at 0.143 correlation was 16.6 Å. f, Top, side and bottom views of the 3D reconstruction after specific refinement of the base.
Extended Data Figure 4 Structural analysis and segmentation.
a, Segmentation of the TssJLM reconstruction. Left: above the base, ten equivalent densities could be defined by segmentation. They are arranged in two concentric rings. The internal ring is represented in green in the top panel and the external ring is represented in blue in the bottom panel. Right: cut-out views of the complex showing the arrangement of the two concentric rings at different levels (grey lines) along the periplasmic portion of the TssJLM complex. The cut-out views are seen from the bottom of the complex. b, Requirement of TssJ lipidation for complex assembly and insertion into the outer membrane. Left: membrane fractionation by differential solubilization followed by immunoblot analysis. Total membrane extracts from cells producing the wild-type TssJLM complex or the TssJLM complex with an unacylated variant of TssJ (Cys1-to-Ser substitution, CS) were solubilized by lauroyl sarcosine to separate inner membranes and outer membranes. HisTssM, FlagTssL and TssJStrep (indicated on the right) were revealed by anti-His, anti-Flag and anti-StrepII antibodies respectively. Controls included immuno-detection of the inner membrane DglA diacylglycrol lipase and the outer membrane OmpF porin. Wild-type TssJLM complex co-fractionates with both the inner and outer membrane fractions whereas the Cys1-to-Ser substitution mutated complex co-fractionates only with the inner-membrane fraction. Top right: negative-stain EM of the mutated TssJCSLM complex. Representative micrograph of the data set used for image processing. Isolated TssJCSLM complexes were clearly visible (white circles). Bottom right: gallery of representative class averages generated after reference-free 2D classification in Relion. End to side views are shown from top left to bottom right. c, Orientation of the TssJLM complex in the cell envelope. Left: schematic representation of the TssJ (J, orange), TssL (L, blue) and TssM (M, green) proteins. Their localization, main characteristics (lipidation or transmembrane segments shown in black) and the location of the 6×His and StrepII tags (red balls) are indicated. The strepII and 6×His tags were introduced at the C terminus and N terminus of TssJ and TssM respectively. Middle: immune and Nanogold labelling coupled to EM. Anti-StrepII or Nanogold-NTA were incubated with the TssJLM complex and visualized by negative-stain EM. A gallery of representative views is presented (top row, StrepII labelling; bottom row, Ni-NTA labelling). StrepII antibodies (a schematic diagram with StrepII antibodies depicted as blue circles is shown on top) and Nanogold-NTA are highlighted in red circles. Right: the positions of the StrepII antibody (targeting TssJ C terminus) and of the Ni-NTA gold particle (targeting TssM N terminus) are indicated on the TssJLM reconstruction. d, SAXS data and low-resolution structure of the TssMp–TssJ complex. Top left: experimental scattering data (green crosses) and the fitting curve (continuous red line) calculated from an ab initio model of the TssMp–TssJ complex. Top right: Guinier plot (dots) with the linear fit (continuous line). Bottom left: distance distribution function of the TssMp–TssJ complex. Bottom right: SAXS envelope (grey surface) of the ‘best representative’ model of the TssMp–TssJ complex. Each view is rotated by 90° around the y-axis. e, Location of the TssMp–TssJ complex SAXS envelope in the 3D reconstruction of TssJLM complex. Left: the volume of the TssMp–TssJ complex determined by SAXS was docked into the EM 3D reconstruction of the TssJLM complex (top). Two optimal docking positions were found, both with 82% correlation with the EM map (coloured magenta and pink). The corresponding volumes in the EM map were extracted (bottom). They correspond to the same volume displayed in Extended Data Fig. 4a. Right: direct comparison of the SAXS (magenta) and EM (blue and green) volumes corresponding to the TssMp–TssJ complex. The volumes are equivalent in size and shape. f, TssL cytoplasmic domain docking into the TssJLM complex base. Fitting of the TssL cytoplasmic domain (TssLcyto)28 dimer in green ribbons in the hooks found in the base. Top and bottom: side and bottom views, respectively.
Extended Data Figure 5 Crystal structure of the TssJ–TssM C-terminal domain complex.
a, Amino-acid sequence of TssM. The different domains as well as the fragments used in this study are indicated (yellow, transmembrane helix; grey, cytoplasmic domain; green, blue and purple, periplasmic domain; blue and purple, C-terminal domain corresponding to the TssM32Ct fragment; purple, C-terminal domain corresponding to the TssM26Ct fragment). b, Crystal structure of the TssM32Ct–nb25 complex. The two proteins are represented as rainbow-coloured ribbons. The complementary determining regions (CDRs 1–3, coloured blue, green and red, respectively) of nb25 are indicated. The inset highlights the TssM32Ct–nb25 interface: the TssM32Ct surface is coloured beige whereas nb25 is represented as rainbow-coloured ribbons; the side chains of the amino acids in contact with TssM32Ct are indicated. The nb25 nanobody binds the TssM C-terminal domain, and covers a surface area of 580 Å2 by inserting its protruding CDR3 between TssM32Ct loops L5–6 and L9–10. The contacts between the two proteins are listed in Extended Data Table 2a. c, Crystal structure of the TssM26Ct–TssJ complex. Left: the two proteins are represented as ribbons and coloured in rainbow mode. Middle: same view rotated by 90°. The TssJ loop 1–2, previously shown to contact TssM25, is indicated. Right: TssM26Ct–TssJ interface. Top panel: the TssM26Ct surface is coloured violet, whereas TssJ is represented as rainbow-coloured ribbons. The TssJ side-chains of the amino acids in contact with TssM are indicated. The loops are numbered according to the flanking β-strands. Bottom panel: the TssJ surface is coloured beige whereas TssM26Ct is represented as rainbow-coloured ribbons. The TssM side-chains of the amino acids in contact with TssJ are indicated. The contacts between the two proteins are listed in Extended Data Table 2b. d, Comparison of the binding sites of nb25 and TssJ on TssM. Left: the structure of the TssM26Ct–TssJ complex (rainbow coloured) has been superimposed to the structure of the TssM32Ct–nb25 complex (only nb25 is shown in grey for clarity). Right: the same partners as in the left panel in surface representation. TssM26Ct (violet), TssJ (green) and nb25 (pink). e, Insertion of the TssJ lipid anchor in the outer membrane. Left: TssJ structure25 with the N-terminal 24 residues (absent in the crystal structure). This N-terminal extension (in magenta), predicted to be disordered, was modelled in Chimera using Modeller. The first cysteine residue is acylated to allow anchorage to the inner leaflet of the outer membrane (orange rectangle). Right: docking of the TssM26Ct–TssJ complex in the EM 3D reconstruction of the TssJLM complex (only the uppermost (tip) part of the TssJLM complex is shown). Left panel: two TssM26Ct–TssJ were docked into the inner and outer pillars of the tip complex. Right panel: docking in each pillar of the TssJLM tip complex (C5 symmetry). f, Hydrophobicity of the TssM26Ct–TssJ complex. Surface representation of the TssM26Ct–TssJ decamer (left, top view; right, side view). The hydrophobicity of the surface residues is displayed (blue to red scale from most hydrophilic to most hydrophobic). No obvious hydrophobic patch is visible at the surface of the complex.
Extended Data Figure 6 Cell surface accessibility of TssM C-terminal domain.
a, Functionality of the TssM cysteine variants. Hcp release was assessed by separating whole cells (C) and supernatant (S) fractions from the wild-type (WT) 17-2 strain and its ΔtssM derivative producing a wild-type allele of TssM or TssM cysteine substitution derivatives (as indicated). A total of 1 × 109 cells and the TCA-precipitated material from the supernatant of 2 × 109 cells were analysed by western blot using anti-HA monoclonal antibody (lower panel) and anti-TolB polyclonal antibodies as a lysis control (upper panel). The molecular mass markers (in kilodaltons) are indicated on the left. b, Cysteine substitution labelling. Accessibility to cysteine residues positioned in TssM domain 4 loops was assessed by treating isolated membranes (M) or whole cells (WC) of the indicated strain (WT, wild-type 17-2; ΔtssBC; ΔtssJ) producing the indicated TssM cysteine derivative (in red letters) with the cysteine-reactive, membrane-impermeant BSA-maleimide (BSA-mal). Samples corresponding to a total of 5 × 109 cells were analysed by western blot using anti-Flag monoclonal antibody. The position of the TssM protein (∼125 kDa) is indicated as well as that of a retarded band corresponding to BSA-maleimide-coupled TssM (∼190 kDa; asterisk). The molecular mass markers (in kilodaltons) are indicated. c, Close-up of the TssM26Ct–TssJ interface. TssM26Ct is represented in blue ribbons. TssJ is represented in orange ribbons and orange transparent surface. TssM residues accessible from the cell exterior when the T6SS is functional are indicated by yellow spheres whereas unaccessible residues are shown by grey spheres. The accessible residues 989 and 1005 are buried at the interface between TssM and TssJ, suggesting that this interface is probably disrupted during T6SS assembly and/or function. Left and right panels are orthogonal views of the same molecule.
Extended Data Figure 7 Comparison with other bacterial secretion systems and model for channel opening.
a, Comparison between the T6SS TssJLM membrane core complex structure and other bacterial secretion systems. From left to right, the E. coli AcrAB-TolC multi-drug efflux pump (EMDB accession number emd-5915)73, the EAEC T6SS membrane core complex (this study, EMDB accession number emd-2927), the Shigella T3SS transmembrane complex (EMDB accession number emd-1617)74 and the E. coli R388 T4SS complex (EMDB accession number emd-2567)75. The positions on the inner membrane (IM) and outer membrane (OM) are indicated (C, cytoplasm; P, periplasm). Scale bar, 10 nm. b, Docking of the Hcp tube/VgrG spike into the TssJLM 3D reconstruction. Left: before sheath contraction. The Hcp tube/VgrG spike (VgrG in yellow and Hcp in green; surface representation) was manually docked in the 3D reconstruction of TssJLM complex (grey surface). The diameter of the channel defined by the closed tip complex is not large enough to allow the passage of the tube/spike, suggesting that large conformational changes probably occur. The cavity at the tip of VgrG could be filled by VgrG-bound PAAR modules or toxin effectors20. Right: during sheath contraction. The diameter of the C10-symmetrized TssM26Ct model (represented as ribbons) is compatible with the passage of the Hcp tube/VgrG spike (same colours as in the left panel). c, Closed and open forms of the TssM26Ct oligomer. Crystal structure of TssM26Ct represented as ribbons and transparent surface. The TssM26Ct α- and β-domains are coloured cyan and blue, respectively. The C-terminal α5-helix and the extended stretch are coloured pink. Cysteines with extracellular accessibility when the T6SS is active are coloured yellow, while the unlabelled ones are coloured red. Left: docking of the TssM26Ct–TssJ crystal structure in the EM 3D reconstruction of the TssJLM tip complex. Top and bottom panels, side and top views, respectively. Right: model of a C10-symmetrized oligomer of the TssM26Ct domain. Top and bottom panels, side and top views, respectively.
Supplementary information
Supplementary Tables
This file contains Supplementary Table 1. (PDF 222 kb)
Rights and permissions
About this article

Cite this article
Durand, E., Nguyen, V., Zoued, A. et al. Biogenesis and structure of a type VI secretion membrane core complex. Nature 523, 555–560 (2015). https://doi.org/10.1038/nature14667
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/nature14667
This article is cited by
-
Cytoplasmic contractile injection systems mediate cell death in Streptomyces
Nature Microbiology (2023)
-
Comparative genomics revealed that Vibrio furnissii and Vibrio fluvialis have mutations in genes related to T6SS1 and T6SS2
Archives of Microbiology (2023)
-
The P. aeruginosa effector Tse5 forms membrane pores disrupting the membrane potential of intoxicated bacteria
Communications Biology (2022)
-
Abiotic factors modulate interspecies competition mediated by the type VI secretion system effectors in Vibrio cholerae
The ISME Journal (2022)
-
Type VI secretion system mutations reduced competitive fitness of classical Vibrio cholerae biotype
Nature Communications (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
