
Abstract
Merlin, the protein product of the Neurofibromatosis type-2 gene, acts as a tumour suppressor in mice and humans. Merlin is an adaptor protein with a FERM domain and it is thought to transduce a growth-regulatory signal. However, the pathway through which Merlin acts as a tumour suppressor is poorly understood. Merlin, and its function as a negative regulator of growth, is conserved in Drosophila, where it functions with Expanded, a related FERM domain protein. Here, we show that Drosophila Merlin and Expanded are components of the Hippo signalling pathway, an emerging tumour-suppressor pathway. We find that Merlin and Expanded, similar to other components of the Hippo pathway, are required for proliferation arrest and apoptosis in developing imaginal discs. Our genetic and biochemical data place Merlin and Expanded upstream of Hippo and identify a pathway through which they act as tumour-suppressor genes.
Similar content being viewed by others

Main
During normal development, the number of cells in growing tissues is controlled by regulating the generation of new cells through cell proliferation and by apoptosis, which eliminates excess or damaged cells that may be harmful to the organism1,2. Deregulation of these processes can lead to tumour formation3. Recently, a novel tumour-suppressor pathway that coordinately regulates cell proliferation and apoptosis was identified via genetic studies in Drosophila4,5. It is composed of the serine/threonine kinases Hippo (Hpo) and Warts (Wts); the adaptor molecule Salvador (Sav); Mats (Mob as tumour suppressor), a regulator of Wts; and Yorkie (Yki), a transcriptional coactivator6,7,8,9,10,11,12,13,14,15,16. Mutations in Hpo, Sav, Wts, Mats or overexpression of Yki result in overgrown tissues containing excess cells. Extra cells in these mutants are generated by deregulated cell proliferation and resistance to apoptotic stimuli that normally eliminate excess cells. All five proteins are highly conserved between flies and vertebrates and, in both systems, Hpo acts upstream of Wts6,7,8,9,10,11,12,13,14,15,16,17. However, factors that act upstream of Hpo remain to be identified.
Here, we identify two upstream components of the Hpo signalling pathway: the Neurofibromatosis type-2 (NF2) encoded protein Merlin (Mer), and a related protein Expanded (Ex). Mutations in the NF2 tumour-suppressor gene underlie neurofibromatosis type-2, a familial cancer syndrome that shows development of tumours in the central nervous system18,19,20. Several lines of evidence further support the idea that Mer acts as a tumour-suppressor gene. For example, heterozygous Nf2 mutant mice spontaneously develop a wide range of highly metastatic tumours21; cultured Nf2-deficient cells fail to undergo contact-dependent growth arrest22, and Mer overexpression suppresses cell proliferation23,24. Mer is a member of the protein 4.1 superfamily of adaptor proteins18,19 and is thought to transduce a signal from membrane receptors to intracellular downstream components20,24,25. However, although Mer has been shown to interact with several proteins, including PAK1, CD44, HRS, PIKE-L, NHE-RF and ERM, how Mer functions as a tumour-suppressor gene is poorly understood20,24,25,26. We further investigated the function of Mer as a growth suppressor in vivo, taking advantage of the genetic tools that are available in Drosophila.
The growth-suppressing function of Mer is conserved in Drosophila, where it acts partially redundant with another FERM domain protein, Ex27,28,29. Mer and Ex colocalize to adherens junctions22,27,30, where they may transduce a growth-suppressing signal from an unknown receptor20,24,25. Null mutations in mer and ex are lethal in Drosophila29,31, but animals that are homozygous for hypomorphic mutations of either mer or ex survive and show slight overgrowth of adult structures, such as wings29,30,31. Mutations in mer and ex show dominant genetic interactions and clones that are doubly mutant for mer and ex have defects in growth control and differentiation27. However, the cellular basis for these phenotypes and the pathway through which mer and ex act as tumour-suppressor genes are not known22,25,27.
Results
Mutations in merlin and expanded cause tissue overgrowth
To define the functions of mer and ex in growth control and to identify the pathway downstream of Mer and Ex, we analysed the phenotypes of mer;ex double-mutant clones during development (see Methods). For this analysis, we used previously identified null alleles of mer and ex28,29, as well as new ex alleles that we isolated in a genetic screen (see Supplementary Information, Fig. S1). We found that clones of cells doubly mutant for mer and ex produced pronounced outgrowths in diverse adult structures, such as antennae, thorax, wings and legs (Fig. 1a–d; and data not shown). Therefore, mer and ex are general growth regulators that are required to restrict the size of adult structures.

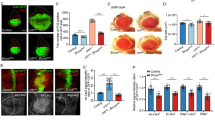
(a, b) Scanning electron micrographs (SEMs) of a wild-type (wt) fly head (a) and a head with mer4;exe1 double-mutant clones (b) in the antenna (arrow), resulting in massive tissue overgrowth. (c, d) SEM images of a wild-type thorax and a thorax with mer4;exe1 double-mutant clones (arrow). (e-j') Mid-pupal retinae stained with anti-Discs large (Dlg) antibodies that localize to apicolateral junctions and visualize cell outlines. (e) Wild-type retina. (f, f') Retina with a mer4;exe1 double-mutant clone marked by the absence of green fluorescent protein (GFP) expression (red in f). (f') shows the Dlg staining only. The mutant area shows excess interommatidial cells. (g) mer4 and (h) exBQ mutants display few extra interommatial cells. (i) hpo42–47 and (j, j') TSC1IQ69 mutant retinae. Absence of GFP (red) marks the mutant clone in (j) and (j') shows Dlg only. The phenotype of mer4;exe1 double-mutant tissues is similar to hpo42–47 but distinct from TSC1IQ69 mutants. Scale bar (j'), 20 μm.
To determine the cause of the overgrowth phenotypes, we studied cell proliferation and apoptosis in tissues lacking Mer and Ex function. We focused our analysis on the developing eye, where defects in the regulation of cell proliferation and apoptosis are readily detectable32. The Drosophila eye develops from the eye imaginal disc, a single-cell-layered epithelial sheet that grows during the larval stages33. After photoreceptor, cone and pigment cells are determined, extra cells are eliminated by apoptosis32, generating a perfect lattice of the different cell types, which can be visualized during the pupal stage (Fig. 1e). In mer;ex double-mutant cell clones, this pattern is disrupted and the retina contains a large excess of interommatidial pigment cells (Fig. 1f, f'). The size of mutant cells appeared normal, although the morphology of photoreceptors and the numbers of cone cells were often abnormal. In contrast to double-mutant clones, mer or ex single-mutant retinae showed only a few extra interommatidial cells (Fig. 1g, h), further supporting that mer and ex can partially substitute for each other. Notably, the mer;ex double-mutant phenotypes are remarkably similar to those of hpo mutants (Fig. 1i)6,8,9,10, but are distinct from the phenotypes of mutations in other growth control genes, such as TSC1 (Tuberous Sclerosis Complex 1), which affects cell size (Fig. 1j, j')34,35. Therefore, Mer and Ex, like Hpo, regulate the number of cells in epithelial tissues.
Merlin and Expanded regulate cell-cycle arrest and Cyclin E transcription
The observation that mer;ex double-mutant retinae have more interommatidial cells raised two questions: How are these extra cells generated and why are they not eliminated by apoptosis? First, we examined the effects of mer and ex on cell proliferation by monitoring bromodeoxyuridine (BrdU) incorporation, which marks cells in S-phase. During the third larval stage, a wave of differentiation — the morphogenetic furrow — sweeps across the eye disc from posterior to anterior32,33. Cells anterior to the furrow are undifferentiated and divide asynchronously. Behind the furrow, cells either differentiate into photoreceptor cells or undergo one additional round of cell division — referred to as the second mitotic wave — before they differentiate into the remaining photoreceptor, cone, pigment and bristle cells32,33. mer;ex mutant cells properly synchronized their cell cycles in the furrow (Fig. 2a', asterisk) and progressed through the second mitotic wave (Fig. 2a', arrow). In contrast to wild-type cells, however, cells in mer;ex mutant clones displayed ectopic BrdU incorporation after the second mitotic wave (Fig. 2a–a'', arrowhead). This ectopic DNA synthesis is followed by cell division, as seen by ectopic phosphorylated histone H3 expression, which marks mitotic chromosomes (not shown). Mutant cells, therefore, failed to arrest the cell cycle after the second mitotic wave and instead continued to proliferate. Cyclin E is a limiting factor for S-phase entry in imaginal-disc cells36, and we found that it was upregulated in mer;ex double-mutant cells (Fig. 2b–b''). This regulation was at the level of transcription, as shown by elevated expression of a Cyclin E reporter gene37 (Fig. 2c–c''). The repression of Cyclin E expression is probably an important downstream effect of Mer and Ex that regulates cell proliferation. We conclude that Mer and Ex are required for proper cell-proliferation arrest.

All panels show third instar eye imaginal discs containing mer−;ex− double-mutant clones marked by the absence of green fluorescent protein (GFP) expression (green in a, b, c, and grayscale in a'', b'', c''). The disc in (a-a'') is labelled for bromodeoxyuridine (BrdU) incorporation (red in a, grayscale in a'). Wild-type cells arrest in G1 in the morphogenetic furrow (asterisk) and non-differentiating cells go through one synchronous S phase in the second mitotic wave (arrows). mer4;exe1 mutant cell clones show ectopic cell proliferation posterior to the second mitotic wave (arrowheads). (b-b'') mer4;exe1 mutant cells have increased levels of Cyclin E (CycE) protein (red in b, grayscale in b') both anterior and posterior to the morphogenetic furrow (asterisk). (c-c'') mer4;exAP50 mutant cells have elevated levels of lacZ expression from the 16.4 lacZ Cyclin E reporter construct (CycE-Z)37 (red in c, grayscale in c'). Anterior is to the left for all discs. All alleles shown are null alleles. Scale bar (c''), 50 μm.
Merlin and Expanded regulate apoptosis
Although the defects in proliferation arrest generate extra cells, this is not sufficient to explain the overgrowth phenotype that is seen in mer;ex mutant tissues. This is because developing tissues compensate by eliminating extra cells by apoptosis. We therefore addressed why the extra interommatidial cells in mer;ex double-mutant retinae survived, whereas extra cells are eliminated by apoptosis in wild-type retinae32. We found that loss of Mer and Ex function suppressed the wave of apoptosis that normally eliminates extra interommatidial cells during the pupal stage (Fig. 3a, a'). We then tested whether Mer and Ex regulate the expression of the anti-apoptotic gene Drosophila Inhibitor of Apoptosis Protein-1 (diap1)38, a target of Hpo signalling6,7,8,9,10,11,12,15. Indeed, mer;ex mutant clones had higher levels of DIAP1 protein (Fig. 3b–b') and cells autonomously upregulated the expression of a lacZ enhancer trap39 insertion in the diap1 gene (Fig. 3c–d'), indicating that this upregulation was at the level of diap1 transcription. This extra DIAP1 may protect cells from apoptosis that is induced during development. We conclude that Mer and Ex are required for developmentally induced apoptosis that normally eliminates extra cells. The combination of the defects in cell-cycle exit and apoptosis observed in mer;ex clones generates extra cells that then evade apoptosis and produce overgrowths in the adult.

All panels show third instar eye imaginal discs containing mer−;ex− double-mutant clones marked by the absence of green fluorescent protein (GFP) expression (green in a', b', c', d'). (a, a') Confocal image showing TUNEL labelling to detect dying cells (grayscale in a, red in a') of a pupal retina 25 h after pupariation with mer4;exe1 mutant cells. Scale bar, 50 μm. (b, b') mer4;exe1 mutant clones upregulated DIAP1 expression (grayscale in b, red in b'). Arrowheads point to a mutant area and asterisks mark the basal levels of DIAP1 expression in a wild-type area. Scale bar, 50 μm. (c, c') mer4;exAP50 mutant clones (arrowheads) upregulated the expression of a lacZ enhancer trap insertion into the DIAP1 gene (grayscale in c, red in c'). Scale bar, 50 μm. (d, d') Close up of a region of the disc in (c, c'), showing that upregulation of DIAP1–lacZ (DIAP1-Z) in mer;ex mutant clones was cell autonomous (arrowhead points to the clone boundary). Anterior is to the left for all discs. All alleles used are null alleles. Scale bar, 20 μm.
Merlin and Expanded act genetically upstream of Hippo
The phenotypes of mer;ex double-mutant tissues are strikingly similar to those of mutations in hpo, sav and wts. Mutations in all of these genes cause extra interommatidial cells, ectopic BrdU incorporation posterior to the morphogenetic furrow and cell-autonomous elevation of Cyclin E and DIAP1 expression within mutant cells. This combination of phenotypes is not observed for mutations in other known growth control genes, and therefore suggests that Mer and Ex interact with Hpo signalling. In addition, we found that the loss of one copy of wts enhanced the imaginal-disc overgrowth phenotypes of hypomorphic mutations in ex (see Supplementary Information, Fig. S2a–d), and that loss of one copy of ex enhanced the larval lethality caused by hypomorphic wts mutations (see Supplementary Information, Fig. S2e). Based on these observations, we hypothesized that Mer and Ex act in Hpo signalling. In the next set of experiments, we tested this hypothesis using overexpression of Ex and Hpo in different mutant backgrounds. We focused on the overexpression of Ex and Hpo because ectopic Mer, Wts or Sav cause only weak phenotypes11 (and data not shown).
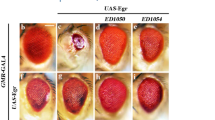
We and others previously reported that overexpression of Hpo hyperactivates Hpo signalling and causes a reduction in cell proliferation and induces apoptosis, phenotypes that are opposite to those of its loss-of function phenotypes (Fig. 4a, a', f, f')6,7,8,10. Similarly, overexpression of Ex during eye development induced apoptosis, reduced cell proliferation and severely disrupted eye development (Fig. 4b, b')30,31. These effects are not caused only by induction of apoptosis, because the phenotypes of ectopic Ex or Hpo are not completely rescued when we inhibited apoptosis by coexpression of the caspase inhibitor p35 or DIAP1 (Fig. 4a–c, a'–c'; and data not shown)8,31. Although coexpression of p35 with Ex gave only a partial rescue, loss of Hpo function reversed the ectopic Ex phenotypes. Therefore, the loss of Hpo function reversed the loss of interommatidial cells, the disruption of ommatidial morphology and the reduction in eye size caused by overexpressed Ex. As a result, hpo mutant eyes that overexpressed Ex showed the same phenotype as hpo mutant eyes (Fig. 4d, d', e, e'). Therefore, Ex requires Hpo for its function. On the other hand, the phenotypes caused by Hpo overexpression were not suppressed by loss of ex or in mer;ex double-mutant clones (Fig. 4f, f', g, g', k, k'). Together, these results indicate that Hpo acts downstream of Ex.

Panels (a-j) show scanning electron microscopy images of adult eyes and panels (a'-j') display mid-pupal retinae stained for anti-Discs large (Dlg). Panels (k-l') show pupal retinae carrying mer;ex (k-k') and wts (l-l') mutant clones that are marked by the absence of green fluorescent protein expression (green in k, l). Retinae are stained for Dlg (red in k, l and gray in k', l'). The genotypes of the animals are indicated above the panels. GMR refers to GMR–gal4-driven overexpression of the indicated transgenes (UAS–ex, UAS–hpo or UAS–p35). GMR–gal4 drives expression of these transgenes in the developing eye posterior to the morphogenetic furrow. Mutant heads were generated via eyFLP-mediated mitotic recombination to generate heads that were entirely mutant for the indicated mutations. For experiments shown in panels (d, d', e, e', g', i, j, j' and k–l'), the mutant chromosome was flipped against a chromosome marked with ubiGFP. (h') shows an exBQ homozygous mutant retina and, in (g, h, i'), ex and wts mutant chromosomes were flipped against chromosomes carrying a cell lethal mutation. The null mutations mer4, hpo42-47, exBQ and wtsx1 were used. Scale bar (a'), 20 μm.
In support of this model, the effects of both overexpressed Ex and overexpressed Hpo were suppressed by loss of wts, which is known to act downstream of Hpo6,8,10,17 (Fig. 4i–j', l, l'). Furthermore, loss of one copy of the wts gene partially suppressed the phenotypes of ectopic Ex, further indicating that Wts acts downstream of Ex and that Wts is required for the effects of Ex (Fig. 5a–c, a'–c').

Panels (a-c) show scanning electron microscopy images of the eyes of adult male flies. The genotypes of the animals are indicated above the panels. GMR–ex refers to GMR–gal4-driven overexpression of a UAS–ex transgene. wt, wild type. (a'-c') show higher magnification images of the panels above them. Removal of one copy of wts partially suppresses the reduced and rough-eye phenotype caused by overexpression of Ex. (d-f') Anti-Drice labelling of third instar eye imaginal discs with indicated genotypes. (d, d') hpo42–47 or (e, e') wtsx1 mutant clones suppress cell death induced by overexpressed Ex, whereas (f, f') mer4;exBQ double-mutant clones do not suppress ectopic Hpo-induced cell death. Panels (d, e, f) show Drice labelling only. Mutant areas are marked by the absence of green fluorescent protein (GFP; red in d', e', f'). Arrowheads point to mutant areas and anterior is to the left for all discs. Scale bar (f'), 50 μm.
In addition to the defects in morphology and interommatidial cell number caused by the overexpression of Ex and Hpo, we used the induction of apoptosis in third instar eye discs as a readout. Overexpression of Ex or Hpo posterior to the morphogenetic furrow induced a stripe of dying cells. The cell death induced by overexpressed Ex was suppressed in hpo or wts mutant clones, but loss of mer and ex had no effect on Hpo-induced cell death (Fig. 5d–f'). Overexpressed Ex therefore requires Hpo to induce cell death but not vice versa. We conclude that Mer and Ex act genetically upstream of Hpo and Wts.
Merlin and Expanded regulate Warts phosphorylation and Yorkie activity
Having established that Mer and Ex act upstream of Hpo genetically, we investigated whether Mer and Ex affected Wts phosphorylation. Previous analyses showed that Hpo induces phosphorylation of Wts, and that the phosphorylation status of Wts is a readout for Hpo pathway activity8. In S2 cells, overexpression of Hpo and Sav induces phosphorylation of Wts, which is visualized as a shift in Wts mobility (Fig. 6a)8. We found that coexpression of Mer and Ex led to a further shift in Wts mobility (Fig. 6a). This shift was due to protein phosphorylation, as it could be reversed by phosphatase treatment (Fig. 6b). In addition, overexpression of Mer and Ex were sufficient to induce phosphorylation of Wts without overexpression of Hpo and Sav (Fig. 6a). Possibly, endogenous Hpo and Sav are sufficient to mediate this effect. Whereas coexpression of Mer and Ex led to a complete shift in Wts mobility, overexpression of Mer without Ex led to an incomplete shift of Wts, and overexpression of Ex alone had no effect on Wts mobility (Fig. 6a). Therefore, Mer and Ex act synergistically to induce phoshorylation of Wts in cultured cells. Although Mer and Ex affect the phosphorylation status of Wts, they did not immunoprecipitate with Hpo, Sav or Wts in vitro or in vivo (data not shown). Thus, Mer and Ex may only transiently associate with Hpo-containing complexes, or other, as yet unidentified, components may link Mer and Ex to Hpo and Wts.

(a) Mer and Ex act synergistically to induce Wts phosphorylation. Western blots of lysates from S2 cells overexpressing different combinations of epitope-tagged proteins, as indicated on the top of the panel. Western blots were probed with indicated antibodies. Wts, Mer and Ex were detected by probing against their tags, whereas Hpo, Sav and αTub were detected with antibodies raised against the native proteins. Note that on the Hpo blot, the endogenous Hpo protein is also detected and migrates faster than the overexpressed Hpo, which carries a Myc tag. (b) Phosphatase (PP2A) treatment reversed the mobility shift of Wts induced by Hpo, Sav, Mer and Ex. Note low phoshorylation levels of overexpressed Wts, which is also diminished by phosphatase treatment (lanes 6 vs. 7). (c) The coactivator activity of Yki is negatively regulated by Mer and Ex. S2 cells were transfected with UAS–luc (Gal4-responsive luciferase reporter) plasmid along with plasmids expressing the Gal4 DNA-binding domain-Yki fusion protein and plasmids expressing Hpo, Sav, Wts, Ex and Mer as indicated. Luciferase activities normalized to that induced by the Gal4 DNA-binding domain (Gal4DBD) alone are plotted. Error bars represent standard deviations from three independent transfections. (d) The transcriptional activator function of the full-length Gal4 (Gal4-FL) is not affected by expression of Hpo, Sav, Wts, Mer and Ex.
Huang et al. recently identified the transcriptional coactivator Yorkie (Yki) as a downstream target of Wts16. Activated Wts phosphorylates Yki and suppresses its transcriptional activator function16. In S2 cells a Gal4-DNA-binding domain–Yki fusion protein can activate a UAS–luciferase reporter construct and this activity is suppressed by coexpression of Hpo, Sav and Wts16. We found that Ex and Mer also downregulated the activity of Yki, and reduced it to a level similar to that achieved by expression of Hpo, Sav and Wts (Fig. 6c). Coexpression of Ex and Mer with Hpo, Sav and Wts led to an even stronger reduction of Yki activity (Fig. 6c). This effect is specific because the expression of Mer, Ex, Hpo, Sav and Wts did not affect the activity of full-length Gal4 (Fig. 6d). These experiments show that Mer and Ex regulate the activity of Yki, which is currently the most downstream component known in the Hpo signal-transduction pathway.
Hippo signalling regulates the expression of Merlin and Expanded
It was previously observed that the levels of Ex protein are elevated in mer mutant cells27. We therefore tested whether this negative regulation was due to Hpo signalling. Indeed, we found that the expression of mer and ex is tightly regulated by Hpo signalling. Cells that are mutant for hpo, sav or wts had highly elevated levels of both Ex and Mer proteins (Fig. 7a–b', and see Supplementary Information, Fig. S3). The subcellular localization of Mer and Ex to the apical-lateral membrane was, however, not affected (Fig. 7c–d'). The upregulation was due to derepressed transcription, because we detected elevated levels of ex transcripts in discs with hpo or wts mutant clones (Fig. 7e–h, and data not shown), and because the expression of a lacZ enhancer trap insertion into the ex locus, which reports the transcription of the ex locus, was strongly induced in wts and sav mutant clones (Fig. 7i–i'; and data not shown). Significantly, Mer and Ex were derepressed in hpo, wts or sav mutant clones in different discs independently of the developmental stage of the discs or the position of the clones. The negative-feedback regulation of mer and ex expression by Hpo signalling is therefore a general effect. Indeed, it is not uncommon that the expression of signalling-pathway components is regulated in a feedback loop by the pathway itself40. This strong and tissue-independent regulation of mer and ex expression by Hpo signalling therefore supports the idea that they act together in a signalling pathway and may provide an important feedback mechanism to keep Hpo signalling in a steady state.

Mer and Ex protein levels are elevated in clones of Hpo pathway mutants, whereas their subcellular localization is not affected. (a-b') hpo42–47 mutant clones in eye-antennal imaginal discs, marked by the absence of green fluorescent protein (GFP; green in a' and b'), have increased levels of Ex (grayscale in a, red in a') and Mer (grayscale in b, red in b') proteins. Upregulation of Ex and Mer is particularly prominent in the morphogenetic furrow (arrows). Arrowheads point to mutant clones. Scale bar (a), 50 μm. (c-d') Z-sections through wing imaginal discs with wtsx1 mutant clones that are marked by the absence of GFP (green in c' and d'). Localization of Ex (grayscale in c, red in c') and Mer (grayscale in d, red in d') is not altered in wtsx1 mutant cells, whereas protein levels are elevated. (e) Third instar eye imaginal disc bearing wtsP2mutant clones marked by the absence of GFP. (f) ex mRNA expression in a wild-type eye-antennal imaginal disc detected by in situ hybridization. (g, h) Increased levels of ex mRNA are detected in discs with wtsP2 mutant clones. Arrows point to the morphogenetic furrow and arrowheads mark patches of upregulated ex expression. (i-j') Eye imaginal discs bearing wtsx1 or mer4;exe1 mutant clones and showing the expression of β-Gal driven by an enhancer trap insertion into the ex gene. Grayscale in (i, j) shows β-Gal expression only. Scale bar (j), 50 μm. (k, k') Higher levels of Ex protein (grayscale in k, red in k') are present in mer4 mutant clones in a wing disc marked by the absence of GFP (arrowhead). (l, l') exe1 mutant clones (GFP-negative) upregulate Mer expression (grayscale in l, red in l'). (m, m') exAP49 mutant clones (GFP-negative, arrowheads) have elevated levels of Ex protein (grayscale in m, red in m') both anterior and posterior to the morphogenetic furrow (arrow). (n, n') Upregulation of Ex (grayscale in n, red in n') in exAP49 mutant clones is suppressed by ectopic expression of Hpo behind the morphogenetic furrow. Asterisk marks Ex upregulation anterior to the furrow. Arrowheads point to the mutant clones with repressed Expanded expression. Hpo overexpression was driven by GMR–gal4, which drives expression posterior, but not anterior to the morphogenetic furrow (arrow). White bar marks the region of GMR–hpo expression. Ex overexpression is therefore only suppressed posterior to the furrow, where Hpo is expressed. (o-o''') Wing imaginal disc with clones overexpressing Hpo and DIAP1 downregulate the expression of Ex (gray in o' and red in o and o'''), although do no affect Dlg (blue in o, gray in o''). Clones overexpressing Hpo and DIAP1 are marked by the coexpression of GFP (green in o and o'''). Scale bar (o'''), 100 μm. Anterior is to the left for all discs.
The identification of mer and ex as transcriptional target genes of Hpo signalling leads to a model in which both Mer and Ex regulate the expression of themselves. Indeed, we corroborated that the levels of Ex are elevated in mer mutant cells (Fig. 7k, k')27 and we found that Mer is upregulated in ex mutant clones (Fig. 7l, l'). In addition, we found elevated levels of Ex in cells mutant for a strong hypomorphic ex allele, exAP49, which encodes a protein that is truncated after the FERM domain and that is still recognized by the anti-Ex antibodies (Fig. 7m, m'; and see Supplementary Information, Fig. S1). Moreover, expression of lacZ from the ex enhancer trap insertion was strongly induced in mer;ex double-mutant cells (Fig. 7j, j'). Therefore, like other components of the Hpo signalling pathway, Mer and Ex negatively regulate the expression of mer and ex, thereby supporting the model in which Mer and Ex act in Hpo signalling.
Hippo hyperactivation rescues transcriptional defects in expanded mutant cells
Once we identified mer and ex as ubiquitous targets of Hpo signalling, we used the expression of mer and ex as readouts for Hpo pathway activity in epistasis experiments. We thus tested whether hyperactivation of Hpo was sufficient to suppress the induction of Ex expression in exAP49 mutant cell clones. To do this experiment, we used the GMR–gal4 driver to overexpress Hpo specifically posterior the morphogenetic furrow. As expected, in such discs, exAP49 mutant clones still upregulated Ex anterior to the furrow where Hpo was not overexpressed (Fig. 7m, m', asterisk). However, the hyperactivation of Hpo posterior to the furrow suppressed the upregulation of Ex in exAP49 mutant clones (Fig. 7n, n', arrowhead). Hpo activation is therefore sufficient to rescue transcriptional defects in ex mutant cells.
Finally, we tested whether ectopic Hpo could suppress endogenous Ex. Clones of cells overexpressing Hpo alone or in combination with DIAP1 (to suppress cell death caused by overexpression of Hpo) showed reduced levels of Ex (Fig. 7o–o'''; and data not shown). These cells, however, showed normal levels of Dlg at the apicolateral junctions indicating that loss of Ex was not due to general defects in cell architecture. The results indicate that in wild-type cells, the activity of the Hpo pathway is in a steady state and can be positively or negatively modulated.
Discussion
Our data functionally link the tumour-suppressor functions of Mer and Ex with Hpo signalling. We propose a model in which Mer and Ex act together to transduce a growth-regulatory signal from an unknown receptor to Hpo and Wts, which in turn regulate the expression of target genes to promote proliferation arrest and apoptosis (Fig. 8). We provided several lines of evidence that place the activities of Mer and Ex upstream of Hpo and Wts. First, the phenotypes induced by overexpression of Ex in imaginal discs are reversed by removing either Hpo or Wts, whereas Hpo-induced phenotypes do not require the activity of Mer and Ex. Second, hyperactivation of Hpo is sufficient to rescue the transcriptional defects of ex mutant cells. Third, Mer and Ex act synergistically to induce phosphorylation of Wts. Fourth, Mer and Ex regulate the activity of Yki, a Wts target, in a cell-culture-based assay. Together, these data support a model in which Mer and Ex act upstream and through Hpo signalling to perform their tumour-suppressor functions in Drosophila (Fig. 8).

Mer and Ex act upstream of Hpo and regulate the expression of mer, ex, diap1 and cyclin E transcription. Mer/Ex may transduce the signal of an, as yet unknown, growth-regulating cell-surface receptor.
Does Mer act through Hpo signalling in vertebrates? All currently known components of the Hpo signalling pathway in Drosophila are highly conserved in vertebrates, where they have been implicated in regulating cell proliferation and apoptosis. Mice and humans have two Wts homologues, Lats1 and Lats2, and mice deficient for Lats1 develop soft-tissue sarcomas and ovarian tumours41,42,43. Therefore, Lats1 acts as a tumour-suppressor gene in vertebrates. Sav may also have an effect on tumorigenesis, because the human orthologue of Sav, Sav1, is mutated in cancer cell lines11. Vertebrates have two Hpo homologues, Mst1 and Mst2 and, as in Drosophila, they promote apoptosis and regulate cell-cycle exit44. Therefore, the Hippo signalling pathway regulates proliferation and apoptosis in vertebrates.
Human Mer has been shown to physically interact with several other proteins in vitro and in cell culture cells20,24,25. However, how these interactions contribute to the tumour-suppressor function of Mer is unclear20,24,25. So far, none of these components have been linked to Mst1/2 signalling, but the Hpo signalling pathway has only recently been discovered. Notably, using BLAST searches, we found two vertebrate homologues of Ex called Ex1 and Ex2 (see Supplementary Information, Fig. S1). Therefore, all known components of the Hpo signalling pathway are conserved in vertebrates. Importantly, not only the genes but also the mechanisms of signal transduction seem to be conserved between Drosophila and vertebrates. Vertebrate Lats1 and Mst2 can rescue the lethality and overgrowth phenotypes of wts and hpo mutants8,43 and Mst1/2 bind to Sav1 and phosphorylate and activate Lats1/2 (ref. 17). Due to this apparent conservation of the components and mechanisms that operate downstream of Mer and Ex in Drosophila and in vertebrates, Mer and the two Ex homologues may function as tumour-suppressor genes through the Hpo homologues Mst1/2. Our data that links Mer to Hpo signalling may therefore have important implications for the study and treatment of neurofibromatosis and other cancers.
Methods
Drosophila stocks.
Mutant clones were induced using the FLP/FRT system45,46. To generate of marked double-mutant clones, we recombined p{w+ mer+}27 with a FRT40A ubiGFP chromosome. Flies with mer;ex double-mutant clones had the following genotype: y w mer4FRT19A/ Y; FRT40A exe1/FRT40A p{w+ mer+} ubiGFP; hsFLP MKRS/+, except for those seen in Figs 2c–c'' and 3c–d', in which we used exAP50 because exe1 is an enhancer trap itself. After mitotic recombination took place, non-green-fluorescent-protein (GFP)-expressing cells were homozygous mutant for ex and lacked the mer rescue construct; they were, therefore, double mutant for mer and ex. To generate ex, mer, hpo, wts, sav or TSC1 mutant clones, the following alleles were flipped against corresponding ubiGFP marked FRT chromosomes: exe1 (null)31, exBQ (null; see Supplementary Information, Fig. S1), exAP49 (hypomorph, see Supplementary Information, Fig. S1), mer4 (null)29, hpo42–47 (null)8, wtsx1 (null)14, wtsP2 (hypomorph)13, savshrp1 (null)1,2 and TSC1IQ69 (ref. 6). Overexpression was achieved using the UAS–GAL4 system47 and the following stocks: UAS–ex (ref. 6), UAS–hpo (ref. 6), UAS–p35 (ref. 38) and GMR–gal4. Overexpression clones of Hpo and DIAP1 were induced using the flp-out technique48 and the following stocks: ywhsflp; act<y+<Gal4, UASGFPnls/CyO and UAS–DIAP1 (obtained from B.Hay, Caltech, USA). Other stocks used were: ex697/CyO (ref. 31), DIAP1–lacZ (ref. 38) and CycE–lacZ (16.4 construct37).
Scanning electron microscopy, immunohistochemistry, in-situ hybridization and cell-death assays.
Scanning electron microscopy (SEM) of adult flies was carried out following the hexamehyldisilazane (HMDS) method12. Antibody stainings of imaginal discs were performed as described previously12. The following antibodies were used (dilutions and source in parentheses): mouse α-Dlg (1:300; DSHB); rabbit α-Drice (1:2000; B. Hay); guinea-pig α-Mer (1:4000; R. Fehon, Duke University, USA); guinea-pig α-Ex (1:2000; R. Fehon); rabbit α-Ex (1:1500; A. Laughon, University of Wisconsin, USA); mouse α-BrdU (Becton-Dickinson, Franklin Lakes, USA; 1:50); mouse α-DIAP-1 (1:200; B. Hay); and mouse α-CycE (1:40). Secondary antibodies were donkey Fab fragments from Jackson Immuno Research (West Grove, PA). BrdU incorporation was carried out as described previously, by incorporating BrdU for 1 h12. For the in situ hybridization to detect expanded transcripts, the Drosophila cDNA clone UG24F8 was used as a template to generate DIG-labelled RNA probes (Roche, Indianapolis, IN). TUNEL labelling was performed on imaginal discs and pupal retinae using an in situ cell-death detection kit (Roche)6.
Cell culture.
N-terminal tagged HA-Ex and HA-Mer were constructed using the pAc5.1 vector. All other constructs were gifts from Duojia Pan, Johns Hopkins University, USA8,16. Drosophila S2 cells, cultured in Schneiders's medium containing 10% fetal bovine serum and antibiotics were transiently transfected using Cellfectin (Invitrogen, Carlsbad, CA) and collected 48 h after transfection. Lysis was performed in RIPA Buffer (150 mM NaCl, 50 mM Tris-HCl, pH 7.4, 1% NP40, 0.5% sodium deoxycholate, 0.1% SDS, 1 mM PMSF, 5 μg ml−1 aprotinin, 5 μg ml−1 leupeptin and phosphatase inhibitors (Roche)), and Western blots were performed according to standard protocols. Antibodies used were: α-V5 (Invitrogen); α-HA (CRP, Cumberland, VA); and α-Hpo, α-Sav and α-αTub (Sigma, St Louis, MO). α-Hpo and α-Sav antibodies were raised in guinea pigs against native His-tagged proteins.
For phosphatase treatments, cells were lysed in IP buffer (150 mM NaCl, 50 mM Tris-HCl, pH 8.0, 0.5% NP40, 1 mM PMSF, 5 μg ml−1 aprotinin, 5 μg ml−1 leupeptin) and V5-Wts proteins were immunoprecipitated. Beads were washed three times in phosphatase treatment buffer (50 mM Tris-HCl, pH 7.5, 0.1 mM EDTA, 0.5 mM MgCl2, 0.5 mM MnCl2, 0.5 mM CaCl2) and 0.3 units of PP2A (Upstate, Charlottesville, VA) were added in a total volume of 75 μl, followed by 30 min incubation at 37°C. The reaction was terminated by addition of an equal volume of 2× SDS sample buffer.
Luciferase reporter gene assays were performed by transfecting 20 ng of Yki–Gal4 or Gal4–FL plasmids with 3 ng of UAS–luc plasmid in triplicates, with or without plasmids expressing Ex, Mer, Hpo, Sav or Wts in 24-well plates. Luciferase assays were performed using the Dual Luciferase Reporter Assay System (Promega, Madison, WI) and a 20/20n Luminometry System with Single Auto-Injector (Promega).
Note: Supplementary Information is available on the Nature Cell Biology website.
References
Johnston, L. A. & Gallant, P. Control of growth and organ size in Drosophila. Bioessays 24, 54–64 (2002).
Conlon, I. & Raff, M. Size control in animal development. Cell 96, 235–244 (1999).
Hanahan, D. & Weinberg, R. A. The hallmarks of cancer. Cell 100, 57–70 (2000).
Ryoo, H. D. & Steller, H. Hippo and its mission for growth control. Nature Cell Biol. 5, 853–855 (2003).
Hay, B. A. & Guo, M. Coupling cell growth, proliferation, and death. Hippo weighs in. Dev. Cell 5, 361–363 (2003).
Udan, R. S., Kango-Singh, M., Nolo, R., Tao, C. & Halder, G. Hippo promotes proliferation arrest and apoptosis in the Salvador/Warts pathway. Nature Cell Biol. 5, 914–920 (2003).
Pantalacci, S., Tapon, N. & Leopold, P. The Salvador partner Hippo promotes apoptosis and cell-cycle exit in Drosophila. Nature Cell Biol. 5, 921–927 (2003).
Wu, S., Huang, J., Dong, J. & Pan, D. hippo encodes a Ste-20 family protein kinase that restricts cell proliferation and promotes apoptosis in conjunction with salvador and warts. Cell 114, 445–456 (2003).
Harvey, K. F., Pfleger, C. M. & Hariharan, I. K. The Drosophila Mst ortholog, hippo, restricts growth and cell proliferation and promotes apoptosis. Cell 114, 457–467 (2003).
Jia, J., Zhang, W., Wang, B., Trinko, R. & Jiang, J. The Drosophila Ste20 family kinase dMST functions as a tumor suppressor by restricting cell proliferation and promoting apoptosis. Genes Dev. 17, 2514–2519 (2003).
Tapon, N. et al. salvador promotes both cell cycle exit and apoptosis in Drosophila and is mutated in human cancer cell lines. Cell 110, 467–478 (2002).
Kango-Singh, M. et al. Shar-pei mediates cell proliferation arrest during imaginal disc growth in Drosophila. Development 129, 5719–5730 (2002).
Justice, R. W., Zilian, O., Woods, D. F., Noll, M. & Bryant, P. J. The Drosophila tumor suppressor gene warts encodes a homolog of human myotonic dystrophy kinase and is required for the control of cell shape and proliferation. Genes Dev. 9, 534–546 (1995).
Xu, T., Wang, W., Zhang, S., Stewart, R. A. & Yu, W. Identifying tumor suppressors in genetic mosaics: the Drosophila lats gene encodes a putative protein kinase. Development 121, 1053–1063 (1995).
Lai, Z. C. et al. Control of cell proliferation and apoptosis by mob as tumor suppressor, mats. Cell 120, 675–685 (2005).
Huang, J., Wu, S., Barrera, J., Matthews, K. & Pan, D. The Hippo signaling pathway coordinately regulates cell proliferation and apoptosis by inactivating Yorkie, the Drosophila homolog of YAP. Cell 122, 421–434 (2005).
Chan, E. H. et al. The Ste20-like kinase Mst2 activates the human large tumor suppressor kinase Lats1. Oncogene 24, 2076–2086 (2005).
Rouleau, G. A. et al. Alteration in a new gene encoding a putative membrane-organizing protein causes neuro-fibromatosis type 2. Nature 363, 515–521 (1993).
Trofatter, J. A. et al. A novel moesin-, ezrin-, radixin-like gene is a candidate for the neurofibromatosis 2 tumor suppressor. Cell 75, 826 (1993).
McClatchey, A. I. Merlin and ERM proteins: unappreciated roles in cancer development? Nature Rev. Cancer 3, 877–883 (2003).
McClatchey, A. I. et al. Mice heterozygous for a mutation at the Nf2 tumor suppressor locus develop a range of highly metastatic tumors. Genes Dev. 12, 1121–1133 (1998).
Lallemand, D., Curto, M., Saotome, I., Giovannini, M. & McClatchey, A. I. NF2 deficiency promotes tumorigenesis and metastasis by destabilizing adherens junctions. Genes Dev. 17, 1090–1100 (2003).
Lutchman, M. & Rouleau, G. A. The neurofibromatosis type 2 gene product, schwannomin, suppresses growth of NIH 3T3 cells. Cancer Res. 55, 2270–2274 (1995).
Xiao, G. H., Chernoff, J. & Testa, J. R. NF2: the wizardry of merlin. Genes Chromosom. Cancer 38, 389–399 (2003).
Bretscher, A., Edwards, K. & Fehon, R. G. ERM proteins and merlin: integrators at the cell cortex. Nature Rev. Mol. Cell. Biol. 3, 586–599 (2002).
Rong, R., Tang, X., Gutmann, D. H. & Ye, K. Neurofibromatosis 2 (NF2) tumor suppressor merlin inhibits phosphatidylinositol 3-kinase through binding to PIKE-L. Proc. Natl Acad. Sci. USA 101, 18200–18205 (2004).
McCartney, B. M., Kulikauskas, R. M., LaJeunesse, D. R. & Fehon, R. G. The Neurofibromatosis-2 homologue, Merlin, and the tumor suppressor expanded function together in Drosophila to regulate cell proliferation and differentiation. Development 127, 1315–1324 (2000).
Boedigheimer, M. & Laughon, A. Expanded: a gene involved in the control of cell proliferation in imaginal discs. Development 118, 1291–1301 (1993).
LaJeunesse, D. R., McCartney, B. M. & Fehon, R. G. Structural analysis of Drosophila merlin reveals functional domains important for growth control and subcellular localization. J. Cell Biol. 141, 1589–1599 (1998).
Boedigheimer, M. J., Nguyen, K. P. & Bryant, P. J. Expanded functions in the apical cell domain to regulate the growth rate of imaginal discs. Dev. Genet. 20, 103–110 (1997).
Blaumueller, C. M. & Mlodzik, M. The Drosophila tumor suppressor expanded regulates growth, apoptosis, and patterning during development. Mech. Dev. 92, 251–262 (2000).
Baker, N. E. Cell proliferation, survival, and death in the Drosophila eye. Semin. Cell Dev. Biol. 12, 499–507 (2001).
Wolff, T. & Ready, D. F. The beginning of pattern formation in the Drosophila compound eye: the morphogenetic furrow and the second mitotic wave. Development 113, 841–850 (1991).
Potter, C. J., Huang, H. & Xu, T. Drosophila Tsc1 functions with Tsc2 to antagonize insulin signaling in regulating cell growth, cell proliferation, and organ size. Cell 105, 357–368 (2001).
Tapon, N., Ito, N., Dickson, B. J., Treisman, J. E. & Hariharan, I. K. The Drosophila tuberous sclerosis complex gene homologs restrict cell growth and cell proliferation. Cell 105, 345–355 (2001).
Richardson, H., O'Keefe, L. V., Marty, T. & Saint, R. Ectopic cyclin E expression induces premature entry into S phase and disrupts pattern formation in the Drosophila eye imaginal disc. Development 121, 3371–3379 (1995).
Jones, L., Richardson, H. & Saint, R. Tissue-specific regulation of cyclin E transcription during Drosophila melanogaster embryogenesis. Development 127, 4619–4630 (2000).
Hay, B. A., Wassarman, D. A. & Rubin, G. M. Drosophila homologs of baculovirus inhibitor of apoptosis proteins function to block cell death. Cell 83, 1253–1262 (1995).
Ryoo, H. D., Bergmann, A., Gonen, H., Ciechanover, A. & Steller, H. Regulation of Drosophila IAP1 degradation and apoptosis by reaper and ubcD1. Nature Cell Biol. 4, 432–438 (2002).
Niehrs, C. & Meinhardt, H. Modular feedback. Nature 417, 35–36 (2002).
Turenchalk, G. S., St John, M. A., Tao, W. & Xu, T. The role of lats in cell cycle regulation and tumorigenesis. Biochim. Biophys. Acta 1424, M9–M16 (1999).
St John, M. A. et al. Mice deficient of Lats1 develop soft-tissue sarcomas, ovarian tumours and pituitary dysfunction. Nature Genet. 21, 182–186 (1999).
Tao, W. et al. Human homologue of the Drosophila melanogaster lats tumour suppressor modulates CDC2 activity. Nature Genet. 21, 177–181 (1999).
Dan, I., Watanabe, N. M. & Kusumi, A. The Ste20 group kinases as regulators of MAP kinase cascades. Trends Cell Biol. 11, 220–230 (2001).
Xu, T. & Rubin, G. M. Analysis of genetic mosaics in developing and adult Drosophila tissues. Development 117, 1223–1237 (1993).
Newsome, T. P., Asling, B. & Dickson, B. J. Analysis of Drosophila photoreceptor axon guidance in eye-specific mosaics. Development 127, 851–860 (2000).
Brand, A. H. & Perrimon, N. Targeted gene expression as a means of altering cell fates and generating dominant phenotypes. Development 118, 401–415 (1993).
Struhl, G. & Basler, K. Organizing activity of wingless protein in Drosophila. Cell 72, 527–540 (1993).
Acknowledgements
We thank R.G. Fehon, A. Laughon, M. Mlodzik, B. Hay, D. Pan, P. Bryant, the Bloomington Drosophila Stock Center and the Developmental Studies Hybridoma Bank (University of Iowa) for fly stocks, antibodies and plasmids. We thank K.K. Norga for his help with the isolation of new ex alleles. We thank K.Dunner for technical help with the scanning electron microscopy analysis, which were performed, along with DNA sequencing, at the M. D. Anderson core facilities supported by a National Cancer Institute Cancer Center support grant. We also thank L. McCord for her help with artwork. Special thanks to M. Acar for his invaluable advice and help with the cell-culture experiments. We thank the members of the X. Chen laboratory for their help with antibody production. We thank R. Behringer, H.J. Bellen, A. Bergmann, K.-W. Choi, V. Dion, N. Giagtzoglou, P.R. Hiesinger, G. Lozano, G. Mardon, R. Johnson, J. Kunz and members of the Halder Lab for discussions. This work was supported by a National Institutes of Health grant to G.H, The Odyssey Fellowship and The Theodore N. Law Award for Scientific Achievement to M.K.S., and a BRASS Scholarship to E.H. H.J.N. is supported by HHMI and by a NIH Medical Genetics Research Fellowship Program grant.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Supplementary Information
Supplementary figures S1, S2 and S3 (PDF 377 kb)
Rights and permissions
About this article
Cite this article
Hamaratoglu, F., Willecke, M., Kango-Singh, M. et al. The tumour-suppressor genes NF2/Merlin and Expanded act through Hippo signalling to regulate cell proliferation and apoptosis. Nat Cell Biol 8, 27–36 (2006). https://doi.org/10.1038/ncb1339
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ncb1339
