
Abstract
MicroRNAs (miRNAs) belong to a class of noncoding, regulatory RNAs that is involved in oncogenesis and shows remarkable tissue specificity. Their potential for tumor classification suggests they may be used in identifying the tissue in which cancers of unknown primary origin arose, a major clinical problem. We measured miRNA expression levels in 400 paraffin-embedded and fresh-frozen samples from 22 different tumor tissues and metastases. We used miRNA microarray data of 253 samples to construct a transparent classifier based on 48 miRNAs. Two-thirds of samples were classified with high confidence, with accuracy >90%. In an independent blinded test-set of 83 samples, overall high-confidence accuracy reached 89%. Classification accuracy reached 100% for most tissue classes, including 131 metastatic samples. We further validated the utility of the miRNA biomarkers by quantitative RT-PCR using 65 additional blinded test samples. Our findings demonstrate the effectiveness of miRNAs as biomarkers for tracing the tissue of origin of cancers of unknown primary origin.
Similar content being viewed by others


Main
Metastatic cancer of unknown primary origin accounts for 3–5% of all new cancer cases and is usually a very aggressive disease with poor prognosis1. The concept of cancer of unknown primary origin comes from the limitation of present methods to identify cancer origin. Recent studies revealed a high degree of variation in clinical management in the absence of evidence-based treatment for cancers of unknown primary origin2. Although many protocols have been evaluated3, they show relatively little benefit4. Determining the origin of tumor tissue is thus an important clinical application of molecular diagnostics5.
Molecular classification studies6 for tumor tissue origin6,7,8,9,10 have generally used classification algorithms that do not use domain-specific knowledge. All cancers were treated as equivalent, ignoring underlying similarities between tissue types with a common developmental origin. An exception of note is one study11 that was based on a pathology classification tree. These studies used machine-learning methods that average effects of biological features (e.g., mRNA expression levels), an approach that is more amenable to automated processing but does not use or generate mechanistic insights.
MiRNAs have emerged as highly tissue-specific biomarkers12,13,14, are postulated to play important roles in differentiation during development and have been tied to the development of specific malignancies15. MiRNAs appear as promising candidates for the construction of a biologically driven classification algorithm for identifying cancer tissue of origin. Previous studies16,17 have paved the way for miRNA-based cancer tissue classification.
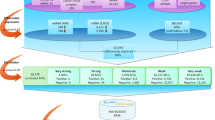
In this study, we construct an miRNA-based tissue classifier to identify the tissue origin of metastatic tumors. We developed an approach that assigns well-defined roles to individual miRNAs in classifying cancer tissue origin. We constructed the classification algorithm as a branched binary tree: in each node of the tree, classification proceeds to one of two possible branches, grouping together tissues with underlying similarities (Fig. 1). This process of coarse-to-fine specification mimics sequential processes of differentiation in embryonic development of tissues. The decision at each node is a simple binary decision that can be performed using the expression levels of a few miRNAs. This scheme is analogous to a pathologist's workup process, wherein a sample is assigned to increasingly finer subgroups through a series of differential diagnosis tests.

Each node is a binary decision between two sets of samples, those to the left and right of the node. A series of binary decisions, starting at node no. 1 and moving downwards, leads to one of the possible tumor types, which are the 'leaves' of the tree. A sample that is classified to the left branch at node no. 1 is assigned to the 'liver' class, otherwise it continues to node no. 2. Decisions are made at consecutive nodes using miRNA expression levels until an end-point (leaf of the tree) is reached, indicating the predicted class for this sample. For example, a sample that is classified as 'breast' must undergo the path through node nos. 1, 2, 3, 12, 16 and 17, taking the left branch at node nos. 3, 16 and 17 and the right branch at node nos. 1, 2 and 12, and no decision is needed at any of the other nodes. In specifying the tree structure, we combined clinico-pathological considerations with properties observed in the training set data. For example, thymus samples are separated into two groups according to their histological types, differing in the expression of epithelial-related miRNAs, ostensibly due to the higher proportion of lymphocytes in B2-type tumors. The first major division (node no. 3) separates tissues of epithelial origin from tissues of other or mixed origin, a biological difference that is reflected in their miRNA expression profiles, especially in expression of the miR-141/200 family. Thymus B2 tumors are grouped here with nonepithelial or mixed tissues (on the right branch) and are separated from these later ones (Supplementary Fig. 6). Liver and testis were placed first in the tree because these tissues contain highly specific expression of miRNAs (hsa-miR-122a and hsa-miR-372, respectively) that can be used to easily identify them, reducing interference later. Subsequent nodes recapitulated the separation of the gastrointestinal tract from other epithelial tissues (node no. 12) using miR-194 (ref. 33) and additional miRNAs (Fig. 2b). Lung carcinoid tumors, as opposed to other types of lung tumors, were found to have high expression of miR-194, which may be related to their distinct biological characteristics. These tumors are therefore grouped with the gastrointestinal tissues at node no. 12 and separated from them at node no. 13 using other miRNAs (Fig. 2a). Cancers of the esophagus differed substantially in the expression of miRNAs used for classification according to their histological types: gastroesophageal junction adenocarcinomas were similar to samples of stomach cancer34, whereas squamous samples had a strong similarity to the highly squamous head and neck cancers. Thus, the 'stomach*' class includes both stomach cancers and gastroesophageal junction adenocarcinomas; the 'head and neck*' class includes cancers of head and neck and squamous carcinoma of esophagus. GIST, gastrointestinal stromal tumors. Additional information such as patient gender or available clinical-pathological information is easy to incorporate into the tree by trimming leaves or branches (Table 2), without need for retraining.
Results
Samples and profiling
Because formalin-fixed paraffin-embedded (FFPE) archival samples are an important source for tumor material, we developed a method for extracting RNA from FFPE blocks that preserves the miRNA fraction. We compared RNA extracted from fresh-frozen, formalin-fixed or FFPE samples, and demonstrated that the RNA quantity and quality was similar for all preservation methods (Supplementary Fig. 1 online). Furthermore, the miRNA profile was stable in FFPE samples stored for as long as 11 years (Supplementary Fig. 2 online).
MiRNA profiling was performed on miRNA microarrays18 (Supplementary Fig. 3 online), containing probes for more than 600 miRNAs19 including all the human miRNAs in the 9th version of miRBase20.
We collected and profiled 333 FFPE samples and 3 fresh-frozen samples, including 205 primary tumors and 131 metastatic tumors, representing 22 different tumor origins or 'classes' (Table 1 and Supplementary Table 1 online). Tumor percentage (area in section) was at least 50% for >90% of the samples. Eighty-three of the samples (∼25% of each class) were randomly selected as a blinded test set. Sixty-five additional primary tumor samples (53 FFPE and 12 fresh-frozen samples, Supplementary Table 2 online) were profiled only by qRT-PCR to validate the selected miRNAs. Overall, 401 samples are included in this study.
Comparison of primary and metastatic tumors
Owing to the difficulty of obtaining sufficient numbers of metastatic samples, this and previous studies7,8,9,10,11,16 have relied on primary tumors to augment the sample set. Differences in expression profiles between primary and metastatic samples can be expected because of underlying biological differences in the tumors, or because of contamination from neighboring tissues. These effects, which were not generally considered in previous studies, can hinder the performance of tumor classifiers on metastatic samples.
For most cancers, such as breast or colon cancer (Supplementary Fig. 4a,b online), we found no significant differences between primary and metastatic tumors (Fig. 2a,b). In other cases, a small set of miRNAs were differentially expressed. For example, in primary tumor samples of the stomach compared to samples of stomach metastases to the lymph node, three miRNAs were significantly differentially expressed (P < 0.001, Supplementary Fig. 4c,d online). Hsa-miR-143, characteristic of epithelial layers12, and hsa-miR-133a, which is characteristic of muscle tissue13, were overexpressed in the primary tumors taken from the stomach; in contrast, hsa-miR-150, which was previously identified as highly expressed in lymphocytes21, was present at higher levels in the metastatic samples taken from lymph nodes. In addition, samples from primary tumors such as prostate or head and neck, which often contain surrounding muscle tissue, showed high expression levels of miR-1, miR-206 and miR-133a, miRNAs that are specific to skeletal muscle13. We concluded that primary tumors can be used in training a classifier for metastases, but must be used with care and with attention to specific markers and to context. To reduce potential biases from these effects, we minimized the use of miRNAs in nodes where cross-contamination may have confounding effects—specifically, we avoided the use of muscle-related miRNAs (miR-1/133/206) and hsa-miR-150.

(a) When training a decision algorithm for a given node, only those sample classes that are possible outcomes ('leaves') of this node are used for training. At node no. 13 (see Fig. 1), lung-carcinoid tumors (green triangles, 7 samples) are easily separated from tumors of gastrointestinal origin (blue and empty squares, 49 samples) using the expression levels of hsa-miR-21 and hsa-let-7e (with one outlier). Other samples that branch out earlier in the tree and are not well separated by these miRNAs (orange circles, 283 samples) are not considered. Notably, metastatic samples of gastrointestinal origin (empty squares, 23 samples) are distributed with the primary tumors. The solid line indicates the values of hsa-miR-21 and hsa-let-7e for which the logistic regression model of node no. 13 assigns a probability P = 0.5 (Supplementary Table 3). Points below the line are assigned a probability P > 0.5 and take the left branch (to node no. 14); points above the line take the right branch and are classified as lung-carcinoid. (b) Expression levels of hsa-miR-194, hsa-miR-145 and hsa-miR-205 at node no. 12 in the tree (Fig. 1). These miRNAs can be used to separate between the left branch of node no. 12 (blue squares, 56 samples, empty squares show metastatic samples), that is, samples from the stomach, pancreas, colon or lung-carcinoid, and other epithelial samples in the right branch of node no. 12 (green triangles, 152 samples, empty triangles show metastatic samples). (c) Validation of the miRNAs used in node no. 1 (Table 2) by qRT-PCR: liver (blue squares, 9 samples) and nonliver samples (green triangles, 71 samples) are easily separated using hsa-miR-122a and hsa-miR-141 with one outlier (Supplementary Fig. 7a,b). The signal shown for each sample is the difference in cycle threshold (Ct) between U6 and the miRNA. A larger difference means higher expression of this miRNA. Liver tumors have higher expression of hsa-miR-122a and lower expression of hsa-miR-141 (Supplementary Table 2). Line indicates the decision threshold of the logistic regression (Supplementary Fig. 7a,b). (d) Validation of the miRNAs used in node no. 12 (Table 2) by qRT-PCR: samples of gastrointestinal tumors (blue squares, 13 samples) show distinct expression levels (Supplementary Fig. 7c,d) of hsa-miR-145, hsa-miR-194 and hsa-miR-205 compared to other epithelial tumors (green triangles, 52 samples). The results obtained by qRT-PCR are very similar to those obtained by the microarray platform at this node (b and Supplementary Fig. 7d) and show similar distributions.
Decision-tree classification algorithm
We built a tumor classifier using the miRNA expression levels by applying a binary tree classification scheme (Fig. 1). This framework is set up to utilize the potential specificity of miRNAs in tissue differentiation and embryogenesis: different miRNAs may be involved in various stages of tissue specification22,23,24 and are used by the algorithm at different decision points or 'nodes'. The tree breaks up the complex multi-tissue classification problem into a set of simpler binary decisions. At each node, classes which branch out earlier in the tree are not considered, reducing interference from irrelevant samples and further simplifying the decision (Fig. 2a). The decision at each node can then be accomplished using only a small number of miRNA biomarkers, which have well-defined roles in the classification (Table 2 and Supplementary Table 3 online).
The structure of the binary tree was based on a hierarchy of tissue development and morphological similarity11, which was modified by prominent features of the miRNA expression patterns (Fig. 1). For example, the expression patterns of miRNAs indicated a significant difference between lung carcinoid and other lung cancer types (P < 10−10 for hsa-miR-194), and these are therefore separated at node no. 12 (Fig. 2a,b) into separate leaves (Fig. 1). Interestingly, an automated algorithm for dividing the data into a binary classification tree generated trees with a similar structure, yet lacked flexibility in structure and in individual node classifiers and resulted in substantially poorer performance (Supplementary Fig. 5 online).
For each of the individual nodes we used logistic regression models, a robust family of classifiers that are frequently used in epidemiological and clinical studies to combine continuous data features into a binary decision (Fig. 2a and Supplementary Fig. 6 online). Because gene expression classifiers have an inherent redundancy in selecting gene features25, we used bootstrapping on the training sample set as a method to select a stable miRNA set for each node. This resulted in a small number (usually 2–3) of miRNA features per node, totaling 48 miRNAs for the full classifier (Table 2 and Supplementary Table 3). Some of these miRNAs were previously identified in similar contexts (Supplementary Table 4 online).
Cross validation and high-confidence classifications
As a first step, we tested the performance of the classifier using leave-one-out cross validation (LOOCV) within the training set. LOOCV simulates the performance of a classification algorithm on unseen samples. In LOOCV the algorithm is repeatedly retrained, leaving out one sample in each round, and testing each sample on a classifier that was trained without this sample (Supplementary Table 1). The decision-tree algorithm reached an average sensitivity, or accuracy, of 78% and specificity of 99%, with notable variation between different classes (Supplementary Table 5 online). We compared the performance to that of the commonly-used K-nearest-neighbors (KNN) classification algorithm8,11,16. The KNN algorithm (at the optimal k = 3) showed poorer performance than the tree (71% accuracy), with different classes having large differences in sensitivity between the algorithms (Supplementary Table 5, root mean square difference 25%).
In clinical practice it is often useful to assess information of different degrees of confidence10,11. In the diagnosis of cancers of unknown primary origin, in particular, a short list of highly probable possibilities is a practical option when no definite diagnosis can be made. Because the decision-tree and the KNN algorithms are designed differently and trained independently, improved accuracy and greater confidence can be obtained by combining and comparing their classifications. The union of the predictions made by the two algorithms included the correct class in 85% of the cases. In 69% of the cases the two algorithms agreed, generating a single, high-confidence prediction. In 93% of these high-confidence predictions the correct class of the sample was accurately identified, with more than half of the 22 tumor classes reaching 100% sensitivity (Supplementary Table 5).
Classifier performance: independent blinded test-set
The most important test of a classification algorithm is on a blinded test-set. We set aside approximately one-quarter of the samples, randomly selected to represent the different classes, as an independent test set, and tested the performance of the classifiers (Table 3). The performance on the test set did not decrease compared to the performance of LOOCV in the training set (Supplementary Table 5), indicating that the classifier is robust and not over-fit. Eighty-six percent of the cases were accurately predicted by the union of the two predictors (most classes had 100% sensitivity). Among high-confidence predictions, which were two-thirds of the cases, 89% were accurately classified. Even in the blinded test-set, 16 of the 22 classes had 100% accuracy in the high-confidence predictions. Finally, we focused on the performance of the classification on the metastatic samples within the blinded test-set. Here, too, the classifier reached 85% sensitivity for high-confidence classifications. The fact that the performance on the blinded metastatic samples reached these levels of accuracy supports the approach of augmenting the training set with primary tumors when concomitantly avoiding potentially confounding markers.
Validation by quantitative RT-PCR platform
The above decision-tree algorithm, which was developed based on an array platform, assigns specific roles to miRNAs in binary decisions between groups of tissues. To rule out effects of a specific platform, we validated the utility of a subset of these miRNAs on a high-sensitivity quantitative RT-PCR platform, using 15 of the original samples plus 65 independent samples (Supplementary Table 2). Even when using a different platform on new samples, the miRNAs maintained their expression distributions and their diagnostic roles (Fig. 2c,d) and could be used for accurate classification (Supplementary Fig. 7 online).
Discussion
Gene expression profiles have recently become a basis for diagnostic, prognostic and predictive information26,27, and for classification of human cancers6. These are particularly important for the diagnosis of cancers of unknown primary origin, which account for 3–5% of all new cancer cases in the United States5. Gene expression signatures of mRNA expression levels have been used for development of molecular classification algorithms to trace tumor origin6,7,8,9,10,11. The 'black-box' support vector machine algorithm6, with >16,000 genes, reached an overall accuracy of 78% in 14 cancer classes. However, the performance of this classifier was not robust and it could not correctly identify poorly differentiated tumors. The use of the large number of data features led to some degree of over-fitting of the classifier, which did not focus on informative genes and was strongly affected by noise or irrelevant variation in gene expression. Furthermore, the design of the algorithm and the large number of genes used made it difficult to extract gene-specific biological information or to make incremental advances to this classifier. Subsequent efforts therefore aimed to use fewer features. These studies generally started with the analysis of tens of thousands of genes, followed by selection of a subset of potential biomarkers.
A pathology-motivated tree reduced the number of mRNAs analyzed, but still required 250 genes to reach accuracy of 83% when classifying up to 14 distinct cancer classes11. The number of mRNAs used could be reduced below 100, but this resulted in a decrease in accuracy below 80%. One group of researchers classified 13 classes with accuracy near 90%, but required ∼600 mRNAs for the task10. They were able to use <100 genes when classifying only five cancer origins. Another group classified 21 cancer classes (from 15 tissue types) with an accuracy of 85% or more using >400 genes, but the accuracy decreased sharply for fewer genes7. These repeated efforts suggest a trade-off between accuracy of classification, number of classes compared and the number of mRNA genes used. The limited sample-sets available for such studies make it difficult to distinguish small sets of informative genes from noise or natural variation owing to the multiple comparisons problem, especially when the initial data set contains tens of thousands of irrelevant genes. Researchers who focused intensively on the issue of feature selection, and included a large training set of nearly 500 samples, were able to substantially outperform these studies, reaching accuracy of ∼90% on a broad spectrum of >30 classes (from 26 tissue origins) using a panel of 92 mRNAs8. This list of genes is probably strongly enriched for tissue-specific genes compared to their initial data set of 22,000 genes. However, all these classifiers used multi-feature algorithms that average effects of biomarkers and provide little insight into the mechanistic or diagnostic role of any individual gene.
MiRNAs possess several features that make them attractive diagnostic biomarkers. MiRNAs are upstream regulators that can target large numbers of protein-coding genes. Unlike measurements of mRNA, which must be translated to protein to have a biological effect, miRNA expression levels represent more closely the functional level of the gene. An added benefit is that emerging miRNA markers can be tested for biological or therapeutic effects by generalized sequence-based methods. Notably, miRNAs show improved stability and maintain their expression profiles in archival FFPE samples28 (Supplementary Figs. 1 and 2). One of the major characteristics of miRNAs is their marked tissue specificity and involvement in organ development16,22,23,24. We thus postulated that a data set of miRNA expression levels would be enriched for tissue-specific markers, and would provide a fruitful starting point for the development of a tissue-of-origin classifier. Our initial data set consisted of the expression levels of several hundred miRNAs, compared to the tens of thousands of protein-coding genes used in other studies. The decision tree we described here performs a systematic search for classification decisions in which the specificity of individual miRNAs may be important. Our classifier used only 48 miRNA markers to reach an overall accuracy of ∼90% among 22 tissue origins, on blinded test samples and on more than 130 metastases. This effort compares favorably with the best result so far using mRNA expression levels8 and will probably continue to improve as larger sample sets are collected and profiled for expression of miRNAs.
The decision-tree classifier follows a diagnostic workup plan for each sample that is based on biological differences. Because a large fraction of the miRNAs used in our classifier are hypothesized to be involved in tissue specification, the classification errors often point to neighboring or related tissues: colon misclassifications pointed to other digestive system organs (pancreas or stomach), whereas female reproductive-system organs (ovary, endometrium and breast) were relatively frequently intermixed, as previously observed11. The tissue of origin that showed the consistently poorest performance, that is, that was most often misclassified, was bladder (Table 3). The most common error was misclassification as lung cancer (Supplementary Table 1), a misclassification that occurs in pathology practice and is further complicated by overlap in immunopositivity of lung and bladder cancer subtypes29. This is likely related to the small number of samples of bladder origin in our study (N = 6).
The roles of specific miRNAs in our classifier are in agreement with previous findings (Supplementary Table 4) but also point to possible new roles and contribute to a broader picture of miRNA function. Our results also suggest that each node in the tree may be used as an independent differential diagnosis tool, for example in the identification of different types of lung cancer (Figs. 1 and 2a,b). The performance of the classifier with a small number of miRNAs highlights the utility of miRNAs as tissue-specific cancer biomarkers and provides an effective means to determine the tissue origin of cancers of unknown primary origin.
Methods
Tumor samples.
Tumor samples were obtained from several sources (Sheba Medical Center, Tel-Hashomer, Israel; Soroka University Medical Center, Beer Sheva, Israel; Beilinson Hospital, Rabin Medical Center, Petah-Tikva, Israel; ABS Inc., Wilmington, Delaware, USA; Tel Aviv Sourasky Medical Center, Tel Aviv, Israel; Bnai-Zion Medical Center, Haifa, Israel; Seoul National University College of Medicine, Seoul, South Korea; Indivumed GmbH, Hamburg, Germany). Institutional review approvals were obtained for all samples in accordance with each institute's institutional review board or IRB-equivalent guidelines. For FFPE samples, initial diagnosis, histological type, grade and tumor percentages were determined by a pathologist on hematoxylin-eosin–stained slides, performed on the first and/or last sections of the sample. Samples included primary tumors, metastatic tumors and two samples of benign prostatic hyperplasia samples (BPH) that showed similar expression profile to prostate tumor samples (not shown). Nondefined samples were not included in this study. Tumor content in 90% of the FFPE samples was >50%.
RNA extraction.
For frozen tissue, a sample ∼0.5 cm3 in dimension was used for RNA extraction. Total RNA was extracted using the miRvana miRNA isolation kit (Ambion) according to the manufacturer's instructions. Briefly, the sample was homogenized in a denaturing lysis solution followed by an acid-phenol:chloroform extraction. Finally, the sample was purified on a glass-fiber filter.
For FFPE samples, total RNA was isolated from seven to ten 10-μm-thick tissue sections using the miRdictorTM extraction protocol developed at Rosetta Genomics. Briefly, the sample was incubated a few times in Xylene at 57 °C to remove paraffin excess, followed by ethanol washes. Proteins were degraded by proteinase K solution at 45 °C for a few hours. The RNA was extracted with acid phenol:chloroform followed by ethanol precipitation and DNAse digestion. Total RNA quantity and quality were checked by spectrophotometer (Nanodrop ND-1000).
miRdicator array platform.
Custom microarrays were produced by printing DNA oligonucleotide probes representing >600 human miRNAs. Each probe, printed in triplicate, carried up to 22-nt linker at the 3′ end of the miRNA's complement sequence in addition to an amine group used to couple the probes to coated glass slides. 20 μM of each probe were dissolved in 2 × SSC plus 0.0035% SDS and spotted in triplicate on Schott Nexterion Slide E coated microarray slides using a Genomic Solutions BioRobotics MicroGrid II according to the MicroGrid manufacturer's directions. Fifty-four negative control probes were designed using the sense sequences of different miRNAs. Two groups of positive control probes were designed to hybridize to miRdicator array: (i) synthetic small RNA were spiked to the RNA before labeling to verify the labeling efficiency and (ii) probes for abundant small RNA (e.g., small nuclear RNAs (U43, U49, U24, Z30, U6, U48, U44), 5.8s and 5s ribosomal RNA) were spotted on the array to verify RNA quality. The slides were blocked in a solution containing 50 mM ethanolamine, 1 M Tris (pH 9.0) and 0.1% SDS for 20 min at 50 °C, then thoroughly rinsed with water and spun dry.
Cy-dye labeling of miRNA for miRdicator array.
Five μg of total RNA were labeled by ligation30 of an RNA-linker, p-rCrU-Cy/dye (Dharmacon), to the 3′ end with Cy3 or Cy5. The labeling reaction contained total RNA, spikes (0.1–20 fmoles), 300 ng RNA-linker-dye, 15% DMSO, 1 × ligase buffer and 20 units of T4 RNA ligase (NEB) and proceeded at 4 °C for 1 h followed by 1 h at 37 °C. The labeled RNA was mixed with 3 × hybridization buffer (Ambion), heated to 95 °C for 3 min and then added on top of the miRdicator array. Slides were hybridized 12–16 h in 42 °C, followed by two washes in room temperature (25 °C) with 1 × SSC and 0.2% SDS and a final wash with 0.1 × SSC.
Arrays were scanned using an Agilent Microarray Scanner Bundle G2565BA (resolution of 10 μm at 100% power). Array images were analyzed using SpotReader software (Niles Scientific).
Array signal calculation and normalization.
Triplicate spots were combined to produce one signal for each probe by taking the logarithmic mean of reliable spots. All data was log-transformed (natural base) and the analysis was performed in log-space. A reference data vector for normalization R was calculated by taking the median expression level for each probe across all samples. For each sample data vector S, a 2nd degree polynomial F was found so as to provide the best fit between the sample data and the reference data, such that R ≈ F(S). Remote data points (outliers) were not used for fitting the polynomial F. For each probe in the sample (element Si in the vector S), the normalized value (in log-space) Mi is calculated from the initial value Si by transforming it with the polynomial function F, so that Mi = F(Si). Data in Supplementary Table 1 and in Figure 2a,b was translated back to linear-space (by taking the exponent). Using only the training set samples to generate the reference data vector did not affect the results.
Logistic regression.
The aim of a logistic regression model is to use several features, such as expression levels of several miRNAs, to assign a probability of belonging to one of two possible groups, such as two branches of a node in a binary decision-tree. Logistic regression models the natural log of the odds ratio, that is, the ratio of the probability of belonging to the first group (P) over the probability of belonging to the second group (1–P), as a linear combination of the different expression levels (in log-space). The logistic regression assumes that

where β0 is the bias, Mi is the expression level (normalized, in log-space) of the ith miRNA used in the decision node, and βi is its corresponding coefficient. βi > 0 indicates that the probability to take the left branch (P) increases when the expression level of this miRNA (Mi) increases, and the opposite for βi < 0. If a node uses only a single miRNA (M), then solving for P results in (Supplementary Fig. 6):

The regression error on each sample is the difference between the assigned probability P and the true 'probability' of this sample, that is, 1 if this sample is in the left branch group and 0 otherwise. The training and optimization of the logistic regression model calculates the parameters β, and the p-values (for each miRNA by the Wald statistic and for the overall model by the χ2 difference), maximizing the likelihood of the data given the model and minimizing the total regression error

The probability output of the logistic model is converted here to a binary decision by comparing P to a threshold, denoted by PTH, that is, if P > PTH then the sample belongs to the left branch ('first group') and vice versa. Choosing at each node the branch that has a P > 0.5, that is, using a probability threshold of 0.5, leads to a minimization of the sum of the regression errors. However, as our goal was the minimization of the overall number of misclassifications (and not of their probability), we used a modification that adjusts the probability threshold (PTH) to minimize the overall number of mistakes at each node. For each node we optimize the threshold to a new probability threshold PTH, such that the number of classification errors is minimized (Supplementary Table 3). Note that this change of probability threshold is equivalent (in terms of classifications) to a modification of the bias β0, which may reflect a change in the prior frequencies of the classes.
Stepwise logistic regression and feature selection.
The original data contain the expression levels of hundreds of miRNAs for each sample, that is, hundreds of data features. In training the classifier for each node, we selected and used only a small subset of these features for optimizing a logistic regression model. In the initial training this was done using a forward stepwise scheme. The features were sorted in order of decreasing log-likelihoods, and the logistic model was started off and optimized with the first feature. The second feature was then added, and the model re-optimized. The regression error of the two models was compared: if the addition of the feature did not provide a significant advantage (χ2 < 7.88, P = 0.005), the new feature was discarded. Otherwise, the added feature was kept. Adding a new feature may make a previous feature redundant (e.g., if they are very highly correlated). To check for this, the process iteratively checks if the feature with the lowest likelihood can be discarded (without losing χ2 difference as above). After ensuring that the current set of features is compact in this sense, the process continues to test the next feature in the sorted list, until features are exhausted. No limitation on the number of features was inserted into the algorithm but in most cases two to three features were selected.
The stepwise logistic regression method was used on subsets of the training set samples by resampling the training set with repetition ('bootstrap') so that each of the 23 runs contained about two-thirds of the samples at least once, and any one sample had >99% chance of being left out at least once. This resulted in an average of ∼2–3 features per node (∼4–8 in more difficult nodes). We selected a robust set of ∼2–3 features per node (Table 2) by comparing features that were repeatedly chosen in the bootstrap sets to previous evidence (Supplementary Table 4) and considering their signal strengths and reliability. To further reduce possible biases from tissue contamination, miRNAs that were specifically high in one tissue (e.g., hsa-miR-145 in gastrointestinal tissues or hsa-miR-122a in liver) were balanced where possible by miRNAs that have an inverse specificity (e.g., hsa-miR-205, which is low in gastric tissues or hsa-miR-141/200c, which is weakly expressed in liver, Fig. 2). When using these selected features to construct the classifier, the stepwise process was not used and the training optimized the logistic regression model parameters only (Supplementary Table 3).
Restriction of classes by gender and liver metastases.
The decision-tree framework allows easy implementation of available clinical information into the classification (Table 2). We used two such data: gender, and liver metastases. Samples from female patients were not allowed to be classified as originating from testis or prostate; thus, samples of female patients that reached node no. 2 were automatically classified to the right branch, and likewise the left branch (= breast) at node no. 17. Samples from male patients were not allowed to be classified as originating from endometrium or ovary and were automatically classified to the left branch at node 20. Samples that were indicated as liver metastases were not allowed to be classified as originating from liver tissue and were classified to the right branch in node no. 1. Thus, additional information is easily used without loss of generality or need to retrain the classifier.
K-nearest-neighbors (KNN) classification algorithm.
The KNN algorithm calculated the distance (Pearson correlation) of any sample to all samples in the training set and classified the sample by the majority vote of the k samples that are most similar (k being a parameter of the classifier). The correlation was calculated on a predefined set of miRNAs (data features), selected by going over all pairs of tissue types (classes) and collecting miRNAs that were significantly differentially expressed between any two classes. Using only the intersection of this list with the 48 miRNAs that were used by the decision tree did not reduce the performance, highlighting the information content of these miRNAs. KNN algorithms with k = 1,3,5 were compared, and the optimal performer was selected, using k = 3 and the smaller set of miRNAs.
qRT-PCR.
One microgram of total RNA was subjected to polyadenylation reaction as described before31. Briefly, RNA was incubated in the presence of poly (A) polymerase (PAP) (Takara-2180A), MnCl2, and ATP for 1 h at 37 °C. Reverse transcription was performed on the poly-adenylated product. An oligo-dT primer harboring a consensus sequence (complementary to the reverse primer) was used for reverse transcription reaction. The primer is first annealed to the poly A–RNA and then subjected to a reverse transcription reaction of SuperScript II RT (Invitrogen). The cDNA was then amplified by real-time PCR reaction, using a miRNA-specific forward primer, TaqMan probe and universal reverse primer. The reactions were incubated for 10 min at 95 °C followed by 42 cycles of 95 °C for 15 s and 600 °C for 1 min. Supplementary Table 2 shows raw signal threshold (Ct) values.
Figure 2c shows data normalized to U6 snRNA32. Data in Figure 2d were normalized by U6, transformed to linear space (by the exponent base 2), and multiplied by a constant (59,000) to shift numeric values to have the same median value as the array signals. Comparing the distributions of the three miRNAs in the two separate sample subsets (six groups in all) between the microarray and the qRT-PCR data, we obtained a mean Kolmogorov-Smirnov statistic of 0.32. Only two (of the six) groups had significantly different distributions (KS-statistic < 0.05); most groups were not significantly different by the Kolmogorov-Smirnov test.
Note: Supplementary information is available on the Nature Biotechnology website.
References
Pimiento, J.M., Teso, D., Malkan, A., Dudrick, S.J. & Palesty, J.A. Cancer of unknown primary origin: a decade of experience in a community-based hospital. Am. J. Surg. 194, 833–7, discussion 837–8 (2007).
Shaw, P.H., Adams, R., Jordan, C. & Crosby, T.D. A clinical review of the investigation and management of carcinoma of unknown primary in a single cancer network. Clin. Oncol. (R. Coll. Radiol.) 19, 87–95 (2007).
Hainsworth, J.D. & Greco, F.A. Treatment of patients with cancer of an unknown primary site. N. Engl. J. Med. 329, 257–263 (1993).
Blaszyk, H., Hartmann, A. & Bjornsson, J. Cancer of unknown primary: clinicopathologic correlations. APMIS 111, 1089–1094 (2003).
Varadhachary, G.R., Abbruzzese, J.L. & Lenzi, R. Diagnostic strategies for unknown primary cancer. Cancer 100, 1776–1785 (2004).
Ramaswamy, S. et al. Multiclass cancer diagnosis using tumor gene expression signatures. Proc. Natl. Acad. Sci. USA 98, 15149–15154 (2001).
Bloom, G. et al. Multi-platform, multi-site, microarray-based human tumor classification. Am. J. Pathol. 164, 9–16 (2004).
Ma, X.J. et al. Molecular classification of human cancers using a 92-gene real-time quantitative polymerase chain reaction assay. Arch. Pathol. Lab. Med. 130, 465–473 (2006).
Talantov, D. et al. A quantitative reverse transcriptase-polymerase chain reaction assay to identify metastatic carcinoma tissue of origin. J. Mol. Diagn. 8, 320–329 (2006).
Tothill, R.W. et al. An expression-based site of origin diagnostic method designed for clinical application to cancer of unknown origin. Cancer Res. 65, 4031–4040 (2005).
Shedden, K.A. et al. Accurate molecular classification of human cancers based on gene expression using a simple classifier with a pathological tree-based framework. Am. J. Pathol. 163, 1985–1995 (2003).
Baskerville, S. & Bartel, D.P. Microarray profiling of microRNAs reveals frequent coexpression with neighboring miRNAs and host genes. RNA 11, 241–247 (2005).
Farh, K.K. et al. The widespread impact of mammalian microRNAs on mRNA repression and evolution. Science 310, 1817–1821 (2005).
Landgraf, P. et al. A Mammalian microRNA Expression Atlas Based on Small RNA Library Sequencing. Cell 129, 1401–1414 (2007).
He, L. et al. A microRNA polycistron as a potential human oncogene. Nature 435, 828–833 (2005).
Lu, J. et al. MicroRNA expression profiles classify human cancers. Nature 435, 834–838 (2005).
Volinia, S. et al. A microRNA expression signature of human solid tumors defines cancer gene targets. Proc. Natl. Acad. Sci. USA 103, 2257–2261 (2006).
Raver-Shapira, N. et al. Transcriptional activation of miR-34a contributes to p53-mediated apoptosis. Mol. Cell 26, 731–743 (2007).
Bentwich, I. et al. Identification of hundreds of conserved and nonconserved human microRNAs. Nat. Genet. 37, 766–770 (2005).
Griffiths-Jones, S., Grocock, R.J., van Dongen, S., Bateman, A. & Enright, A.J. miRBase: microRNA sequences, targets and gene nomenclature. Nucleic Acids Res. 34, D140–D144 (2006).
Xiao, C. et al. MiR-150 controls B cell differentiation by targeting the transcription factor c-Myb. Cell 131, 146–159 (2007).
Hornstein, E. et al. The microRNA miR-196 acts upstream of Hoxb8 and Shh in limb development. Nature 438, 671–674 (2005).
Lee, Y.S., Kim, H.K., Chung, S., Kim, K.S. & Dutta, A. Depletion of human micro-RNA miR-125b reveals that it is critical for the proliferation of differentiated cells but not for the down-regulation of putative targets during differentiation. J. Biol. Chem. 280, 16635–16641 (2005).
Sempere, L.F. et al. Expression profiling of mammalian microRNAs uncovers a subset of brain-expressed microRNAs with possible roles in murine and human neuronal differentiation. Genome Biol. 5, R13 (2004).
Ein-Dor, L., Kela, I., Getz, G., Givol, D. & Domany, E. Outcome signature genes in breast cancer: is there a unique set? Bioinformatics 21, 171–178 (2005).
Paik, S. et al. Gene expression and benefit of chemotherapy in women with node-negative, estrogen receptor-positive breast cancer. J. Clin. Oncol. 24, 3726–3734 (2006).
van de Vijver, M.J. et al. A gene-expression signature as a predictor of survival in breast cancer. N. Engl. J. Med. 347, 1999–2009 (2002).
Li, J. et al. Comparison of miRNA expression patterns using total RNA extracted from matched samples of formalin-fixed paraffin-embedded (FFPE) cells and snap frozen cells. BMC Biotechnol. 7, 36 (2007).
Parker, D.C. et al. Potential utility of uroplakin III, thrombomodulin, high molecular weight cytokeratin, and cytokeratin 20 in noninvasive, invasive, and metastatic urothelial (transitional cell) carcinomas. Am. J. Surg. Pathol. 27, 1–10 (2003).
Thomson, J.M., Parker, J., Perou, C.M. & Hammond, S.M. A custom microarray platform for analysis of microRNA gene expression. Nat. Methods 1, 47–53 (2004).
Shi, R. & Chiang, V.L. Facile means for quantifying microRNA expression by real-time PCR. Biotechniques 39, 519–525 (2005).
Thomson, J.M. et al. Extensive post-transcriptional regulation of microRNAs and its implications for cancer. Genes Dev. 20, 2202–2207 (2006).
Hino, K., Fukao, T. & Watanabe, M. Regulatory interaction of HNF1α to microRNA194 gene during intestinal epithelial cell differentiation. Nucleic Acids Symp. Ser. (Oxf.), 415–416 (2007).
van Duin, M. et al. High-resolution array comparative genomic hybridization of chromosome 8q: evaluation of putative progression markers for gastroesophageal junction adenocarcinomas. Cytogenet. Genome Res. 118, 130–137 (2007).
Acknowledgements
We thank Jung-Hwan Yoon of Seoul National University College of Medicine, Seoul, South Korea. N.R. dedicates this work to the memory of Yasha (Yaakov) Rosenfeld.
Author information
Authors and Affiliations
Contributions
R.A., A.A., I. Bentwich, Z.B., D.C., A.C. and I. Barshack directed research; N.R., R.A., E.M., S.R., Y.S., S.G., A.C. and I. Barshack designed experiments; N.S.-V., A.T., M.F., O.K., O.N., D.N., M.P., A.Y., B.S., S.P.-C., E.F. and I. Barshack provided samples and performed pathological analysis; E.M., M.Z., N.S., S.T., D.L. and S.G. performed experiments; N.R., R.A., S.R., Y.G. and E.S. developed algorithms; N.R., S.R., H.B. and Y.G. analyzed data; Y.S., A.L., N.T. and A.B.-A. provided bioinformatic and database support; N.R., R.A., A.C. and I. Barschack wrote the paper.
Corresponding authors
Ethics declarations
Competing interests
All authors affiliated with Rosetta Genomics, except E.S., are full-time employees of Rosetta Genomics Ltd. and hold equity in the company, the value of which may be influenced by this publication. E.S. was engaged as an external consultant to Rosetta Genomics. O.N. is a paid consultant to Rosetta Genomics. All other authors declare that they have no competing interests.
Supplementary information
Supplementary Text and Figures
Supplementary Figures 1–7, Table 4 (PDF 515 kb)
Supplementary Table
Supplementary Table 1 (XLS 186 kb)
Supplementary Table
Supplementary Table 2 (XLS 25 kb)
Supplementary Table
Supplementary Table 3 (XLS 21 kb)
Supplementary Table
Supplementary Table 5 (XLS 29 kb)
Rights and permissions
About this article
Cite this article
Rosenfeld, N., Aharonov, R., Meiri, E. et al. MicroRNAs accurately identify cancer tissue origin. Nat Biotechnol 26, 462–469 (2008). https://doi.org/10.1038/nbt1392
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/nbt1392
This article is cited by
-
Prognostic value of plasma microRNAs for non-small cell lung cancer based on data mining models
BMC Cancer (2024)
-
Role of microRNA-363 during tumor progression and invasion
Journal of Physiology and Biochemistry (2024)
-
RNA therapy
Experimental & Molecular Medicine (2023)
-
Expression analysis of circulating miR-22, miR-122, miR-217 and miR-367 as promising biomarkers of acute lymphoblastic leukemia
Molecular Biology Reports (2023)
-
A novel microRNA signature for the detection of melanoma by liquid biopsy
Journal of Translational Medicine (2022)
