Abstract
The ability to generate lung and airway epithelial cells from human pluripotent stem cells (hPSCs) would have applications in regenerative medicine, modeling of lung disease, drug screening and studies of human lung development. We have established, based on developmental paradigms, a highly efficient method for directed differentiation of hPSCs into lung and airway epithelial cells. Long-term differentiation of hPSCs in vivo and in vitro yielded basal, goblet, Clara, ciliated, type I and type II alveolar epithelial cells. The type II alveolar epithelial cells were capable of surfactant protein-B uptake and stimulated surfactant release, providing evidence of specific function. Inhibiting or removing retinoic acid, Wnt and BMP—agonists to signaling pathways critical for early lung development in the mouse—recapitulated defects in corresponding genetic mouse knockouts. As this protocol generates most cell types of the respiratory system, it may be useful for deriving patient-specific therapeutic cells.
Similar content being viewed by others


Main
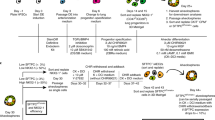
Lung and airway epithelial cells generated from human pluripotent stem cells (either embryonic stem cells (ESCs) or induced pluripotent stem cells (iPSCs)) would have multiple applications, including recellularization of decellularized lung scaffolds to provide an autologous graft for transplantation, study of human lung development, modeling of diseases that primarily affect airway epithelial cells and drug screening1. Trachea and bronchi are lined by a pseudostratified epithelium. The alveoli consist of alveolar epithelial type I (ATI) cells, which are essential for gas exchange, and alveolar epithelial type I (ATII) cells, which produce surfactant, critical for the maintenance of alveolar integrity2. The respiratory system is derived from lung buds on the anterior ventral aspect of the definitive endoderm, which grow and branch in a stereotyped pattern driven by renewing progenitors on the tips3,4. Directed differentiation of PSCs into pulmonary tissue should therefore proceed by first differentiating into definitive endoderm, followed by ventral anterior foregut endoderm and specification of lung and airway lineages.
We have previously demonstrated that anterior foregut endoderm can be generated from hPSCs by exposing Activin A–induced definitive endoderm to dual transforming growth factor (TGF)-β and bone morphogenic protein (BMP) inhibition5. The anterior foregut endoderm cells could be partially specified toward a putative lung bud fate, as suggested by expression of the lung marker NKX2.1. However, the fraction of NKX2.1+FOXA2+ cells was <40%, and expression of specific markers of lung and airway epithelial cells was not detected. A recent report described differentiation of hPSCs to lung progenitors at low efficiency; only a few percent of NKX2.1+p63+ putative airway progenitors was obtained, and the cells did not express markers of mature airway epithelial cells6. In mouse studies7, a NKX2.1:GFP reporter ESC line was used to isolate NKX2.1+ cells after differentiation into anterior foregut endoderm by a strategy similar to our previously published protocol5. The cells were committed to a lung and thyroid fate, and amenable to further differentiation, although expression of markers of ATI and ATII cells remained sporadic7. Wong et al.8 showed differentiation of hPSCs into proximal airway cells expressing cystic fibrosis transmembrane conductance regulator (CFTR). However, the efficiency of this protocol is unclear, and generation of distal lung epithelial cells was not documented at the protein or functional level.
Here we describe a strategy to achieve high yields of progenitor cells committed to a lung fate and to differentiate these in vivo and in vitro into functional respiratory epithelial cells. The cells expressed markers of at least six types of lung and airway epithelial lineages and were particularly enriched in distal ATII cells capable of surfactant protein-B (SP-B) uptake and release. Notably, a high degree of similarity in terms of marker expression was observed between in vitro–differentiated hPSC-derived cells and cultured fetal human lung, and between in vivo–differentiated hPSC-derived lung progenitor cells and adult human lung.
Results
Induction of highly enriched lung and airway progenitors
We have previously shown that definitive endoderm, induced using established protocols9,10,11,12, can generate anterior foregut endoderm (FOXA2+SOX2+CDX2−) when BMP and TGF-β signaling are inhibited5. Application of a 'ventralization cocktail' containing WNT, FGF10, keratinocyte growth factor (KGF), BMP4 and retinoic acid13,14,15,16,17,18—factors involved in dorsoventral patterning of the anterior foregut endoderm and lung bud specification—yielded cultures containing FOXA2+NKX2.1+ cells that corresponded to the lung field (progenitors) of the anterior foregut endoderm5. However, the enrichment in FOXA2+NKX2.1+ cells never exceeded 35–40%, and specific lung and airway epithelial cell markers were absent.
To improve lung field specification efficiency from anterior foregut endoderm we first refined the anterior foregut endoderm induction approach. In the mouse embryo, definitive endoderm cells fated to become anterior foregut endoderm pass through a zone where the Nodal/Activin inhibitor Lefty and the BMP4 inhibitor Noggin are expressed19,20, likely explaining why blocking TGF-β and BMP signaling is required for anterior foregut endoderm specification. Subsequently, the cells are exposed to the Wnt inhibitor, Dkk1 (ref. 21). Indeed, in the present study, we found that sequential inhibition of these pathways after definitive endoderm induction yielded efficient lung field induction. We first exposed ESC-derived definitive endoderm to small-molecule signaling inhibitors, including dorsomorphin (DSM)22, which inhibits BMP; SB431542 (ref. 23), which inhibits TGF-β; and IWP2, which inhibits endogenously produced Wnts by blocking porcupine-mediated Wnt palmitoylation24. The cells were then cultured until day 15 in the presence of the ventralization factors CHIR99021 (a small-molecule glycogen synthase kinase inhibitor that mimics Wnt signaling)25, FGF10, KGF, BMP4 and retinoic acid (a condition designated CFKB + RA) (Fig. 1a). Compared to continuous supplementation of DSM/SB431542 only, supplementation of DSM/SB431542 from days 4.5–5.5, followed by SB431542/IWP2 from days 5.5–6.5, significantly increased the fraction of FOXA2+NKX2.1+ cells (from 51.2 ± 1.6% to 70.1 ± 1.2%, P = 0.004, n = 3) and of NKX2.1 mRNA (Fig. 1a) at day 15. Reversing the DSM/SB431542 before SB431542/IWP2 sequence to SB431542/IWP2 before DSM/SB431542 or using SB431542/IWP2 alone was detrimental to NKX2.1 protein expression (22.0 ± 4.8% and 18.8 ± 5.0% FOXA2+NKX2.1+ cells, respectively) and mRNA expression (Fig. 1a). These data suggest that the way anterior foregut endoderm is induced determines its subsequent lung potential.

(a) Expression of NKX2.1 mRNA at day 15 in the four culture conditions shown at top of the panel. *P < 0.01 (Student's t-test), compared with the IWP2/SB431542 before DSM/SB431542 group, experiments done in triplicate. 'Liver' represents definitive endoderm cultured in 'hepatic conditions'41, data for definitive endoderm were samples analyzed at day 4.5 of the differentiation protocol. (b) Expression of FOXA2, SOX2 and NKX2.1 in RUES2 cells cultured according to the protocol at top of the panel. (n = 3 independent experiments, ten 10 × fields quantified per experiment). (c) 10 × tile scan of 25 (5 × 5) contiguous fields showing expression of FOXA2, SOX2 and NKX2.1 in RUES2 cells cultured according to the protocol at top of panel b. (d) 20 × tile scan of 4 (2 × 2) contiguous fields showing expression of TUJ1, NKX2.1 and FOXA2.1 in RUES2 cells cultured according to the protocol at top of panel b. (e) 20 × tile scan of 4 (2 × 2) contiguous fields showing expression of p63, NKX2.1 and FOXA2.1 in RUES2 cells cultured according to the protocol at top of panel b. Immunofluorescence images from Figure 1b–e) represent reproducible results from three independent experiments.
We further modified this protocol by optimizing the timing of dissociation of definitive endoderm (day 4, Supplementary Fig. 1a) and the concentration of retinoic acid in the ventralization stage (50–100 nM, Supplementary Fig. 1b). These manipulations resulted in cultures in which 95.7 ± 3.2% of the cells were FOXA2+ and 86.4 ± 1.7% were FOXA2+NKX2.1+ (n = 3 independent experiments, ten 10× fields quantified per experiment) (Fig. 1b,c). Expression of NKX2.1 mRNA was 3,170-fold ± 120-fold higher compared to that in liver-specified cells and >60,000-fold higher than that in definitive endoderm (day 4 in our protocol). However, the efficiency of lung progenitor induction was lower in two iPSC lines generated by Sendai virus (sv)26 (sviPSC; FOXA2+: 87.3 ± 4.4%; FOXA2+NKX2.1+: 37.0 ± 1.6%) and mRNA transfection27 (mRNA iPSC; FOXA2+: 69 ± 2.7%; NKX2.1+FOXA2+: 36.1 ± 4.3%), respectively (Supplementary Fig. 2). Notably, the optimal definitive endoderm dissociation time point in both iPSC lines was days 4.5–5 (data not shown).
At day 15 of the culture, no markers of mature lung or airway epithelial cells were detected by immunofluorescence (Supplementary Fig. 3a; positive staining controls using adult human lung, Supplementary Fig. 3b). Furthermore, qPCR (Supplementary Fig. 4) or immunofluorescence (data not shown) evidence for differentiation into the thyroid (expression of PAX8, TG, TSHR), which also expresses NKX2.1 (refs. 7,28), was absent. However, other types of contaminating cells were present, and these varied depending on the cell lines used. In RUES2 cells, clusters of FOXA2+NKX2.1− cells that stained weakly for the neural marker TUJ1 (Fig. 1d, arrows), but not for PAX6 (data not shown), were observed, a phenotype suggestive of midbrain floorplate neuronal precursors29. Cultures of RUES2 cells (Fig. 1e) and sviPSCs (data not shown) also contained islands of cells that were negative for NKX2.1, FOXA2, (Fig. 1e, arrows), SOX2, TUJ1 and PAX6 (data not shown), but expressed the epithelial markers p63 (Fig. 1e, arrows) and EPCAM (data not shown). Their identity is unknown. In the mRNA iPSCs, contamination with PAX6+ neuronal cells was observed (data not shown).
We next examined which factors in the ventralization cocktail were essential. Removing BMP4 or WNT agonists reduced the numbers of NKX2.1+ cells, and blocking these pathways or removing retinoic acid (Supplementary Fig. 1b) virtually abolished generation of these cells (Fig. 2). These data are consistent with mouse genetic models, where BMP on the one hand and retinoic acid and Wnt signaling on the other hand have nonredundant roles in dorsoventral patterning of the anterior foregut endoderm and lung bud specification, respectively13,16,17,18,30. However, removing either FGF10 (Fig. 2), FGF7 (Fig. 2) or both (data not shown), or inhibition of FGF signaling (data not shown) had no effect, suggesting that FGF signaling is dispensable in the human system in contrast to the mouse system.

Effect of removing individual factors or blocking signaling pathways during the ventralization stage (days 6–15) on the expression of FOXA2 and NKX2.1 in RUES2 cells cultured according to the protocol shown on the upper left of the figure. 10 × tile scans of 9 (3 × 3) contiguous fields. Immunofluorescence images represent reproducible results from three independent experiments.
Differentiation of NKX2.1+FOXA2+ cells in vivo
To determine the in vivo differentiation potential of the NKX2.1+FOXA2+ cells, we transplanted 106 day-15 RUES2 cells under the kidney capsule of immunodeficient NOD/SCID/Il2rg−/− (NSG) mice. After 6 months, we observed multiple macroscopic growths, which contained cystic and tubular structures (Fig. 3a) lined by a uniformly FOXA2+SOX2+NKX2.1+ (Fig. 3b) epithelium, which ranged from pseudostratified, containing cells consistent with basal, ciliated, Clara and goblet cells, to a monolayer consisting of flatter cells (Fig. 3a). Glandular structures resembling submucosal glands were also present (Fig. 3a).

(a) Representative examples of H&E stains of growths removed 6 months after transplantation of RUES2 cells differentiated according to the protocol shown in Figure 1b, under the kidney capsule of NSG mice (BC: basal cell; Ca: cartilage; CC: Clara cell; Ci: ciliated cell; GC: goblet cell; Ki: mouse kidney; PSE: pseudostratified epithelium; SM: smooth muscle; SMG: submucosal glands). (b) Representative examples of the expression of markers of mature lung and airway epithelial cells in the growths from a. Representative of four animals each in two independent experiments.
All tested markers of mature lung and airway epithelial cells were detected. These included mucins (MUC1, MUC5AC, MUC2; goblet cells), FOXJ1 (ciliated cells), CC10 (Clara cells), p63 and nerve growth factor receptor (NGFR; basal cells)31, pro-SP-C, SP-C and SP-B (ATII cells) as well as AQP5, HOPX and PDN (ATI cells) (Fig. 3b). These staining patterns were remarkably similar to those observed in fetal human lung (Supplementary Fig. 5) and adult human lung (Supplementary Fig. 3b). The structures were surrounded by smooth muscle and cartilage, in addition to areas containing looser connective tissue (Fig. 3a). All cells, including the mesodermal cells, were of human origin, as determined by staining with antibodies specific for human nuclei (Supplementary Fig. 6).
Together, these data indicate that day-15 cells in our protocol almost exclusively gave rise in vivo to lung and airway endoderm, in addition to mesodermal cells that adopted cell fates consistent with the mesodermal elements surrounding trachea and large airway.
Differentiation of NKX2.1+FOXA2+ cells in vitro
BMP4, FGF10, KGF, Wnt and retinoic acid are involved in the differentiation of respiratory epithelium13,14,15,17,32,33,34,35,36. However, upon initiation of branching morphogenesis in mouse embryos, retinoic acid signaling inhibits distal lung and favors proximal airway development35,36. We have shown that removal of BMP4, the role of which is controversial and model-dependent16,32, from the growth factor cocktail increased the expression of SFTPC mRNA (which is translated to SP-C), although pro-SP-C protein was not detected5. Therefore, we focused subsequent experiments mostly on cells cultured in the presence of the Wnt agonist CHIR99021, FGF10 and KGF (a condition designated CFK).
Day-15 lung-specified anterior foregut endoderm cells were replated after mild trypsinization, brief sedimentation and collection of larger cell clumps for replating (Fig. 4a). Preliminary experiments indicated that these were depleted of P63+FOXA2−NKX2.1− cells and of most neural elements, which were present in smaller aggregates in the supernatant (data not shown). In these cultures, cells grew as large colonies that were virtually entirely positive for FOXA2 (data not shown), NKX2.1 and SOX2 at day 25 (Fig. 4b,c), and contained p63+NKX2.1+ cells at the periphery (Fig. 4b,d). In cells derived from sviPSCs, 86.8 ± 2.2% were FOXA2+ and 75.2 ± 2.9% were FOXA2+NKX2.1+, whereas in cells derived from mRNA iPSCs, 88.5 ± 1.0% were FOXA2+, and 79.1 ± 1.7% were FOXA2+NKX2.1+ (Supplementary Fig. 2). In cells derived from both iPSC lines, but not from RUES2 cells, contaminating neuronal cells were observed. Notably, whereas these were TUJ1+PAX6− in sviPSC-derived cells, a detectable number of PAX6+ cells were observed in the mRNA iPSC–derived cells (Supplementary Fig. 2).

(a) Culture protocol of RUES2 cells shown in panels b–d. (b,c) 10 × tile scans of the expression of p63, SOX2 and NKX2.1 in representative colonies obtained after culturing RUES2 cells according to the protocol shown in a. (d) Expression of p63 and MUC5AC after culturing RUES2 cells according to the protocol shown in a. Immunofluorescence images represent reproducible results from four independent experiments.
Except for sporadic expression of MUC5AC (Fig. 4d), other markers of respiratory epithelial cells were absent at day 25 (data not shown). At day 48, however, expression of all markers detected in growths arising after transplantation in NSG mice were observed (Fig. 5a,b; whole-culture tile scans shown in Fig. 5c and Supplementary Fig. 7; data from sviPSCs shown in Supplementary Fig. 8). In addition, we noticed that staining for the secreted mucins (MUC5AC (data not shown), MUC2 and MUC5B, Supplementary Fig. 9) and CC10 (Supplementary Fig. 9) extended outside cell boundaries as defined by EPCAM expression and occurred in structures that were discernable in bright-field microscopy. These findings suggest secretory activity of both goblet and Clara cells. The cells positive for PDN and AQP5 displayed flat, crescent-shaped nuclei at the periphery of the cells (Fig. 5b, insets), a morphology typical of ATI cells, whereas SP-B expression occurred in a punctate pattern, suggestive of lamellar bodies of ATII cells where surfactant accumulates (Fig. 5b, inset).

(a) Culture protocol of RUES2 cells shown in panels b–d. (b) Representative examples of the expression of markers of mature lung and airway epithelial cells after culturing RUES2 cells according to the protocol shown in a. Immunofluorescence images represent reproducible results from four independent experiments. (c) Representative 10 × whole culture tile scan of SP-B and SP-C expression in RUES2 cells cultured according to the protocol shown in a, without (left) and with (right) addition of DCI at day 25. (d) Cellular expansion of RUES2 cells during the culture according to the protocol shown at top of panel a (n = 4 independent experiments).
Next, we examined the effect of adding dexamethasone, 8-bromo-cAMP and isobutylmethylxanthine (DCI) (Fig. 5a,c), factors that induce alveolar maturation in fetal mouse lung explants and enhance surfactant protein expression in mouse ESC–derived lung progenitors7,37. In the presence of DCI, SP-B+ cells became the predominant cell type (Fig. 5c). Furthermore, only under these conditions was expression of mature SP-C detected, although at a much lower frequency than SP-B (Fig. 5c). More than 50% of the cells expressed SP-B, whereas the frequency of all other cell types ranged between 2% and 5% (n = 3). It has been shown in fetal rat lung explant cultures that SP-C expression is more responsive to mechanical stretch than is SP-B expression38, consistent with the lower SP-C expression in cultured cells. Addition of DCI at day 15 did not result in SP-B expression at day 25, however (data not shown). Most likely, the cells respond to DCI if and when they reach a differentiation stage that allows them to respond with increased surfactant production. Cell number increased 35-fold up to day 25, was similar in the presence or absence of DCI, and then plateaued (Fig. 5d). These data attest to the high efficiency of this protocol and suggest cessation of cellular expansion coinciding with expression of lineage-specific epithelial markers.
As hPSC-derived cells are cultured on fibronectin-coated plastic, marker protein expression and cell morphology may differ from that observed in native lung tissue in vivo. Therefore, we cultured dissociated cultured fetal human lung under the same conditions (CHIR, FGF7, FGF10 and DCI) for 3 weeks. Both by immunofluorescence (Supplementary Fig. 10a) and by transmission electron microscopy (TEM) (Fig. 6a), cultured fetal human lung was very similar to differentiated hESCs. Notably, p63+ cells expressed NGFR, a marker of basal cells31, in adult human lung (Supplementary Fig. 10b) and in day-15 differentiated ESCs 6 months after grafting under the kidney capsule of NSG mice (Fig. 3b); however, in native and cultured fetal human lung and in hPSCs differentiated in vitro (Supplementary Fig. 10b), p63+ cells did not express NGFR. These findings suggest that NGFR expression on basal cells is developmentally regulated, and that hPSCs differentiated in vitro are more similar to fetal human lung than to adult human lung. TEM showed structures consistent with lamellar bodies of ATII cells and with multivesicular bodies, precursors of lamellar bodies, in both fetal human lung and differentiated hPSCs (Fig. 6a). Together, these findings suggest that hPSCs differentiated into lung and airway are nearly indistinguishable from cultured fetal human lung.

(a) Representative transmission electron micrographs of cultured fetal human lung or RUES2 cells differentiated according to the protocol shown in Figure 5a with DCI (LB: lamellar body; MVB: mulitvesicular body). (b) Fluorescence micrographs and flow cytometric analysis of uptake of BODIPY-SP-B by cells cultured according to the protocol shown in Figure 5a. Immunofluorescence images and flow cytometry represent reproducible results from three independent experiments. (c) Culture in the presence of decellularized human lung matrix. (c, i,ii) Expression of p63 and NKX2.1 at day 25 of cultures of RUES2 cells seeded on slices of decellularized human lung matrix at day 15 of the protocol in Figure 5a. (c, iii,iv) Expression of endogenous SP-B at day 48 of culture with DCI according to the protocol in Figure 5a after being seeded on decellularized human lung matrix. (c, v) Confocal fluorescence micrograph of the uptake of BODIPY-SP-B at day 48 in the same conditions. Scale bar, 5 μm. (c, vi) Morphology of mouse ATII cells as observed by two-photon microscopy of live mouse lung after instillation of BODIPY-SP-B. Scale bar, 5 μm. (d) qPCR of the expression of proximal and distal lung markers at days 15, 23 and 55 of culture in the conditions in Figure 5a in the presence of DCI. Cultures in the presence or absence of BMP4 and retinoic acid (+BMP/RA vs. −BMP/RA) from days 15–55. (ECM, human decellularized extracellular lung matrix) (representative of three independent experiments).
To assess the function of the cells expressing SP-B, we examined their capacity to take up surfactant proteins, a distinguishing feature of ATII cells39. By flow cytometry, only 0.6 ± 0.1% of the cells showed detectable uptake of fluorescent BODIPY-labeled recombinant human SP-B at day 15, whereas at day 48, 17 ± 10% and 52 ± 6% showed uptake in the absence and presence of DCI (P = 0.04, n = 3), respectively (Fig. 6b). The fact that functional ATII cells could be detected by flow cytometry also allows their further purification from the cultures. Next, we monitored release of BODIPY-SP-B from the cells after addition of a cell-permeable analog of diacylglycerol (DAG), a surfactant secretagogue40. Addition of DAG caused a rapid decrease in BODIPY-SPB fluorescence, indicative of increased release (Supplementary Fig. 11). Together, these data indicate that the ATII cells generated in our cultures are functional, and that their maturation is enhanced by DCI.
We also cultured the cells from day 15 on in the presence of decellularized slices of human lung. Initially, NKX2.1+p63+ cells were seen to be adhering to the matrix (Fig. 6c, i,ii). Subsequently, cells overgrew the matrix slices and showed positive staining for SP-B (Fig. 6c, iii,iv) and uptake of BODIPY-SP-B at day 48 (Fig. 6c, v). The morphology of the BODIPY SP-B+ cells was very similar to that seen using two-photon microscopy after uptake of BODIPY-SP-B in live mouse lung (Fig. 6c, vi). These data show the ability of the NKX2.1+p63+ cells to attach to native lung matrix, grow and express distinct functional properties of pulmonary cells, an important feature for potential applications in which lung progenitors are used to treat lung disease.
Finally, we confirmed our protein expression data by qPCR, using definitive endoderm and definitive endoderm differentiated into the hepatic lineage41 as controls (Fig. 6d). We observed early induction of mRNAs for distal markers without protein expression at day 15, when no protein expression is detected. These mRNAs subsequently disappear, and then reappear as the cells mature, this time accompanied by ample protein expression. It is possible that upon initial lung specification, several lung-specific genes are temporarily de-repressed and subsequently controlled in a lineage-specific fashion. Differentiation on human lung matrix or on plastic coated with fibronectin was similar in terms of mRNA expression of lung and airway markers. Furthermore, these experiments also confirmed that better distal lung differentiation is achieved in the absence of BMP4 and retinoic acid after lung field induction.
Discussion
We report differentiation of hPSCs into lung field progenitors that can differentiate into at least six identifiable types of lung and airway cells in vivo and in vitro. Notably, a high degree of similarity was observed between in vitro–differentiated hPSC-derived cells and cultured fetal human lung, and between in vivo differentiated hPSC-derived lung field cells and adult human lung. The only endodermal cells detected after transplantation in NSG mice were lung and airway epithelial cells. However, a large mesodermal component consisting of smooth muscle, cartilage and loose connective tissues was present, despite the very high enrichment of lung field endoderm in the transplanted cells. These cells likely originated from small amounts of contaminating mesoderm. The fact that these mesodermal cells appeared to differentiate into the appropriate mesodermal elements surrounding the airways may suggest that pulmonary endoderm has an instructive role in the development of appropriate mesodermal elements in this model.
The efficiency of our differentiation protocol and the nature of contaminating lineages varied across hPSC lines, a reflection of the well-documented variability in the lineage-specific differentiation potential of ESC42 and iPSC43,44 lines. Among the ESC lines tested (RUES1, RUES2, H1, H9, HES2), HES2 and RUES2 were most efficient for anterior foregut endoderm induction. We focused on RUES2, as it is approved by the US National Institutes of Health. Furthermore, morphogen concentration and timing of culture stages may require optimization for each individual line. This notion is supported by our observation that the optimal timing of definitive endoderm dissociation is cell line–dependent and by the observation that ESC lines are heterogeneous with respect to Wnt signaling45.
Wong et al.8 generated CFTR-expressing proximal airway cells from hiPSCs by exposing definitive endoderm to SHH and FGF2 to induce anterior foregut endoderm and by specifying lung progenitors through addition of FGF10, FGF7 and a low concentration of BMP4 (5 ng/ml), followed by air-liquid interphase culture. We systematically compared the anterior foregut endoderm and lung field induction protocols described in that study with ours, for the hPSC lines used here (Supplementary Fig. 12). Applying the anterior foregut endoderm induction protocol of Wong et al.8 (SHH + FGF2) or ours (DSM/SB431542 before SB431542/IWP2), followed by lung field induction using the protocol of Wong et al.8 yielded only sporadic NKX2.1+ cells. However, anterior foregut endoderm induction using SHH and FGF2, followed by our CFKB + RA lung field induction strategy yielded approximately 50% NKX2.1+ cells (Supplementary Fig. 12). Similar data were obtained using our sviPSCs (data not shown). Taken together, these data confirm that Wnt, BMP4 and retinoic acid are essential for efficient induction of lung progenitors from hPSCs (Fig. 2). It is possible that the protocol of Wong et al.8 is oriented toward the generation and selection of precursors of proximal airway cells in air-liquid interphase cultures, whereas our protocol generates a wider array of respiratory epithelium that is biased toward distal cells. The two protocols clearly provide alternative strategies to induce anterior foregut endoderm, suggesting that complex and hierarchical relationships between TGF-β, BMP, Wnt, SHH and FGF2 signaling in the specification of anterior foregut endoderm from definitive endoderm require further examination.
The ability to generate ATII cells from hPSCs will allow modeling of diseases such as congenital surfactant deficiency syndromes39. Furthermore, ATII cells have recently been identified as alveolar stem cells46 and may be at the origin of lung adenocarcinoma47. As the current protocol yields most cell types of the respiratory system, it will allow studies of lineage determination and may help to address a central challenge in lung tissue engineering using autologous, iPSC-derived cells—generating sufficient numbers of cells with the variety and ratio of epithelial cells and their progenitors normally found in the lung.
Methods
Maintenance of hPSCs.
RUES2 (Rockefeller University Embryonic Stem Cell Line 2, NIH approval number NIHhESC-09-0013, Registration number 0013; passage 13-24) and Sendai Virus and modified mRNA generated human dermal fibroblasts iPSC lines (derived from healthy fibroblasts, passage 12–25, purchased from the Mount Sinai Stem Cell Core facility) were cultured on mouse embryonic fibroblasts as previously described5. Mouse embryonic fibroblasts (GlobalStem, Rockville, MD) were plated at a density of ∼25,000 cells/cm2. hPSCs were cultured in a medium of DMEM/F12, 20% knockout serum replacement (Gibco (Life Technologies, Grand Island, NY)), 0.1 mM β-mercaptoethanol (Sigma-Aldrich, St. Louis, MO) and 20 ng/ml FGF-2 (R&D Systems, Minneapolis, MN). Medium was changed daily and cells were passaged with Accutase/EDTA (Innovative Cell Technologies, San Diego, CA) every 4 d at 1:24 dilution. Cultures were maintained in an undifferentiated state in a 5% CO2/air environment. hPSC differentiations were maintained in a 5% CO2/5% O2/95% N2 environment unless indicated elsewhere.
Induction of endoderm.
24-h primitive streak formation and 3–4 d of endoderm induction were performed in serum-free differentiation (SFD) media of DMEM/F12 (3:1) (Life Technologies) supplemented with N2 (Gibco (Life Technologies)), B27 (Gibco), ascorbic acid (50 μg/ml, Sigma), Glutamax (2 mM, Life Technologies), monothioglycerol (0.4 μM, Sigma), 0.05% bovine serum albumin (BSA) (Life Technologies), 1% penicillin-streptomycin (Thermo Fisher Scientific, Waltham, MA), as previously described with slight modification5. hPSCs were treated with Accutase (2 min at 37 °C) and plated onto Matrigel-coated (Life Technologies) 10-cm tissue culture dish (1:2 dilution) for 12–24 h to deplete residual mouse embryonic fibroblasts. Cells were then briefly trypsinized (0.05%, 1 min at 37 °C) into 3–10 small cell clumps and plated onto low attachment 6-well plates (Costar (Corning Incorporated, Tewksbury MA)) (1:1 dilution) to form embryoid bodies in serum-free differentiation media. For primitive streak formation, Y-27632, 10 μM (Tocris (R&D Systems)) and human BMP4, 3 ng/ml were added in the media for 24 h. Embryoid bodies were then collected, resuspended in endoderm induction media containing Y-27632, 10 μM, human BMP4, 0.5 ng/ml; human bFGF, 2.5 ng/ml (R&D Systems); human Activin A, 100 ng/ml (R&D Systems) for 72, 84 or 96 h on low-adherence plates. Cells were fed every 36–48 h (depending on the density) by removing half of the old media and adding half fresh media.
Induction of anterior foregut endoderm.
On day 4, 4.5 or 5, embryoid bodies were dissociated into single cells using 0.05% Trypsin/EDTA (cellgro (Corning)) (2–4 min). For anterior foregut endoderm induction, endodermal cells were plated on fibronectin-coated (Sigma), 48-well tissue culture plates (∼50,000–75,000 cells/well) in serum-free differentiation media supplemented with 1.5 μM Dorsomorphin dihydrochloride (Tocris, R&D Systems) and 10 μM SB431542 (Tocris (R&D Systems)) for 24 h, and then switched to 24 h of 10 μM SB431542 (Sigma) and 1 μM IWP2 (Tocris (R&D Systems)) treatment. In some experiments, the anterior foregut endoderm was specified by 48 h of Dorsomorphin (1.5 μM) and SB431542 (10 μM) treatment only, without switching to SB431542 and IWP2. Hepatic conditions contained BMP-4, 50 ng/ml; HGF, 10 ng/ml; dexamethasone, 40 ng/ml; bFGF, 10 ng/ml; VEGF, 10 ng/ml; TGF-α, 20 ng/ml; murine EGF, 20 ng/ml (all from R&D, except dexamethasone from Sigma)5,41.
Induction of lung progenitors.
For day 6–15 lung progenitor induction, the resulting anterior foregut endoderm was treated with a ventralization cocktail containing CHIR99021, 3 μM (WNT signaling agonist), human FGF10, 10 ng/ml; human FGF7, 10 ng/ml; human BMP4, 10 ng/ml; murine EGF, 20 ng/ml (optional) and all-trans retinoic acid (ATRA), 0–1 μM (all from R&D, except CHIR99021 from (Stemgent, Cambridge, MA) and ATRA from Sigma) in SFD media for 8–10 d. For RUES2 differentiation, the day 8–15 cultures were maintained in a 5% CO2/air environment. For sviPSC differentiation, the day 12–15 cultures were maintained in a 5% CO2/air environment. In some experiments, each of the factors was withdrawn from the ventralization cocktail (i.e., CHIR99021, FGF10, FGF7, BMP4 and EGF), or blocked by specific inhibitors (i.e., NOGGIN as biological inhibitor of BMP4 signaling and IWP2 as pharmacological inhibitor of WNT signaling), or added at different concentrations (i.e., RA 0, 0.05, 0.1, 0.5, 1 μM), to examine the contribution of each of the factors to the specification of lung and airway progenitors.
Induction of lung/airway epithelial maturation.
On days 15 and 16, the lung field progenitor cells were replated after brief trypsinization onto fibronectin-coated plates at 1:5 dilution. The cells were incubated in 0.05% warm trypsin/EDTA for 1 min. Trypsin was then removed by suction and wash media (IMDM+5% FCS) was added into the well, and the cell clumps were gently removed off the plate using 1-ml pipette tips and transferred to a 15-ml tube. After gently mixing with 1-ml pipette tips, the clumps were allowed to settle for 2 min. Supernatant (containing single cells and small clumps of cells (<10 cells/clump)) was removed. The remaining cell clumps were replated into fibronectin-coated plates at 1:5 dilutions in the presence of SFD containing either a combination of five factors (CHIR99021, 3 μM; human FGF10, 10 ng/ml; human FGF7, 10 ng/ml; human BMP4, 10 ng/ml; and ATRA, 50 nM), or three factors (CHIR99021, 3 μM, human FGF10, 10 ng/ml; human FGF7, 10 ng/ml). Day 15–25 cultures were maintained in a 5% CO2/air environment. From day 25 to 48, cultures were carried further in either of these two conditions with or without the addition of maturation components containing 50 nM dexamethasone, 0.1 mM 8-bromo-cAMP (Sigma) and 0.1 mM IBMX (3,7-dihydro-1-methyl-3-(2-methylpropyl)-1H-purine-2,6-dione) (Sigma)37.
Immunofluorescence staining.
Day-15, 25 or 48 cultures in 48-well tissue culture plates were fixed with 4% paraformaldehyde for 15 min at room temperature and washed twice with PBS. The cells were permeabilized in PBS with 0.25% triton and 5% fetal donkey serum (Jackson ImmunoResearch, West Grove, PA) for 15–30 min (depending on the thickness of the cultures) and blocked in 5% fetal donkey serum for 2 h at room temperature. OCT-embedded differentiation cultures or human fetal and adult lung tissues were sectioned into 6-μM slices, put on microscopic slides, briefly fixed with 95% ethanol for 2 min and further fixed with 4% paraformaldehyde for 15 min at room temperature and washed twice with PBS. The sections were permeabilized for 15 min and blocked for 1 h in the solutions described above. The cell cultures and sections were stained with one, or a combination of two or three of the following primary antibodies: FOXA2/HNF-3β (goat, Santa Cruz Biotechnology, Inc., Santa Cruz, CA, Cat# sc-6554, clone M-20, 1:50), TTF-1/Nkx2.1 (mouse, Invitrogen, 18-0221, clone 8G7G3/1, 1:100), TTF-1/Nkx2.1 (Rabbit, Seven Hills Bioreagents, Cincinnati, OH, Cat# WRAB-1231, 1:1,000), p63α (rabbit, Santa Cruz, sc-8344, clone H-129, 1:100), NGFR (EMD Millipore, Billerica, MA. Cat# 05-446, clone ME20.4, 1:100), Sox2 (rabbit, Stemgent, 09-0024, 1:100), Sox2 (goat, Santa Cruz, Cat# sc-17320, clone Y-17, 1:100), Pax 6 (rabbit, Covance, Princeton, NJ, Cat# PRB-278P, 1:300), Pax8 (mouse, Abcam, Cambridge, MA, Cat# ab53490, 1:100), EpCAM (APC conjugated, mouse, BD Biosciences, BDB347200, 1:100), Tuj1 (mouse, Sigma, T8578, Clone 2G10, 1:4,000), Mucin5AC (mouse (Biotin), Abcam, ab79082, clone 45M1, 1:100), Mucin5B (rabbit, Santa Cruz, sc-20119, clone H-300, 1:100), Mucin2 (rabbit, Santa Cruz, sc-15334, clone H-300, 1:100), Foxj1 (mouse, eBioscience, San Diego, CA, Cat#14-9965-82, Clone: 2A5, 1:100), cc-10 (goat, Santa Cruz, sc-9770, clone C-20, 1:100), pro-SPC (rabbit, Seven Hills, WRAB-9337, 1:2,000), mature-SPC (rabbit, Seven Hills, WRAB-76694, 1:1,000), mature SPB (rabbit, Seven Hills, WRAB-48604, 1:1,000), ABCA3 (rabbit, Seven Hills, WRAB-70565, 1:1,000), Mucin1 (Armenian Hamster, NeoMarkers, Fremont, CA, Cat# HM-1630-P1ABX, clone MH1, 1:100), Podoplanin (rabbit, Santa Cruz, sc-134482, FL-162, 1:100), AQP5 (goat, Santa Cruz, sc-9890, clone G-19, 1:100), HOPX (Rabbit, Santa Cruz, sc-30216, 1:250), Anti-Nuclei Antibody (EMD Millipore, MAB1281, clone 235-1, 1:200). Secondary antibodies were donkey anti-mouse whole IgG-Alexa Fluor 488, 715-545-150, donkey anti-mouse whole IgG-Alexa Fluor 647, 715-605-150, donkey anti-rabbit whole IgG-Alexa Fluor 488, 711-545-152, donkey anti-rabbit whole IgG-Cy3, 711-166-152, donkey anti-goat whole IgG-Alexa Fluor 488, donkey anti-goat whole IgG-Cy3, 705-165-147, donkey anti-goat whole IgG-Alexa Fluor 647, 705-605-147, all from Jackson ImmunoResearch.
After blocking, all the triple, double or single staining (except “d25 Nkx2.1 (mouse)/p63(rabbit)/FOXA2(goat)” and “Tuj1/TTF-1/FOXA2”) were performed by incubating primary antibody(ies), according to the dilution factors indicated in Supplementary Table 1 in staining/wash buffer (5% fetal donkey serum in PBS) at 4 °C overnight (minimum 12 h), followed by 3 × 10 min wash. The cultures were then incubated with the corresponding secondary antibodies at 1:300 dilutions in staining/wash buffer at room temperature for 2 h, washed twice for 10 min and incubated with DAPI for 5 min at room temperature. The stained cultures can be preserved in antibiotics supplemented PBS in dark at 4 °C for 2–3 months. The day-48 Mucin2, CC-10 SPB and pro-SPC can be preserved up to 6 months or longer. For better maintenance, we preserved the cultures in VECTASHIELD Mounting Media (Vector laboratories, Inc. Burlingame, CA, Cat# H-1000).
Immunofluorescence.
On day-25 differentiation culture, “Nkx2.1(mouse)/p63/FOXA2” and “Nkx2.1(mouse)/Sox2/FOXA2” triple stains were processed as following: after blocking nonspecific binding, the cultures were incubated in a cocktail of Nkx2.1 and Foxa2 primary antibodies for 12 h at 4 °C, followed by 3 × 10-min wash at room temperature, cultures were incubated in donkey anti-mouse Alexa Fluor 488 and donkey anti-goat Alexa Fluor 647 for 2 h and washed three times. The cultures were then blocked in 5% fetal donkey serum for 2 h at room temperature, followed by p63 primary antibody incubation for 12h; after 3 × 10-min wash, the cells were stained with donkey anti-rabbit Cy3 for 2 h at room temperature, washed and stained with DAPI and preserved in dark at 4 °C for imaging.
“Tuj1/TTF-1(Rb)/FOXA2” triple stain was processed by incubating the cultures in Nkx2.1 and Foxa2 primary antibody cocktails for 12 h at 4 °C, wash 3 × for 10 min at room temperature; donkey anti-rabbit Alexa Fluor 488 and donkey anti-goat Alexa Fluor 647 were added and incubated for 2 h at room temperature; after three washes, the cultures were then blocked in 5% fetal donkey serum overnight at 4 °C, followed by 1h Tuj1 primary antibody incubation at room temperature. The cultures were washed an additional three times, and donkey anti-mouse Alexa Fluor 488 secondary was added for 2 h at room temperature. The cultures were washed, incubated in DAPI and preserved in PBS for imaging.
Samples were visualized and imaged using motorized Leica DMI 6000B fluorescence microscope coupled with Leica DFC365 FX digital camera and operated by LAS AF 6.2 software (Leica Microsystems GmbH, Wetzlar, Germany). All the pictures were imaged with HCX PL S-APO 10 × /NA 0.3 or HCX PL FL L 20 × /NA 0.4 objectives. The tile scan images were taken with the image tiling module coupled with either autofocus or z-stack scanning (1.5 μm/stack) module, and auto-stitched by the LAS AF 6.2 software. The images were exported as JPG files and processed (contrast and brightness adjustments) with Photoshop CS5.1 (New York, NY).
Mice and kidney capsule transplantation.
NSG mice purchased from The Jackson Laboratory (Bar Harbor, ME) were kept in a specific, pathogen-free mouse facility and used at 8–12 weeks of age. Experiments were performed in accordance with the protocols approved by The Columbia University Institutional Animal Care And Use Committee. About one million of day-15 lung progenitor cells were implanted under the kidney capsule. The outgrowths were excised 6 months after transplantation. Outgrowths were embedded in OCT for immunofluorescence as described above, or fixed with 10% formalin and then processed for paraffin section and hematoxylin and eosin (H&E) stains to visualize the morphology. Samples were visualized and imaged using motorized Leica DMI 6000B fluorescence microscope as described above. H&E stains were visualized with the same microscope coupled with Leica DFC450 digital camera; the pictures were imaged with HCX PL FLUOTAR 5 × /0.15, HC PLAN APO 20 × /0.70, or HCX PL APO 100 × /1.4–0.70 oil objectives.
Cell cultivation on decellularized slices of human lung.
Human lungs rejected for transplantation were procured from the New York Organ Donor Network (NYODN) under a protocol approved by the Institutional Review Board at Columbia University. The lower left lobes of the lungs were sectioned to 2-mm-thick sheets that were decellularized using our previously described method with 16-mM 3-((3-cholamidopropyl) dimethylammonio)-1-propanesulfonate (CHAPS)48. This method effectively removed all cellular material while preserving the structure, mechanical properties and much of the molecular composition of the native lung matrix48. Decellularized lung slices were cored into 7-mm discs using a dermal punch and attached by fibrin to the wells in 96-well plates. All procedures were done under sterile conditions. Prior to cell seeding, the lung matrix was incubated in SFD medium for 48 h to test sterility. Day-15 lung progenitors were replated after brief trypsinization onto the lung matrix at 1:5 dilution. Cultures on the lung matrix were maintained under conditions identical to those of fibronectin-coated cultures. At day 48, the cultures on the matrix were embedded in OCT and processed for immunofluorescence of specific antigens as described above, or processed for live imaging of fluorescent-SPB protein localization as described below.
Quantitative real-time PCR.
Total RNA was extracted using Trizol (Invitrogen), phase lock tubes (5′ Prime) and RNeasy kit (Qiagen, Valencia, CA). RNA concentration was measured with a NanoDrop 2000 fluorospectrometer (Thermo Fisher Scientific). RNA quality was verified using an Agilent microfluidic RNA 6000 Nano Chip kit on the 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA). cDNA was generated by reverse transcription of a total of 1 μg RNA with random hexamers and Superscript III (Invitrogen (Life Technologies)) following the manufacturer's instructions. The reaction was carried out in a 20-μl volume. Real-time quantitative PCR was performed on ABI vii7A Thermocycler (Applied Biosystems (Life Technologies)) using ABI Power SYBR Green PCR Master Mix (Applied Biosystems (Life Technologies)). The Real-time PCR conditions were 50 °C for 2 min and 95 °C for 10 min followed by 40 cycles of 95 °C for 15 s and 60 °C for 1 min, dissociation/melt curves were obtained for each of the genes. Absolute quantification of each gene was obtained using a standard curve of serial diluted genomic DNA and normalized to housekeeping genes β-ACTIN and TBP (Tata Box Binding protein). Quantitative PCR for each sample was performed in triplicates; the input of template per triplicate was cDNA transcribed from 5–10 ng of RNA. Primer sequences are listed in Supplementary Table 2.
Live cell imaging of fluorescent-SPB protein localization in day-48 cultures.
Day-15 and day-48 differentiation cultures on fibronectin-coated plates or decellularized human lung matrix were loaded with 8 μg/ml purified, fluorescent human SPB (BODIPY-SPB) protein in SFD media, and incubated in a 5% CO2/air environment for 1 h. Cultures were rinsed twice with SFD. Cultures on fibronectin-coated plates were imaged on motorized Leica DMI 6000B fluorescence microscope with HCX PL FL L 20X/NA 0.4 objective. Cultures on decellularized human lung matrix were imaged by laser scanning confocal microscope (LSM 510, Carl Zeiss Microscopy) using a 40 × water immersion objective (numerical aperture 0.80).
Flow cytometry.
Day-4, 4.5 or 5 embryoid bodies were dissociated into single cells with 0.05% trypsin/EDTA. The cells were stained directly with PE-conjugated CXCR4 (Cat# MHCXCR404, Invitrogen) (1:200), and APC conjugated c-KIT (Cat# CD11705, Invitrogen) or EpCAM (Cat# 347200, BD Biosciences, San Jose, CA) (1:100) in PBS supplemented with 0.1% BSA and 0.2 mM EDTA for 45 min at 4 °C. Stained cells were analyzed on a LSRII (BD Biosciences) and results were analyzed by Flowjo software (Tree Star, Ashland, OR).
For quantification of ATII cells in differentiated culture, day-15 and day-48 cultures were incubated with BODIPY-SPB for 1 h as described above, in the presence of 10 ng/ml Hoechst 33342 (Sigma). Cultures were rinsed with DPBS, trypsinized (0.05%) for 2 min at 37 °C and passed through a 40-μM cell strainer. The single-cell suspensions were stained with APC-conjugated EpCAM in PBS supplemented with 0.1% BSA and 0.2 mM EDTA for 45 min at 4 °C. Stained cells were analyzed on a LSRII (BD Biosciences) and results were analyzed by Flowjo software (Tree Star, Ashland, OR).
Electron microscopy.
Day-15 and day-48 differentiation cultures and human fetal lung cultures grown on 35-mm dishes (Thermo Fisher Scientific (Nunc), Cat#153066) were fixed with 2.5% glutaraldehyde in 0.1 M Sorensen's buffer for 2 h at room temperature. Samples were further processed, sectioned and stained by Columbia University Department of Pathology and Cell Biology Electron Microscope Facility. Sections were examined under a JEOL JEM-1200EXII electron microscope. Images were acquired on an ORCA-HR digital camera (Hamamatsu) and recorded with an AMT Image Capture Engine.
Statistical analysis.
Statistical analysis was done using unpaired two-tailed Student's t-test. For multiple group comparison (more than two), results were analyzed using one-way analysis of variance followed by Dunnett's multiple comparison test. Results were shown as mean ± s.e.m., p values < 0.05 were considered statistically significant. Variances in all groups compared were similar.
For animal studies, no randomization was required. The investigator was not blinded to any group allocation. Animals showing signs of distress (weight loss >20%, inactivity, ruffled fur) are in principle euthanized. This did not occur in this study.
Isolation and culture of human fetal lung cells.
Human fetal lungs (gestational age of 22–23 weeks) were purchased from Advanced Bioscience Resources (INC, Alameda, CA). The tissues were sliced into small pieces, homogenized and passed through 100 μM cell strainers. The resulting cell suspensions were treated with RBC lysis buffer (Affymetrix/eBioscience, San Diego, CA) for 5 min and washed twice with DPBS, suspended in SFD media containing CHIR99021, 3 μM; human FGF10, 10 ng/ml; human FGF7, 10 ng/ml onto 10-mm tissue culture dishes. The nonepithelial cells attached to the bottom of the dish within 24 h; small cell mass formed by epithelial cells appear as suspension in the media. 48 h later, the suspension epithelial cell mass was collected and plated onto new fibronectin-coated tissue culture plates or 35-mm culture dishes in SFD media containing CHIR99021, human FGF10 and human FGF7. We allowed the cell mass to attach for a few days. On day 6–post isolation, maturation components were added in SFD media in the presence of CHIR99021 human FGF10, and human FGF7. The cultures were maintained for an additional 21 days before being processed for immunofluorescence or electron microscopy.
Live cell imaging of fluorescent-SPB protein secretion in day-48 cultures.
For assaying the secretion of fluorescent-SP-B protein by day-48 cultures, the cells were incubated with BODIPY-SPB as described above; the cultures were counterstained with CellTrace calcein red-orange AM (Life Technologies) for 10 min. The baseline BODIPY-SPB fluorescence was imaged for 20 min with Zeiss LSM 510 confocal microscope; the cells was then incubated in 10 μM of a DAG analog, 1-oleoyl-2-acetyl-sn-glycerol (Sigma, cat# O6754) for 10 min. Cultures were rinsed with DPBS and imaged for another 20 min. The minus DAG controls were processed identically and imaged for the same length of time. The fluorescent intensity of chosen cells was analyzed with Metamorph (Molecular Devices, LLC. Sunnyvale, CA).
References
Green, M.D., Huang, S.X. & Snoeck, H.W. Stem cells of the respiratory system: from identification to differentiation into functional epithelium. Bioessays 35, 261–270 (2013).
Rock, J.R. & Hogan, B.L. Epithelial progenitor cells in lung development, maintenance, repair, and disease. Annu. Rev. Cell Dev. Biol. 27, 493–512 (2011).
Morrisey, E.E. & Hogan, B.L. Preparing for the first breath: genetic and cellular mechanisms in lung development. Dev. Cell 18, 8–23 (2010).
Rawlins, E.L., Clark, C.P., Xue, Y. & Hogan, B.L. The Id2+ distal tip lung epithelium contains individual multipotent embryonic progenitor cells. Development 136, 3741–3745 (2009).
Green, M.D. et al. Generation of anterior foregut endoderm from human embryonic and induced pluripotent stem cells. Nat. Biotechnol. 29, 267–272 (2011).
Mou, H. et al. Generation of multipotent lung and airway progenitors from mouse ESCs and patient-specific cystic fibrosis iPSCs. Cell Stem Cell 10, 385–397 (2012).
Longmire, T.A. et al. Efficient derivation of purified lung and thyroid progenitors from embryonic stem cells. Cell Stem Cell 10, 398–411 (2012).
Wong, A.P. et al. Directed differentiation of human pluripotent stem cells into mature airway epithelia expressing functional CFTRTR protein. Nat. Biotechnol. 30, 876–882 (2012).
Kubo, A. et al. Development of definitive endoderm from embryonic stem cells in culture. Development 131, 1651–1662 (2004).
Nostro, M.C. & Keller, G. Generation of beta cells from human pluripotent stem cells: potential for regenerative medicine. Semin. Cell Dev. Biol. 23, 701–710 (2012).
Nostro, M.C. et al. Stage-specific signaling through TGFbeta family members and WNT regulates patterning and pancreatic specification of human pluripotent stem cells. Development 138, 861–871 (2011).
D'Amour, K.A. et al. Efficient differentiation of human embryonic stem cells to definitive endoderm. Nat. Biotechnol. 23, 1534–1541 (2005).
Goss, A.M. et al. Wnt2/2b and beta-catenin signaling are necessary and sufficient to specify lung progenitors in the foregut. Dev. Cell 17, 290–298 (2009).
Bellusci, S., Grindley, J., Emoto, H., Itoh, N. & Hogan, B.L. Fibroblast growth factor 10 (FGF10) and branching morphogenesis in the embryonic mouse lung. Development 124, 4867–4878 (1997).
Bellusci, S., Henderson, R., Winnier, G., Oikawa, T. & Hogan, B.L. Evidence from normal expression and targeted misexpression that bone morphogenetic protein (Bmp-4) plays a role in mouse embryonic lung morphogenesis. Development 122, 1693–1702 (1996).
Domyan, E.T. et al. Signaling through BMP receptors promotes respiratory identity in the foregut via repression of Sox2. Development 138, 971–981 (2011).
Li, Y., Gordon, J., Manley, N.R., Litingtung, Y. & Chiang, C. Bmp4 is required for tracheal formation: a novel mouse model for tracheal agenesis. Dev. Biol. 322, 145–155 (2008).
Chen, F. et al. A retinoic acid-dependent network in the foregut controls formation of the mouse lung primordium. J. Clin. Invest. 120, 2040–2048 (2010).
Yamamoto, M. et al. Nodal antagonists regulate formation of the anteroposterior axis of the mouse embryo. Nature 428, 387–392 (2004).
Perea-Gomez, A. et al. Nodal antagonists in the anterior visceral endoderm prevent the formation of multiple primitive streaks. Dev. Cell 3, 745–756 (2002).
del Barco Barrantes, I., Davidson, G., Grone, H.J., Westphal, H. & Niehrs, C. Dkk1 and noggin cooperate in mammalian head induction. Genes Dev. 17, 2239–2244 (2003).
Yu, P.B. et al. Dorsomorphin inhibits BMP signals required for embryogenesis and iron metabolism. Nat. Chem. Biol. 4, 33–41 (2008).
Inman, G.J. et al. SB-431542 is a potent and specific inhibitor of transforming growth factor-beta superfamily type I activin receptor-like kinase (ALK) receptors ALK4, ALK5, and ALK7. Mol. Pharmacol. 62, 65–74 (2002).
Chen, B. et al. Small molecule-mediated disruption of Wnt-dependent signaling in tissue regeneration and cancer. Nat. Chem. Biol. 5, 100–107 (2009).
Bennett, C.N. et al. Regulation of Wnt signaling during adipogenesis. J. Biol. Chem. 277, 30998–31004 (2002).
Fusaki, N., Ban, H., Nishiyama, A., Saeki, K. & Hasegawa, M. Efficient induction of transgene-free human pluripotent stem cells using a vector based on Sendai virus, an RNA virus that does not integrate into the host genome. Proc. Jpn. Acad., Ser. B, Phys. Biol. Sci. 85, 348–362 (2009).
Warren, L. et al. Highly efficient reprogramming to pluripotency and directed differentiation of human cells with synthetic modified mRNA. Cell Stem Cell 7, 618–630 (2010).
Kimura, S. et al. The T/ebp null mouse: thyroid-specific enhancer-binding protein is essential for the organogenesis of the thyroid, lung, ventral forebrain, and pituitary. Genes Dev. 10, 60–69 (1996).
Kriks, S. et al. Dopamine neurons derived from human ES cells efficiently engraft in animal models of Parkinson's disease. Nature 480, 547–551 (2011).
Harris-Johnson, K.S., Domyan, E.T., Vezina, C.M. & Sun, X. beta-Catenin promotes respiratory progenitor identity in mouse foregut. Proc. Natl. Acad. Sci. USA 106, 16287–16292 (2009).
Rock, J.R. et al. Basal cells as stem cells of the mouse trachea and human airway epithelium. Proc. Natl. Acad. Sci. USA 106, 12771–12775 (2009).
Weaver, M., Yingling, J.M., Dunn, N.R., Bellusci, S. & Hogan, B.L. Bmp signaling regulates proximal-distal differentiation of endoderm in mouse lung development. Development 126, 4005–4015 (1999).
Shu, W. et al. Wnt/beta-catenin signaling acts upstream of N-myc, BMP4, and FGF signaling to regulate proximal-distal patterning in the lung. Dev. Biol. 283, 226–239 (2005).
Post, M. et al. Keratinocyte growth factor and its receptor are involved in regulating early lung branching. Development 122, 3107–3115 (1996).
Malpel, S., Mendelsohn, C. & Cardoso, W.V. Regulation of retinoic acid signaling during lung morphogenesis. Development 127, 3057–3067 (2000).
Wongtrakool, C. et al. Down-regulation of retinoic acid receptor alpha signaling is required for sacculation and type I cell formation in the developing lung. J. Biol. Chem. 278, 46911–46918 (2003).
Gonzales, L.W., Guttentag, S.H., Wade, K.C., Postle, A.D. & Ballard, P.L. Differentiation of human pulmonary type II cells in vitro by glucocorticoid plus cAMP. Am. J. Physiol. Lung Cell. Mol. Physiol. 283, L940–L951 (2002).
Sanchez-Esteban, J. et al. Mechanical stretch promotes alveolar epithelial type II cell differentiation. J. Appl. Physiol. 91, 589–595 (2001).
Whitsett, J.A., Wert, S.E. & Weaver, T.E. Alveolar surfactant homeostasis and the pathogenesis of pulmonary disease. Annu. Rev. Med. 61, 105–119 (2010).
Rooney, S.A. Regulation of surfactant secretion. Comp. Biochem. Physiol. A Mol. Integr. Physiol. 129, 233–243 (2001).
Gouon-Evans, V. et al. BMP-4 is required for hepatic specification of mouse embryonic stem cell-derived definitive endoderm. Nat. Biotechnol. 24, 1402–1411 (2006).
Osafune, K. et al. Marked differences in differentiation propensity among human embryonic stem cell lines. Nat. Biotechnol. 26, 313–315 (2008).
Bock, C. et al. Reference maps of human ES and iPS cell variation enable high-throughput characterization of pluripotent cell lines. Cell 144, 439–452 (2011).
Boulting, G.L. et al. A functionally characterized test set of human induced pluripotent stem cells. Nat. Biotechnol. 29, 279–286 (2011).
Blauwkamp, T.A., Nigam, S., Ardehali, R., Weissman, I.L. & Nusse, R. Endogenous Wnt signalling in human embryonic stem cells generates an equilibrium of distinct lineage-specified progenitors. Nat. Commun. 3, 1070 (2012).
Barkauskas, C.E. et al. Type 2 alveolar cells are stem cells in adult lung. J. Clin. Invest. 123, 3025–3036 (2013).
Xu, X. et al. Evidence for type II cells as cells of origin of K-Ras-induced distal lung adenocarcinoma. Proc. Natl. Acad. Sci. USA 109, 4910–4915 (2012).
O'Neill, J.D. et al. Decellularization of human and porcine lung tissues for pulmonary tissue engineering. Ann. Thorac. Surg. 96, 1046–1056 (2013).
Acknowledgements
S.X.L.H. is a Druckenmiller Fellow of the New York Stem Cell Foundation. The authors wish to thank J. Sonnet and the Columbia Lung Regeneration Team, D. Farber and J. Thome for providing samples of human lung tissue; K. Brown for kind help with electron microscopy; S.-H. Ho for assistance with microscopy.
Author information
Authors and Affiliations
Contributions
S.X.L.H. performed most experiments, developed this protocol and co-wrote the manuscript; M.N.I. performed functional analysis of ATII cells supervised by J.B.; M.M., M.D.G. and Y.-W.C. gave technical advice and assisted S.X.L.H. experimentally; Z.H. performed kidney capsule transplantations supervised by Y.-G.Y.; J.O.N. provided human lung human decellularized extracellular lung matrix and these experiments were supervised by G.V.-N.; H.-W.S. developed the concept, co-analyzed primary data and co-wrote the manuscript with S.X.L.H.
Corresponding author
Ethics declarations
Competing interests
The authors have filed patent application IRCU13340.
Supplementary information
Supplementary Text and Figures
Supplementary Figures 1–12 and Supplementary Tables 1 and 2 (PDF 2840 kb)
Rights and permissions
About this article
Cite this article
Huang, S., Islam, M., O'Neill, J. et al. Efficient generation of lung and airway epithelial cells from human pluripotent stem cells. Nat Biotechnol 32, 84–91 (2014). https://doi.org/10.1038/nbt.2754
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/nbt.2754
This article is cited by
-
Directed differentiation of mouse pluripotent stem cells into functional lung-specific mesenchyme
Nature Communications (2023)
-
A multi-organoid platform identifies CIART as a key factor for SARS-CoV-2 infection
Nature Cell Biology (2023)
-
Alveolar epithelial-like cell differentiation in a dynamic bioreactor: a promising 3D-approach for the high-throughput generation of lung cell types from human induced pluripotent stem cells
In vitro models (2023)
-
Derivation of Human Salivary Epithelial Progenitors from Pluripotent Stem Cells via Activation of RA and Wnt Signaling
Stem Cell Reviews and Reports (2023)
-
Harnessing three-dimensional (3D) cell culture models for pulmonary infections: State of the art and future directions
Naunyn-Schmiedeberg's Archives of Pharmacology (2023)
