
Abstract
Intracellular tumor antigens presented on the cell surface in the context of human leukocyte antigen (HLA) molecules have been targeted by T cell–based therapies, but there has been little progress in developing small-molecule drugs or antibodies directed to these antigens. Here we describe a bispecific T-cell engager (BiTE) antibody derived from a T-cell receptor (TCR)-mimic monoclonal antibody (mAb) ESK1, which binds a peptide derived from the intracellular oncoprotein WT1 presented on HLA-A*02:01. Despite the very low density of the complexes at the cell surface, ESK1-BiTE selectively activated and induced proliferation of cytolytic human T cells that killed cells from multiple leukemias and solid tumors in vitro and in mice. We also discovered that in an autologous in vitro setting, ESK1-BiTE induced a robust secondary CD8 T-cell response specific for tumor-associated antigens other than WT1. Our study provides an approach that targets tumor-specific intracellular antigens without using cell therapy and suggests that epitope spreading could contribute to the therapeutic efficacy of this BiTE.
Similar content being viewed by others

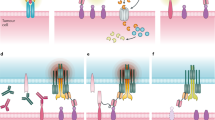
Main
Tumor-specific antigens are in most cases intracellular proteins, inaccessible to classical mAb therapy. These intracellular proteins are, however, degraded, processed and presented by major histocompatibility complex (MHC) class I molecules as peptide-MHC complexes that can be recognized by the TCR of cytotoxic T lymphocytes (CTLs). Immunotherapies involving CTLs have been central to cancer immunotherapy1,2. However, given the intrinsic complexity of cell-based therapies, alternative approaches using molecular agents would be desirable. Although TCR-mimic mAbs3,4,5 against tumor-specific intracellular antigens have been developed, their potency will be limited by very low epitope density on the cell surface, which reduces efficacy. In contrast, BiTE antibodies—heterodimers of IgG single-chain fragment variable regions (scFv) with dual specificities for a mAb-defined tumor-associated antigen and for CD3 T cells6,7,8,9,10,11—may be a more promising approach because of their greater potency by virtue of their recruiting of cytolytic T cells. BiTE molecules have been shown to redirect both CD4 and CD8 T cells to kill tumor cells independent of the T cells' intrinsic antigen-specific TCR recognition. Therapeutic BiTE mAbs that have been developed to date are directed to well-known, high-density, cell surface proteins that are not tumor-specific. An example is blinatumomab (Blincyto), which is reactive with the pan B-cell antigen CD19; it has approved by the US Food and Drug Administration for the treatment of B-cell neoplasms8,9.
Here we describe the generation and therapeutic efficacy of a tumor-specific BiTE derived from the high-affinity TCR-mimic antibody ESK1, which specifically binds the Wilms' tumor protein (WT1) epitope RMF, in the context of HLA-A*02:01, the most common HLA-A allele in people of European descent3,4,5. Despite the ultra-low density of expression of the peptide-MHC complex, ESK1-BiTE effectively treated BV173 Ph+ acute lymphocytic leukemia (ALL), primary ALL, SET-2 acute myeloid leukemia (AML) and JMN mesothelioma in mouse models. Notably, ESK1-BiTE induced a long-lasting, autologous T-cell response in vitro to non-WT1 epitopes, including HER2/Neu, in cells from patients with HER2/Neu+ ovarian cancer. Our study suggests that ESK1-BiTE induces epitope spreading—the expansion of T cells against various tumor antigens not targeted by the original therapy—which could provide a broader, more effective and long-term response than the original BiTE-mediated short-term therapy against a single antigen originally targeted.
Results
ESK1-BiTE induces activation of T cells that kill WT1+ cancer cells
The full-length ESK1 mAb binds to cancers and cell lines in a WT1- and HLA-A*02:01-restricted manner3,4. The ESK1-BiTE, a scFv construct, had the expected binding specificity (Supplementary Fig. 1). We did not observe binding of ESK to any CD34+ cells from a HLA-A*02:01+ healthy donor (Supplementary Fig. 2a). The activation of T cells by BiTE constructs depends on the proximal contact between T cells and target cells expressing the target antigens. This proximity also avoids possible unwanted inflammatory responses caused by activation through the invariant CD3 signaling complex10,12. Incubation of ESK1-BiTE with target WT1+ SET-2 AML cells caused a dose-dependent interferon (IFN)-γ release in human T cells (Fig. 1a). CD3 T cells incubated with control-BiTE in the presence of SET-2 cells were not stimulated. When T-cell activation was further evaluated by intracellular cytokine staining, only peripheral blood mononuclear cells (PBMCs), incubated with SET-2 cells in the presence of ESK1-BiTE, showed elevated expression of CD107, CD137, IFN-γ and tumor necrosis factor (TNF)-α, which was sustained over at least 3 d (Supplementary Fig. 2b). In a HLA-A*02:01- WT1− ALL cell line, BA-45, such T-cell activation was not elicited. Both CD4 and CD8 T cells were similarly activated in all experimental groups, as expected for CD3 engagement. NK T cell-like cells (CD3+CD56+) were activated, but no changes were observed before or after ESK1-BiTE engagement for CD4+CD25+Foxp3+ T-regulatory (Treg) cells monitored on a daily basis over 3 d.

(a) IFN-γ secretion in the presence of WT1+/HLA A*02:01+ tumor cells. Purified human CD3 T cells and SET-2 cells at a 15:1 ratio were incubated with or without ESK1-BiTE or control BiTE at concentrations of 3, 1, 0.3, 0.1 or 0.03 μg/ml overnight. The cultures of T cells alone or T cells with SET-2 cells, plus control BiTE did not show any detectable IFN-γ. Their values were therefore subtracted from the data shown here. The data show the average of duplicate cultures and represent one of two similar experiments from two donors. (b) T-cell cytotoxicity was measured in parallel against SET-2 after overnight culture by LDH release assay. (c–i) T-cell cytotoxicity against WT1+/HLA A*02:01+ tumor cells. Purified T cells were incubated with primary ALL cells at an E/T ratio of 10:1 (c), SET-2 AML (d), BV173 Ph+ ALL (e) or HL-60 cells (f). (b–f) E/T ratios are 50:1 (red and blue lines) or 5:1 (green and purple lines) in the presence or absence of ESK1 or control BiTEs at the indicated concentrations. The cytotoxicity was measured by 5 h-51Cr-release assay. Similarly, ESK1-BiTE-mediated cytotoxicity by EBV-primed human T cells was titrated by a 5-h 51Cr-release assay against primary ovarian cancer cells (g), BV173 Ph+ ALL (h) or JMN mesothelioma (i) at the indicated E/T ratio. BiTEs were used at 0.1 μg/ml. All the data points are averages of triplicate cultures and represent one of multiple similar experiments. (g–i) Red lines, ESK1-BiTE; blue lines, BiTE control; yellow lines, cultures without BiTEs.
We next assessed the efficacy and potency of ESK1-BiTE-directed T-cell cytotoxicity in 5-h 51Cr− release assays with multiple target cancer cell types. In a dose-dependent manner, ESK1-BiTE induced fresh T-cell cytotoxicity against WT-1+ SET-2 AML and BV173 Ph+ ALL cells, even at a low effector to target cell (E/T) ratio of 5:1, and a low concentration of 30 ng/ml of ESK1-BiTE, but not against WT1+HLA-A*02:01-HL-60 cells (Fig. 1b–f), or WT1− HLA-A*02:01+cells (B-JAB and SKLY-16), which was consistent with the binding specificity. When T-cell cytotoxicity was further titrated on SET-2 and BV173 targets at an E/T ratio of 1:1, the ESK1-BiTE-mediated cytotoxicity was observed to concentrations down to 1 ng/ml (Supplementary Fig. 2c). The cytotoxicity increased up to 90% after longer incubation (Fig. 1b). Cytotoxicity was also observed against primary ALL blasts (Fig. 1c). However, despite the specificity seen in these models, because the BiTE may have high enough potency to kill cells that express very few cell surface target sites, a full understanding of the possible on-target, off-tumor toxicity may not be known until human trials.
ESK1-BiTE could also redirect the cytotoxicity of T cells that had previously been repeatedly primed with autologous Epstein-Barr virus (EBV)-transduced B cells containing different viral antigens. Using such EBV-specific T cells allowed us to avoid xenogenic graft-versus-host disease in the xenograft animal model, as the T cells were specific for EBV antigens not found in mice. ESK1-BiTE lysed WT-1+HLA A*02:01+ fresh ovarian cancer cells, Ph+ ALL BV173 and JMN mesothelioma in the presence of EBV-specific T cells (Fig. 1g–i), even more effectively than with fresh PBMCs. Effector cells alone or with control BiTEs did not induce any great cytotoxicity, indicating that the target specificity is required for the T-cell activation. Therefore, ESK1-BiTE specifically redirected potent cytotoxicity of both polyclonal resting T cells and previously sensitized T cells specific for an antigen other than WT1, to lyse tumor cells that were WT-1+HLA A*02:01+.
We also investigated the ESK1-BiTE-mediated activation and cytotoxicity of T cells in an autologous setting, thus more closely mimicking the human situation in vivo, using PBMCs from a patient with ovarian cancer with her autologous tumor cells. Dose-dependent killing was observed in the cultures with ESK1-BiTE, even with as low as 1,000 PBMCs at the start of the assay (Supplementary Fig. 3a). No killing was observed in the control groups. In parallel, T-cell proliferation was measured by 3H-thymidine incorporation at the end of 8 d of culture (Supplementary Fig. 3b). Proliferation of T cells was seen only in the cultures with ESK1-BiTE and irradiated, nonproliferating tumor cells and T cells together. PBMCs alone or PBMCs with ESK1-BiTE or control BiTE alone, the irradiated tumor cells alone or with ESK- or control BiTE, co-cultures of PBMCs with autologous irradiated tumor cells alone, or with tumor and control BiTE, showed no T-cell proliferation. This shows that without the BiTE, T cells were not responsive to the self-tumor antigens and that specific activation of T cells by ESK1-BiTE requires the presence of tumor cells. That the ESK1-BiTE could induce autologous T-cell proliferation and killing of its target cancer cells was further illustrated in a second autologous model evaluating T-cell responses with or without the ESK1-BiTE to autologous AML blasts from a patient with AML (Supplementary Fig. 3c–f).
Treatment of human leukemias expressing WT1/HLA-A*02:01 in NSG mice
The anti-leukemic activity of ESK1-BiTE in vivo first was tested using an aggressive AML line, SET-2, which migrates to bone marrow rapidly upon engraftment. Dosing and schedule were guided by pharmacokinetics studies that showed a short, 5 h, beta plasma half-life of the BiTE (Supplementary Fig. 4). Luminescence imaging of a luciferase-tagged tumor was used in all therapy experiments to allow precise quantification of the cancer cells throughout the entire mouse, without euthanizing them, thereby also showing the kinetics of the response, and possible regional differences. Such analyses avoid the sampling biases of tumor biopsies when animals are euthanized.
Massive infiltration of leukemia was seen in the bone marrow of NSG (nonobese diabetic (NOD) severe combined immunodeficient (SCID) gamma) mice that received SET-2 cells or SET-2 with allogeneic EBV-specific T cells, or SET-2 with control BiTE (Fig. 2). However, no detectable leukemia was seen in the group treated with EBV-specific T cells and ESK1-BiTE for up to 14 d and only a minimum leukemia burden was observed up to 25 d, more than a week after treatment was stopped. The control-BiTE group showed slightly delayed leukemia growth on day 14, possibly caused by activation of EBV-specific T cells by the anti-CD3 arm of the control-BiTE. Whereas all the mice in the ESK1-BiTE+ T-cell-treated group were still alive after a month, there was little survival in the control groups. In addition, the mice in the ESK1-BiTE-treated group showed no sign of central nervous system (CNS) paralysis caused by leukemia infiltration into vertebral bone marrow for up to 40 d, whereas nearly all animals in the control group developed this leukemic sequela.

(a) Tumor burden is shown by posterior bioluminescence imaging (BLI) of mice to show vertebral leukemia infiltration. The BLI scale was increased in all animals tenfold on the images from day 7 onward to show the leukemia burden in all groups equally normalized. (b) Tumor burden was calculated by summing the luminescent signal of each mouse in the back and front two positions. Mean signal for each group (n = 5) is plotted. Leukemia infiltration was also assessed by day of appearance of limb paralysis caused by damage to the spinal cord central nervous system.
ESK1-BiTE mediates T-cell retention at cancer cell sites
Although it is known that BiTE mAbs effectively engage T cells to kill targets in vitro, no studies have reported this mechanism in vivo. We investigated whether the therapeutic efficacy of ESK1-BiTE was the result of its ability to attract T cells and retain them at the sites of leukemia in live animals. Ten million EBV-specific T cells transduced with Renilla luciferase were injected into NSG mice 3 d after firefly luciferase–positive SET-2 AML cells were engrafted. ESK1-BiTE was injected 4 h later. T-cell migration was monitored by bioluminescence imaging. Differential luminescence imaging of luciferase-tagged tumor and T cells simultaneously was used to allow determination of the time course of quantitative localization of both types of cells throughout the entire mouse. T cells alone migrated into the lungs immediately after the injection, then became distributed into other organs including liver, spleen and bone marrow at 8–24 h (Supplementary Fig. 5a). T-cell signal gradually declined over 72 h. T-cell injection to tumor-bearing mice showed a similar distribution pattern and time course as that of T cells alone. However, mice treated with ESK1-BiTE showed a significant (P < 0.02) increase in T-cell signals in lungs and bone marrow from 8–24 h, which lasted up to 72 h. Cells declined in the lungs substantially, but remained in the liver, spleen and bone marrow. Monitoring the leukemia progression at the same time in these mice revealed an inverse correlation between T-cell retention and leukemia burden (Supplementary Fig. 5b). Quantification of bioluminescence intensity showed that 4 h after the ESK1-BiTE injection, there was approximately threefold more T-cell accumulation in the liver, spleen and bone marrow compared to the other two control groups (Supplementary Fig. 5c). These results provided evidence in vivo that ESK1-BiTE could mediate prolonged retention of T cells at the leukemia sites, and effectively redirect T cells to the tumor cells expressing WT1-RMF and HLA-A*02:01. However, although the numbers of T cells at the target sites are increased, these data do not address whether the number or the activation state of these cells was critical for the effects observed.
We also tested the therapeutic efficacy in vivo of ESK1-BiTE in NSG mice xenografted intravenously with primary ALL cells. T cells were injected intravenously into mice followed by BiTE injection. All mice in the control groups showed increasing tumor growth and massive tumor burdens in the bone marrows and other organs. Dramatic BiTE-dependent tumor inhibition was observed, which was especially prominent in the bone marrow of mice, persisting more than 3 weeks, 9 d after treatment was stopped (Fig. 3a). The average of photon intensity from five mice showed an approximately 10- to 20-fold tumor reduction in the ESK1-BiTE-treated group when compared to control groups (Supplementary Fig. 6a). Almost no sign of graft-versus-host disease was observed clinically for up to 49 d after inoculation with tumor cells. These results demonstrated that ESK1-BiTE could efficiently engage and redirect potent T-cell cytotoxicity to kill primary leukemia cells.

(a) ESK1-BiTE inhibits primary ALL cell growth in NSG mice. Tumor inhibition day 18 and 23 after tumor inoculation is shown by bioluminescence imaging (BLI) in prone and supine views for each time point. (b) ESK1-BiTE eliminates peritoneal JMN mesothelioma cells in NSG mice. JMN cells were mixed with EBV-specific T cells and injected into mice. Tumor development was monitored by supine firefly BLI at the indicated time points. Day 1 (1 d after treatment) showed no visible tumor in the mice treated with ESK1-BiTE, suggesting the elimination of tumor cells. Tumor inhibition was seen up to 23 d of monitoring.
Therapy of mesothelioma in NSG mice
We next investigated if ESK1-BiTE is effective in treating an aggressive solid tumor using the JMN peritoneal-cavity mesothelioma. Bioluminescence imaging showed no visible tumor in the mice treated with ESK1-BiTE until more than 2 weeks after treatment, whereas mice in three controls groups showed tumor progression by day 4 (Fig. 3b). The tumor suppression by ESK1-BiTE persisted until day 23, 18 d after the treatment was stopped, with only a minimum tumor burden seen in the mice. Averaging the bioluminescence intensity of five mice per group showed more than a 20-fold tumor suppression on day 23 (Supplementary Fig. 6b) when compared to control groups including tumor cells alone, tumor cells with T cells or tumor cells, T cells and control bite. In addition, therapeutic efficacy was also obtained in disseminated xenograft of Ph+ ALL BV173 cells (Supplementary Fig. 6c).
ESK1-BiTE induced a secondary T-cell response to a HER2/Neu epitope
The mechanism of action for BiTE mAbs has been attributed exclusively to their capacity to bridge the cancer cell targets and T cells to form a cytolytic synapse. We asked whether such a proximal contact could also directly activate pre-existing T cells in the polyclonal population that were specific for other tumor antigens expressed by the autologous tumor cells. That is, could the BiTE exert an epitope spreading effect? We tested this hypothesis in vitro using T-cells from a HLA-A*02:01+ patient with ovarian cancer. The patient's PBMCs were co-cultured with her autologous tumor cells in the presence of low-dose ESK1-BiTE. A week later, cells were washed and tested for epitope-specific, HLA-A*02:01-restricted T-cell responses by IFN-γ ELISPOT assay. Control groups, including the T cells alone with tumor or with control BiTE and tumor did not show any specific IFN-γ release. The patient's tumor cells express both WT1-RMF and HER2/Neu on their cell surface (Supplementary Fig. 7a–c). Notably, a strong secondary T-cell response was induced against the HER2/Neu-369 epitope13 presented by HLA A*02:01 in T2 cells and against the autologous tumor cells alone, as well as against SET-2 AML cells, long after ESK1-BiTE-mediated interactions had ceased (Fig. 4a,b). No response in these autologous cells was measurable against 56 pooled WT1-derived peptides, WT1 epitopes RMF, AILDF or LDF14; p53-derived epitope 264-273 (ref. 15); Prame-300 (ref. 16), Prame-435 (ref. 17); or Ewings sarcoma epitope EW18 that are also presented by HLA-A*02:01, or to A*02:01− HL60 cells. The T-cell response against SET-2 showed recognition of other yet-to-be defined epitopes that are neither WT1 nor HER2/Neu. These results indicate that ESK1-BiTE can induce secondary T-cell responses to multiple tumor antigens, thereby providing a postulated epitope spreading effect.

(a) PBMCs from a patient with ovarian cancer were stimulated with autologous tumor cells at an E/T ratio of 5:1, in the presence of ESK1-BiTE or control BiTE, human IL-5 and human IL-2 for a week. Epitope-specific response, as measured by IFN-γ ELISPOT assay, against T2 cells, pulsed with indicated peptides is shown. (b) Remaining PBMCs from the experiment in a was restimulated in the same manner, at an E/T ratio of 9:1, and epitope-specific T-cell response was measured by IFN-γ ELISPOT assay. (c,d) The same stimulation protocol and IFN-γ ELISPOT assay were conducted to compare epitope-specific T-cell response between purified CD3+ T cells versus PBMCs depleted of NK and macrophage (indicated as T+B) (c), or PBMCs versus purified CD3+ T cells, or T cells stimulated with dead autologous tumor cells (d). Dead tumor cells were generated by frequent freeze thawing without DMSO. The data represent the average of triplicate culture ±s.d. Supernatant from the co-cultures of PBMCs or purified T cells with autologous ovarian cancer cells in the presence or absence of BiTEs at 0.1 μg/ml, were collected after 3 h, 3 or 6 d and IFN-γ (e) and TNF-α (f) were measured by ELISA kits.
In principle, a secondary T-cell response should require antigen-presenting cells (APCs) among the PBMCs that could take up and present the intracellular antigens released after tumor cell death. Alternatively, tumor cells could directly activate pre-existing antigen-specific T cells through proximate contact between T cells and tumor. To clarify the mechanism, we asked whether the epitope spreading effect could be elicited with purified T cells, or NK cell-depleted and macrophage-depleted PBMCs (T plus B cells). T cells alone when cultured with autologous ovarian carcinoma cells and the ESK1-BiTE also elicited a T-cell response to HER2/Neu, autologous tumor cells and SET-2 cells, of a similar magnitude as that produced by T plus B cells (Fig. 4c) or whole PBMCs (Fig. 4d). To exclude the possibility that activated T cells themselves present these self-cancer antigens, purified T cells were co-cultured with autologous tumor cell lysates, generated by repetitive freeze/thaw, in the presence or absence of ESK1-BiTE. No T-cell response was seen against the HER2/Neu epitope or tumor cells in this setting (Fig. 4d). These results suggest that BiTE-mediated interactions between T cells and tumor cells expressing the peptide-HLA complex targeted by the BiTE may be sufficient to re-activate pre-existing T cells specific for other peptide-MHC complexes on the surface of tumor cells.
We also examined if tumor cell antigen presentation depends on co-stimulatory molecules. Whereas CD14+ monocytes from a healthy donor showed strong CD86 expression, ovarian cancer cells had little expression of the CD86. The results indicated that a key co-stimulatory molecule CD86 is unlikely to be involved in the tumor cell antigen presentation (Supplementary Fig. 8). No CD86 or ICOSL expression was detected in resting PBMCs or in the tumor cells. Lack of co-stimulatory molecules is one of the reasons that tumor cells are poor APCs; however, certain cytokines may provide co-stimulatory signals to fulfill the requirements for tumor-specific T-cell activation. ESK1-BiTE induced IFN-γ and TNF-α secretion by both PBMCs and T cells (Fig. 4e,f).
To test if ESK1-BiTE has any long-term effect on the T-cell population, we expanded T cells in vitro in low-dose IL-15 and IL-2, after the first activation by ESK1-BiTE. Whereas T cells in the control groups did not survive more than 1 week in culture, T cells activated by ESK1-BiTE continued to grow for 2 months, without further activation. Phenotype analysis revealed that 7 weeks after activation by ESK1-BiTE and tumor, CD8+ T cells increased to 89% of the population and CD4+ T cells decreased to 6% (Fig. 5a,c). Among the CD8+CCR7− cells, CD45RA−CD45RO+ cells increased, indicative of an effector memory phenotype. No TCR gamma delta or invariant NK T cells were detected. However, T cells co-expressing NK marker CD56 increased from 1% to 69%, after the activation, further support of the effector memory formation, as this population indicates a highly specialized effector memory phenotype19 (Fig. 5b). Even at this late time point, these T cells still retained their original specificity to autologous tumor and SET-2 AML cells (Fig. 5d). A reversal of the percentage of CD4 versus CD8 T cells was already evident 3 weeks after ESK1-BiTE activation. Only CD8 T cells acquired the CD56 marker, which was consistent with its cytotoxicity against autologous tumor and SET-2 AML cells (Supplementary Fig. 9). These results suggest that the secondary T-cells induced by ESK1-BiTE-mediated tumor interactions might be able to elicit long-lasting, secondary, specific CD8 T-cell responses.

(a,b) PBMCs from the patient in Figure 4 were stained with CD4, CD8, CD45RA, CD45RO, CCR7, TCR-α/β, V-24/J-18 and CD56 before and 7 weeks after activation with ESK1-BiTE (0.1 μg/ml) in the presence of autologous tumor cells. CD45RA and CD45RO versus CCR7 were shown on gated CD8 T cells. (b) The large selective increase in CD8 T cells 7 weeks after BiTE activation as measured by flow cytometry and cell counting. (c,d) Effector cells from the experiments shown in Figure 4 were expanded by weekly supplement of IL-15 and IL-2 for 5 weeks (c), and cytotoxicity was measured against autologous tumor, SET-2 and HL-60 cells at an E/T ratio 50:1 by standard 51Cr-release assay (d). The data represent the average of triplicate wells.
To test the generalizability of this observation, we performed the same experiments in three additional HLA-A*02:01 positive patients with ovarian cancer. We detected a similar T-cell response to the HER2/Neu epitope, autologous tumor cells and ovarian cancer cells in the second patient (Fig. 6a), whose tumor cells were positive for HER2/Neu, HLA-A*02:01 and WT1-RMF/HLA-A*02:01. PBMCs activated by ESK1-BiTE in the presence of autologous tumor also showed cytotoxicity against autologous tumor cells, primary ovarian cancer cells from the first patient (K-ovarian tumor), and SET-2 cells (which are HER2/Neu−) but not HL-60 (Fig. 6b). A similar response was detected in the third patient (HLA-A*02:01+, ESK1+ and HER2/Neu+) as well. In this case, a lower concentration of ESK1-BiTE (0.01 μg/ml) did not restore T-cell response to the RMF epitope, but it did induce a T-cell response to the HER2/Neu epitope (Fig. 6c). T cells from the same culture also showed cytotoxicity against the target cells, as seen in the two other patients (Fig. 6d). No T-cell response was detected in a fourth patient whose tumor cells were not WT1-RMF positive. These results confirmed again that ESK1-BiTE-mediated interaction between T cells and tumor cells was required for eliciting both the primary and secondary response.

(a) Ascites cells (ratio of T cells to tumor cells are approximately 5:1) were activated with ESK1-BiTE or control BiTE at 0.1 μg/ml, in the presence of human IL-15 and human IL-2, at an approximate E/T ratio of 5:1 for a week. The epitope-specific response was measured by IFN-γ ELISPOT assay, against T2 cells, pulsed with indicated peptides or unpulsed cancer cell targets. (b) PBMCs from the same patient in a were stimulated with ESK1-BiTE in the same manner, at an E/T ratio of 4:1, for 8 d, and the cytotoxicity against indicated target cells was measured by 51Cr release assay. The data in a and b represent one of two similar experiments from the same patient. (c) Ascites cells (T cells/tumor ratio, 7:1) from a third patient were activated with ESK1-BiTE or control BiTE at 0.01 μg/ml or 0.1 μg/ml, in the presence of human IL-15 and human IL-2, for a week and the epitope-specific response was measured by IFN-γ ELISPOT assay, against T2 cells, pulsed with indicated peptides. (d) Cells from the culture with ESK1-BiTE at 0.1 μg/ml (third patient from c) were tested for cytotoxicity against indicated target cells and measured by 51Cr release assay. All the data represent the average of triplicate microwell cultures.
To test if the ESK1-BiTE-induced T-cell responses to HER2/Neu was a result of priming of naive T cells or reactivation of pre-existing T cells specific for the HER2/Neu epitope, we performed the same experiments with PBMCs from HLA-A*02:01+ cord blood as effectors and the ovarian cancer cells from the first patient. Cord blood T cells are considered to be antigen-naive and also have lower allo-reactivity, which would confound our results. No T-cell response to the HER2/Neu epitope or other epitopes in the context of HLA-A*02:01 were detected. The results are consistent with a process in which ESK1-BiTE rapidly activates HER2/Neu epitope-specific T cells that are already present in the patients. Therefore, this secondary T-cell response may be biased toward the cancer present in the patient.
Discussion
In this study, we demonstrated the feasibility and potential applications of a BiTE construct derived from a TCR-mimic mAb, targeting an intracellular tumor-specific protein, WT1 (refs. 20,21,22). Our data suggest that the BiTE format can be applied to a much larger universe of targets. By use of an autologous system, we also discovered a BiTE mechanism of action—to our knowledge not previously described—in which the BiTE induced long-lived, secondary, specific T-cell responses against other tumor antigens, such as HER2/Neu. Further, the secondary T-cell responses do not appear to require cross-presentation effected by classical APCs, but rather result from the direct and physically close interaction between T cells and tumor cells, fostered by the BiTE.
Therapeutic TCR-mimic mAbs offer the combined features of T-cell and mAb therapies, joining the functionalities of TCR-like recognition of specific, intracellular tumor antigens and the pharmacologically useful mAb therapeutic activities3,4,5,23. We have demonstrated in our previous study that it is feasible for a TCR-mimic mAb to specifically bind and kill cells expressing a peptide-MHC complex at ultra-low density on the cell surface3,4,5. We have now demonstrated that a TCR-mimic BiTE construct is capable of potent therapeutic activity in several mouse models of both hematopoietic and solid tumors, providing the evidence for vastly broadening the development of BiTE mAbs targeting other cancer-specific intracellular tumor antigens. Although it is known from in vitro assays that BiTEs bring T cells in contact with cancer cells9,10, the dual bioluminescence studies here are consistent with such redirecting in vivo.
The mechanisms of BiTE action to date have been attributed to a direct interaction between T cells and the mAb-specified cellular target that induces short-term T-cell activation and cytotoxicity9,10,12. The upregulation of CD107 and CD137 expression and increased IFN-γ and TNF-α secretion by fresh T cells from normal donors over 24 and 48 h after ESK1-BiTE activation, with no difference in the ratio between CD4 versus CD8 T cells, suggested broad activation of T cells through CD3 signaling. Notably, in patients with ovarian cancer, we discovered not only this short-term activation and cytotoxicity of T cells, but a secondary T-cell response highly specific for other, non-WT1 cancer antigens. We tested several possible alternate targets that might be expressed by ovarian cancer cells, including multiple epitopes from WT1, PRAME and P53, among others, and identified one, the HER2/Neu epitope 369, known to be both expressed on ovarian cancer cells and highly immunogenic. As SET-2 cells do not express HER2/Neu, there is at least one other recognized target, but there may be many more. Other putative targets may also be presented by other class 1 and class 2 molecules and identifying them would be difficult.
We showed that proximate contact between T cells and live target cells could directly reactivate pre-existing T cells from the patients to react with other cancer antigens, without cross-presentation. The fast, robust and long-lasting HER2/Neu epitope–specific T-cell response, the reversal of the CD4 versus CD8 T-cell ratio, and the dramatic increase of CD8 T cells with effector memory phenotype is evidence that ESK1-BiTE induces CD8 epitope-specific memory T-cell responses. The CD8 T cells were activated to proliferate in response to additional HLA-A*02:01-restricted epitopes. In addition, the epitope-spreading response appears to be a result of reactivation and amplification of pre-existing T cells in patients, because naive T cells from HLA-A*02:01+ cord blood did not respond to ESK1-BiTE in the presence of the same ovarian cancer cells that were both WT1+ and HER2/Neu+. ESK1-BiTE did not induce significant T-cell responses against the original target epitope, WT1-RMF, even though the first patient may have had WT1-reactive T cells in her circulation as a consequence of prior adaptive cell therapy. These findings are consistent with our hypothesis that the BiTE is bringing the T cell's TCRs close to the tumor cell to recognize new peptide-MHC epitopes directly on the tumor. We speculate that one explanation for the lack of WT1 reactivity is that the binding of the ESK1-BiTE to its cognate peptide-MHC during this T-cell stimulation phase may be blocking any recognition of the RMF/HLA-A*02:01 complex from a cognate TCR in the population of T cells.
Although the effects seen here with the ESK1-BiTE have not yet been confirmed in vivo, owing to the inability to generate a non-human model that could accomplish this, nor seen yet with other BiTEs, epitope spreading induced by antigen-specific T-cell vaccines or T-cell infusion is a well-documented phenomenon. In the responding patients or regressing tumors, a considerable expansion of pre-existing and new T cells against various tumor antigens have been detected. It is postulated that vaccine-specific T cells kill the tumor cells and APCs uptake and process the apoptotic tumor cells to activate a variety of T cells, thereby broadening the immune response24,25. An epitope-spreading effect of mAb to CD20 has also been reported; the effect appears to be mediated by antibody-mediated opsonization of the target cells into APCs26,27,28, mechanistically not similar to what we describe here.
T cells directed against various tumor antigens have been found in the blood and tumor sites in patients1,24,25. Similarly, checkpoint blockade therapy promotes T-cell activation, amplifies pre-existing anti-tumor response and eliminates immunosuppression, including of Treg cells29,30. In addition, whole exomic sequencing approaches to identify neo-antigens from cancer patient's tumor cells and adoptive transfer of T cells generated against such antigens have shown promise31,32. ESK1-BiTE-induced secondary T-cell response may have an effect similar to checkpoint blockade therapy in terms of reactivating anergic or tumor-suppressed T cells, but in the case of BiTE, the response may be tumor specific. We speculate that the activated T cells are likely directed to neo-antigens that have not been identified yet and that this mechanism of ESK1-BiTE action could be highly dependent on the tumor and the patient. It is also interesting to speculate that such effects would not be seen with chimeric antigen receptor T cells, because the T cells have had their specificity redirected by the dominant chimeric antigen receptor.
We chose WT1 peptide RMF as our target for these studies as WT1 has been extensively studied as a oncofetal cancer antigen, with limited expression in adult tissues, outside of kidney podocytes and testes3,4,20,33,34. WT1 is found overexpressed in many human cancer cell types, including leukemias, ovarian cancers and mesothelioma, among others, and was ranked by the National Cancer Institute as the top cancer target21,33,34. The ESK1-BiTE-mediated cytotoxic T-cell clonal expansion for a variety of tumor-associated epitopes, could contribute greatly to its long-term therapeutic efficacy by preventing the escape of WT1− cancer cell variants or cells with low WT1 antigen density, as well as by promoting long-lived immunity. In addition, the epitope-spreading effect could be used for ex vivo expansion in advance of specific adoptive T-cell therapy.
Methods
Cell samples, cell lines and antibodies.
After informed consent on Memorial Sloan-Kettering Cancer Center Institutional Review Board approved protocols, peripheral blood mononuclear cells (PBMCs) from HLA-typed healthy donors and patients were obtained by Ficoll density centrifugation. The sources for obtaining human leukemia and solid tumor cell lines are described previously3. The cell lines for this study include: TAP-deficient T2 cells, AML lines HL60, SET-2, Ph+ ALL line BV173, mesothelioma cell lines JMN and MSTO. All tumor cells were HLA typed by the Laboratory of Cellular Immunology at Memorial Sloan-Kettering Cancer Center. The cell lines were cultured in RPMI 1640 supplemented with 5% FCS, penicillin, streptomycin, 2 mmol/L glutamine, and 2-mercaptoethanol at 37 C/5% CO2. Cells were checked regularly for mycoplasma. Cell identities were confirmed by phenotype or genotype. Primary ovarian cancer cells obtained from a patient's ascites fluid were expanded in NSG mice as ascites cells. Tumor cells for all animal studies were transduced with GFP/luciferase as described previously3. ESK1 and its control human IgG1 were produced by Eureka Therapeutics Inc. and APC conjugation was done according to the instructions of the manufacturer3. mAb against human HLA A*02 (clone BB7.2), its isotype control mouse IgG2b (clone MPC-11), human CD3 (clone HIT3A or OKT3), CD4 (clone RPA-T4), CD8 (clone RPA-T8), CD19 (clone HIB19), CD25 (clone 2A3), CD33 (clone WM53), CD34 (clone 581), CD45RA (clone HI100), CD45RO (clone UCHL1), CD56 (clone B159), CCR-7 (clone 3D12), CD127 (clone HIL-7R-M21), TCR-γ/γ (clone WT31), TCR-V delta 1 (clone 11F2), V delta 2 (clone B6), TCR-Vγ24-Jγ18 (clone 6B11) conjugated to various florophores, mouse anti-His tag mAb (clone F24-796) conjugated to FITC or PE, were purchased from BD Biosciences, (San Diego, CA). Trastuzumab and rituximab were obtained from the hospital pharmacy at MSKCC. ELISA kits for human IFN-γ and tumor necrosis factor (TNF)-α were purchased from Invitrogen (NY). Renilla luciferase substrate ViviRen was purchased from Promega (Madison, MI).
Peptides.
All peptides were purchased and synthesized by Genemed Synthesis, Inc. (San Antonio, TX). Amino acid sequences for HER2/Neu-369-377: KIFGSLAFL; p53 264-272: LFEVRVCAC; WT1-RMFPNAPYL. WT1-NQM, AILDF, LDF and total pooled peptides were previously described13,14,15,16,17. Prame-300: ALYVDSLFFL, p435: NLTHVLYPV. Control HLA-A2-binding peptide was derived from Ewing's sarcoma: qlqnpsydk.
Construction, expression and purification of ESK1-BiTE.
ESK1 BiTE is a single-chain bispecific antibody comprising ESK1 scFv at the N-terminal end and an anti-human CD3ɛ scFv of a mouse mAb at the C-terminal end35. The DNA fragments coding for the ESK1 scFv antibody and the anti-human CD3ɛ scFv antibody were synthesized by GeneArt (Invitrogen) and subcloned into Eureka's mammalian expression vector pGSN-Hyg using standard DNA technology. A hexhistamine (His) tag was inserted downstream of the ESK1-BiTE antibody at the C-terminal end for antibody purification and detection.
Chinese hamster ovary (CHO) cells were transfected with the ESK1-BiTE expression vector and stable expression was achieved by standard drug selection with methionine sulfoximine (MSX), a glutamine synthetase (GS)-based method36. CHO cell supernatants containing secreted ESK1-BiTE molecules were collected. ESK1-BiTE was purified using HisTrap HP column (GE healthcare) by FPLC AKTA system. Briefly, CHO cell culture was clarified and loaded onto the column with low imidazole concentration (20 mM), and then an isocratic high imidazole concentration elution buffer (500 mM) was used to elute the bound ESK1-BiTE protein. A negative control BiTE antibody, was constructed from an irrelevant human IgG1 antibody (Cat#ET901, Eureka Therapeutics,) replacing ESK1 scFv.
Flow cytometry analysis.
For ESK1-BiTE staining, human T cells or cancer cells were incubated with different concentrations of ESK1-BiTE or control BiTE for 30 min on ice, washed, and incubated with secondary mAbs against His-Tag. HER2/Neu expression on primary ovarian cancer cells was measured by staining the tumor cells with trastuzumab, followed by secondary goat anti-human IgG (clone 1140017A). HLA-A*02 expression and ESK1 binding was determined by direct staining of the cells with respective mAbs. Phenotype of PBMCs or T cells from patient samples were characterized by direct staining of the cells with mAbs for CD3, CD4, CD8, CD45RA, CD45RO, CCR7, CD19 or CD33 conjugated to various fluorophores. Flow cytometry data were collected on a FACS Calibur (Becton Dickinson) and analyzed with FlowJo 9.8.1 software.
ESK1-BiTE-mediated T-cell activation.
CD3 T cells were isolated from PBMCs by negative immunomagnetic cell separation using a pan T-cell isolation kit (Miltenyi Biotec). The ESK1-BiTE or its control BiTE at various concentrations were incubated with target cells and purified resting human CD3 T cells at different effector: target (E/T) ratio for different time periods. The supernatant fluids were harvested and cytokine release was measured by ELISA for IFN-γ. T-cell activation was also assessed by flow cytometric analysis on the expression of CD107 (clone H4A3), CD137 (clone 4B4-1), Ki67 (clone B56) and intracellular cytokines IFN-γ (clone B27) and TNF-γ (clone MAB11) (BD Biosciences). Briefly, 18 h before harvesting the cells, 10 mg/ml BrefeldinA (Sigma) plus 1 μl/ml Monensin BD GolgiStop (BD Pharmingen) was added to the cell cultures. Cytoplasmic staining was performed using the Cytofix/Cytoperm kit (BD Biosciences) according to the manufacturer's instructions. T regulatory cells (Treg) were assessed by intracellular staining of the Foxp3 protein (eBiosciences) on the CD4+ CD25+ and CD127low cells. In addition, ESK1-BiTE-mediated T-cell activation in the presence of autologous tumor cells from a patient with ovarian cancer was evaluated by cell proliferation, measured by overnight 3H-thymidine incorporation after 7 d of incubation.
ESK1-BiTE-induced secondary T-cell response.
PBMCs, PBMCs depleted of NK cells and macrophages or purified CD3 T cells from a patient with ovarian cancer were cultured with irradiated (3,000 rad) autologous ovarian cancer cells at an E/T ratio of 4–5:1, in the presence or absence of ESK1-BiTE, or control-BiTE at 0.1 μg/ml, and presence of human IL-5 (5 ng/ml) and human IL-2 (10 U/m) in RPMI1640 medium supplemented with 10% autologous plasma (AP) for 6 d. On day 7, the cells were harvested and washed and used as effectors for IFN-γ ELISPOT assay. In the case of ascites derived from patients with ovarian cancer, the percentage of CD3 T cells and tumor cells were estimated by flow cytometric analysis, based on the CD3+ cells and forward scatter. Ascites cells were then stimulated with BiTEs in the same manner as PBMCs. For ELISPOT assay, HA-Multiscreen plates (Millipore) were coated with 100 μl of mouse anti-human IFN-γ antibody (10 μg/ml; clone 1-D1K; Mabtech) in PBS, incubated overnight at 4 °C, washed with PBS to remove unbound antibody, and blocked with RPMI 1640/10% autologous plasma (AP) for 2 h at 37 °C. Effector cells were plated with either T2 cells (4:1 E: APC ratio) or irradiated autologous tumor cells or other tumor cell lines. Various test peptides were added to the wells at 20 μg/ml. Negative control wells contained APCs and T cells without hemagglutinin (PHA, Sigma). All conditions were done in triplicate. Microtiter plates were incubated for 20 h at 37 °C and then extensively washed with PBS/0.05% Tween and 100 μl/well biotinylated detection antibody against human IFN-γ (2 μg/ml; 7-B6-1; Mabtech) was added. Plates were incubated for an additional 2 h at 37 °C and spot development was done as described18. Spot numbers were automatically determined with the use of a computer-assisted video image analyzer with KS ELISPOT 4.0 software (Carl Zeiss Vision).
The remaining T cells were expanded by adding fresh medium with IL-15 and IL-2 once in a week, up to 7–8 weeks. In some cases, remaining T cells were restimulated with autologous tumor at an E/T ratio of 9:1 for a week in the same conditions as for the first stimulation and T-cell response was measured by IFN-γ ELISPOT as described18.
ESK1-BiTE-redirected T-cell cytotoxicity.
The ESK1-BiTE or its control BiTE at various concentrations were incubated with target cells and PBMCs, purified CD3 T cells or EBV-specific T cells at different E/T ratios for 5 h or overnight. The cytotoxicity was measured by 51Cr-release assay (after 5 h incubation) or lactate dehydrogenase (LDH) release assay (after overnight incubation) using Cytotox 96 nonradioreactive kit from Promega following their instructions. In one case of an AML patient, PBMCs and autologous blasts were co-incubated in the presence or absence of ESK1-BiTE or control BiTE at 20 γg/ml and the cells were harvested and dual stained with CD33 for leukemia blasts and CD3 for T cells on day 3 and 4. For the case of an ovarian cancer patient, PBMCs were incubated with autologous ovarian cancer cells for a week and the cytotoxicity was measured by 51Cr-release assay.
EBV-specific T-cell expansion and reporter gene transduction.
T-cells were enriched from PBMCs by depletion of monocytes by adhesion. Non-adhering cells were stimulated with irradiated autologous EBV-transformed B cells (EBV-BLCLs) generated by transformation with the B95.8 strain of EBV at a 20:1 responder/stimulator (R/S) ratio and cultured in Yssel's medium, containing 5% HS(YH5; Gemini). Beginning on day 7, interleukin (IL)-2 at 10 to 30 units/ml was added to the T-cell cultures every 2–3 d (Collaborative Biomedical Products, Bedford, MA), and were restimulated weekly with the same EBV-BLCLs at a 4:1 R/S ratio.
EBV-specific T lymphocytes were transduced with retroviral vector tdrrsRLuc, expanded and enriched by sorting for phycoerythrin as previously described37. Transduced EBV-specific T lymphocytes were cultured in G-rex flask (Wilson Wolf Manufacturing Corporation). For T-cell tracing study in vivo, ten million cells were in injected into mice and 4 h later, Renilla luciferase substrate ViviRen was given intravenously.
Pharmacokinetic and biodistribution studies.
All animal studies were conducted under an IACUC approved protocol. ESK1-BiTE or control BiTE were labeled with 125I (PerkinElmer) using the chloramine-T method. 100 μg of antibody was reacted with 1 mCi 125I and 20 μg chloramine-T, quenched with 200 μg Na metabisulfite, then separated from free 125I using a 10DG column equilibrated with 2% bovine serum albumin in PBS. Specific activity of the product was about 6 mCi/mg. Radiolabeled BiTE (2 μg) was diluted with unlabeled BiTE to 20 μg per dose, and injected into mice retro-orbitally. Blood was collected at various time points, weighed and measured on a gamma counter. At 24 h, organs were harvested, weighed and measured for activity on a gamma counter.
Therapeutic trials of the ESK1-BiTE in human tumor xenograft NSG models.
Human EBV-specific T cells were used for all xenograft models, as their antigenic specificity had been heavily skewed toward EBV antigens, wherein they should not induce graft-versus-host disease. For the SET-2 AML model, one million cells were intravenously injected into mice and the tumor engraftment was confirmed on day 3 by bioluminescence imaging and mice were randomized into treatment groups. On day 4, ten million EBV-specific T cells were injected intravenously and 6 h later, 20 μg ESK1-BiTE or its control BiTE was injected intravenously. Over the treatment course, T cells were given twice a week and BiTEs were given every day for 6 d. In the primary ALL model, five million ALL cells were injected intravenously into NSG mice. On day 6, tumor engraftment was confirmed by firefly luciferase imaging in all mice that were to be treated; mice were then randomly divided into different treatment groups. Thirty million EBV-specific T cells were injected intravenously into mice followed by injection intravenously of 20 μg ESK1-BiTE or its control BiTE. BiTE injection was given daily and T cells were given twice a week for total 2 weeks. For the mesothelioma JMN model, three hundred thousand tumor cells were mixed with six hundred thousand EBV-specific T cells and injected intraperitoneally (ip) and 1 h later, 20 μg ESK1-BiTE or control BiTE was intravenously injected into mice. The BiTEs were given every day for total 5 d. Tumor growth was monitored by firefly luciferase imaging at least twice a week, for all the animal models.
References
Coulie, P.G., Van den Eynde, B.J., van der Bruggen, P. & Boon, T. Tumour antigens recognized by T lymphocytes: at the core of cancer immunotherapy. Nat. Rev. Cancer 14, 135–146 (2014).
Morris, E. et al. Generation of tumor-specific T-cell therapies. Blood Rev. 20, 61–69 (2006).
Dao, T. et al. Targeting the intracellular WT1 oncogene product with a therapeutic human antibody. Sci. Transl. Med. 5, 176ra33 (2013).
Veomett, N. et al. Therapeutic efficacy of an Fc-enhanced TCR-like antibody to the intracellular WT1 oncoprotein. Clin. Cancer Res. 20, 4036–4046 (2014).
Dubrovsky, L. et al. A TCR-mimic antibody to WT1 bypasses tyrosine kinase inhibitor resistance in human BCR-ABL+ leukemias. Blood 123, 3296–3304 (2014).
Curran, K.J., Pegram, H.J. & Brentjens, R.J. Chimeric antigen receptors for T cell immunotherapy: current understanding and future directions. J. Gene Med. 14, 405–415 (2012).
Sadelain, M., Brentjens, R. & Rivière, I. The basic principles of chimeric antigen receptor design. Cancer Discov. 3, 388–398 (2013).
Frankel, S.R. & Baeuerle, P.A. Targeting T cells to tumor cells using bispecific antibodies. Curr. Opin. Chem. Biol. 17, 385–392 (2013).
Nagorsen, D. & Baeuerle, P.A. Immunomodulatory therapy of cancer with T cell-engaging BiTE antibody blinatumomab. Exp. Cell Res. 317, 1255–1260 (2011).
Brischwein, K. et al. Strictly target cell-dependent activation of T cells by bispecific single-chain antibody constructs of the BiTE class. J. Immunother. 30, 798–807 (2007).
Brentjens, R.J. et al. CD19-targeted T cells rapidly induce molecular remissions in adults with chemotherapy-refractory acute lymphoblastic leukemia. Sci. Transl. Med. 5, 177ra38 (2013).
Hoffmann, P. et al. Serial killing of tumor cells by cytotoxic T cells redirected with a CD19-/CD3-bispecific single-chain antibody construct. Int. J. Cancer 115, 98–104 (2005).
Mittendorf, E.A., Holmes, J.P., Ponniah, S. & Peoples, G.E. The E75 HER2/neu peptide vaccine. Cancer Immunol. Immunother. 57, 1511–1521 (2008).
Doubrovina, E. et al. Mapping of novel peptides of WT-1 and presenting HLA alleles that induce epitope-specific HLA-restricted T cells with cytotoxic activity against WT-1(+) leukemias. Blood 120, 1633–1646 (2012).
Pedersen, A.E. et al. Wild type p53-specific antibody and T-cell responses in cancer patients. J. Immunother. 34, 629–640 (2011).
Kessler, J.H. et al. Efficient identification of novel HLA-A(*)0201-presented cytotoxic T lymphocyte epitopes in the widely expressed tumor antigen PRAME by proteasome-mediated digestion analysis. J. Exp. Med. 193, 73–88 (2001).
Quintarelli, C. et al. High-avidity cytotoxic T lymphocytes specific for a new PRAME-derived peptide can target leukemic and leukemic-precursor cells. Blood 117, 3353–3362 (2011).
Dao, T. et al. Identification of a human cyclin D1-derived peptide that induces human cytotoxic CD4 T cells. PLoS One 4, e6730 (2009).
Peralbo, E., Alonso, C. & Solana, R. Invariant NKT and NK-like lymphocytes: Two different T cell subtypes that are differentially affected by aging. Exp. Gerontol. 42, 703–708 (2007).
Oka, Y. et al. WT1 peptide cancer vaccine for patients with hematopoietic malignancies and solid cancers. Scientific World Journal 7, 649–665 (2007).
Cheever, M.A. et al. The prioritization of cancer antigens: a national cancer institute pilot project for the acceleration of translational research. Clin. Cancer Res. 15, 5323–5337 (2009).
Chapuis, A.G. et al. Transferred WT1-reactive CD8+ T cells can mediate antileukemic activity and persist in post-transplant patients. Sci. Transl. Med. 5, 174ra27 (2013).
Sergeeva, A. et al. An anti-PR1/HLA-A2 T-cell receptor-like antibody mediates complement-dependent cytotoxicity against acute myeloid leukemia progenitor cells. Blood 117, 4262–4272 (2011).
Corbière, V. et al. Antigen spreading contributes to MAGE vaccination-induced regression of melanoma metastases. Cancer Res. 71, 1253–1262 (2011).
Hunder, N.N. et al. Treatment of metastatic melanoma with autologous CD4+ T cells against NY-ESO-1. N. Engl. J. Med. 358, 2698–2703 (2008).
Abès, R., Gélizé, E., Fridman, W.H. & Teillaud, J.L. Long-lasting antitumor protection by anti-CD20 antibody through cellular immune response. Blood 116, 926–934 (2010).
Selenko, N. et al. CD20 antibody (C2B8)-induced apoptosis of lymphoma cells promotes phagocytosis by dendritic cells and cross-priming of CD8+ cytotoxic T cells. Leukemia 15, 1619–1626 (2001).
Hilchey, S.P. et al. Rituximab immunotherapy results in the induction of a lymphoma idiotype-specific T-cell response in patients with follicular lymphoma: support for a “vaccinal effect” of rituximab. Blood 113, 3809–3812 (2009).
Ott, P.A., Hodi, F.S. & Robert, C. CTLA-4 and PD-1/PD-L1 blockade: new immunotherapeutic modalities with durable clinical benefit in melanoma patients. Clin. Cancer Res. 19, 5300–5309 (2013).
Tumeh, P.C. et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 515, 568–571 (2014).
Gubin, M.M. et al. Checkpoint blockade cancer immunotherapy targets tumour-specific mutant antigens. Nature 515, 577–581 (2014).
Tran, E. et al. Cancer immunotherapy based on mutation-specific CD4+ T cells in a patient with epithelial cancer. Science 344, 641–645 (2014).
Inoue, K. et al. Aberrant overexpression of the Wilms tumor gene (WT1) in human leukemia. Blood 89, 1405–1412 (1997).
Oka, Y. et al. WT1 peptide cancer vaccine for patients with hematopoietic malignancies and solid cancers. Scientific World Journal 7, 649–665 (2007).
Brischwein, K. et al. MT110: a novel bispecific single-chain antibody construct with high efficacy in eradicating established tumors. Mol. Immunol. 43, 1129–1143 (2006).
Fan, L. et al. Improving the efficiency of CHO cell line generation using glutamine synthetase gene knockout cells. Biotechnol. Bioeng. 109, 1007–1015 (2012).
Doubrovina, E. et al. Adoptive immunotherapy with unselected or EBV-specific T cells for biopsy-proven EBV+ lymphomas after allogeneic hematopoietic cell transplantation. Blood 119, 2644–2656 (2012).
Acknowledgements
The study was supported by US National Institutes of Health grant R01 CA 55349, P01 CA23766, MARF, P30 CA008748, Memorial Sloan Kettering Cancer Center's (MSKCC's) Experimental Therapeutics Center and the Lymphoma Foundation and Tudor and Glades funds. We thank D Levine, F. Dao and M. Mattar for their efforts and help in collecting clinical samples. We also thank the MSKCC Small-Animal Imaging Core Facility, R. Gejman for statistical analyses, T.-Y. Kuo for helpful discussions for Renilla transduction, A. Selvakumar and A. Yeh for their expert HLA typing.
Author information
Authors and Affiliations
Contributions
T.D., D.A.S. and R.J.O'R. designed the experiments, interpreted the data and wrote the manuscript. D.P., E.D. and M.D.d.M.G. participated in the design of some experiments. T.D., D.P., A.S., T.K., V.Z., N.V., L.D., M.C., V.P. and M.D.d.M.G. performed the experiments. Y.X., J.X., S.Y. and C.L. engineered T-BiTEs. D.A.S. is the principal investigator.
Corresponding author
Ethics declarations
Competing interests
T.D., L.D. and D.A.S. are inventors of technology described in this paper and licensed by Memorial Sloan Kettering Cancer Center to Novartis.
Supplementary information
Supplementary Text and Figures
Supplementary Figures 1–9 (PDF 1945 kb)
Rights and permissions
About this article

Cite this article
Dao, T., Pankov, D., Scott, A. et al. Therapeutic bispecific T-cell engager antibody targeting the intracellular oncoprotein WT1. Nat Biotechnol 33, 1079–1086 (2015). https://doi.org/10.1038/nbt.3349
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/nbt.3349
This article is cited by
-
T cell receptor therapeutics: immunological targeting of the intracellular cancer proteome
Nature Reviews Drug Discovery (2023)
-
Facile repurposing of peptide–MHC-restricted antibodies for cancer immunotherapy
Nature Biotechnology (2023)
-
Immunotherapy in hematologic malignancies: achievements, challenges and future prospects
Signal Transduction and Targeted Therapy (2023)
-
Dual targeting ovarian cancer by Muc16 CAR T cells secreting a bispecific T cell engager antibody for an intracellular tumor antigen WT1
Cancer Immunology, Immunotherapy (2023)
-
CD19-targeted BiTE expression by an oncolytic vaccinia virus significantly augments therapeutic efficacy against B-cell lymphoma
Blood Cancer Journal (2022)
