
Abstract
The anaphase promoting complex or cyclosome (APC/C) is a ubiquitin protein ligase that, together with Cdc20 or Cdh1, targets cell-cycle proteins for degradation. APC/C–Cdh1 specifically promotes protein degradation in late mitosis and G1. Mutant embryos lacking Cdh1 die at E9.5–E10.5 due to defects in the endoreduplication of trophoblast cells and placental malfunction. This lethality is prevented when Cdh1 is expressed in the placenta. Cdh1-deficient cells proliferate inefficiently and accumulate numeric and structural chromosomal aberrations, indicating that Cdh1 contributes to the maintenance of genomic stability. Cdh1 heterozygous animals show increased susceptibility to spontaneous tumours, suggesting that Cdh1 functions as a haploinsufficient tumour suppressor. These heterozygous mice also show several defects in behaviour associated with increased proliferation of stem cells in the nervous system. These results indicate that Cdh1 is required for preventing unscheduled proliferation of specific progenitor cells and protecting mammalian cells from genomic instability.
Similar content being viewed by others


Main
Targeted protein destruction during the cell cycle is achieved mainly by ubiquitination and subsequent degradation of key cell-cycle regulators, such as cyclins or cyclin-dependent kinase (Cdk) inhibitors1,2. Two major E3 ubiquitin ligases that have a role in cell-cycle control are SCF (Skp1/Cullin/F-box protein complex) and APC/C3,4,5,6. SCF mainly controls protein levels during S-phase, whereas APC/C is thought to regulate mitotic proteins, reducing their levels after mitosis.
APC/C is composed of at least a dozen different structural subunits5 and requires the association of either Cdc20 or Cdh1 (encoded by the Fzr1 gene in mammals), two additional subunits that recognize and recruit specific substrates. Cdc20 is thought to activate APC/C in early mitosis and Cdh1 in late mitosis and during G1 (refs 7, 8). APC/C substrates include mitotic cyclins (cyclins A and B), mitotic kinases and their regulators (Aurora A/B, TPX2, survivin, Plk1, Nek2A), proteins involved in chromosome segregation (securin, sororin, Sgo1) and replication proteins (geminin or thymidine kinase). In addition, both Cdc20 and Cdh1 subunits are themselves substrates of APC/C-mediated degradation2,5. Skp2, a Leu-rich-repeat F-box protein that recruits SCF targets is also an APC/C target9,10. On the other hand, Emi1, an APC/C inhibitor, is a target of the SCF complex, indicating an interdependence between APC/C and SCF in controlling cell-cycle progression11,12. Some APC/CCdh1 substrates, such as cyclin A, cyclin B, Aurora A, Plk1, Tpx2, Cdc6 or Spk2, are overexpressed in human cancers associated with chromosomal instability and poor prognosis13.
To understand the physiological function of mammalian Cdh1, we have generated conditional knockout mice with a targeted mutation in the Fzr1 locus. Complete inactivation of Cdh1 leads to embryonic lethality caused by specific aberrations in the placenta. Ablation of Cdh1 also causes slight defects in the proliferation of mouse embryonic fibroblasts (MEFs) in culture and accumulation of chromosomal aberrations, suggesting that Cdh1 has a function in preserving genome integrity. Fzr1+/− mice develop a variety of tumours late in life without losing the Fzr1 wild-type allele. This suggests a haploinsufficient role for Cdh1 in tumour suppression. Fzr1+/− mice also show behavioural abnormalities accompanied by hyperproliferation of specific progenitor cells in the brain.
Results
Ablation of Cdh1 results in placental defects and embryonic lethality
Two recombinant Fzr1+/loxfrt embryonic stem (ES) cell clones (Fig. 1) were isolated and used to generate Fzr1+/loxfrt mice. Expression of the Flp and Cre recombinases produced the corresponding Fzr1lox and Fzr1− alleles (Fig. 1). Fzr1+/− mutants are fertile and do not develop gross anatomical abnormalities during the first year of life. We found the expected fractions of homozygous Fzr1−/− embryos until embryonic day (E) 9.5 (Fig. 1d; Supplementary Information, Table S1). However, by E10.5–E11.5, the proportion of live Fzr1−/− embryos was lower than expected (about 10% versus the expected 25%) and all of these embryos seemed to be growth-retarded, compared with wild-type littermates (Fig. 1e). There were no live Fzr1−/− embryos at E12.5 or later stages of development.

(a) Schematic representation of Fzr1 alleles used in this study. The mouse Fzr1 locus encoding Cdh1 contains 14 exons (boxes) spanning 12kbp. Noncoding sequences (open boxes), loxP (open triangles) and frt (shaded triangles) sites are indicated. The neoR cassette used for selection of homologous recombinant ES clones is indicated by an open arrow. The wild-type and different mutated Fzr1 alleles used in this work are shown. (b) Southern blot analysis of recombinant ES cell clones carrying the indicated alleles. The origin of the probe is indicated in a (probe a). Sizes of the diagnostic DNA fragments are indicated. (c) Analysis of Cdh1 expression in wild-type and mutant mice. β-actin was used as a loading control. (d) Appearance of viable Fzr1+/+ and Fzr1−/− embryos at day 9.5 of gestation (E9.5). (e) By E11.5 most of the embryos were dead and the few viable embryos were growth-retarded. (f) The corresponding extra-embryonic tissues were smaller and their blood content was reduced by E11.5. Scale bars, 1mm. Uncropped images of the scans are shown in Supplementary Information, Fig. S5.
Histological examination of E10.5–E11.5 mutant embryos revealed normal morphogenesis and no signs of gross pathological lesions. In contrast, mutant placentas had a pale appearance (Fig. 1f), and a significant decrease in the size of the labyrinth and the number of fetal erythroblasts (Fig. 2a). In addition, trophoblast giant cells were underdeveloped and contained much smaller nuclei. These giant cells represent a unique population that undergoes repeated rounds of DNA synthesis without intervening mitoses (endoreduplication). This leads to formation of giant nuclei14. In wild-type placentas, trophoblast giant cells contained up to 1500 genome copies (N), with most of the cells containing over 100 N. In contrast, the DNA content of Fzr1−/− trophoblasts was greatly reduced and virtually all cells had fewer than 100 N (Fig. 2b), suggesting a specific defect in endoreduplication.

(a) Histological sections of placentas derived from Fzr1+/+ and Fzr1−/− embryos at E10.5 (S, spongiotrophoblast; L, labyrinth). Primitive nucleated red blood cells (open arrowheads) were significantly reduced in mutant placentas. Trophoblast giant-cell nuclei (filled arrowheads) were also smaller in mutant placentas. Both phenotypes were rescued in the Fzr1Δ/−; Sox2–Cre+/T placentas. Original magnification: ×40 (top panels), ×400 (middle and bottom panels). (b) Ploidy distribution of trophoblast giant cells in wild-type, Fzr1−/− and Fzr1Δ/−; Sox2–Cre+/T placentas. (c) Fzr1−/− embryos die before E11.5, whereas Fzr1Δ/−; Sox2–Cre+/T embryos, in which Cdh1 is specifically eliminated in the embryo, but not in the placenta, develop to term. Two representative pictures of Fzr1+/Δ and rescued Fzr1Δ/−; Sox2–Cre+/T E18.5 embryos are shown. Scale bars are 200 μm (a) and 1mm (c).
To analyse whether these placental defects cause lethality in Fzr1−/− embryos, we crossed Fzr1lox/lox mice with transgenic mice expressing the Cre recombinase under the Sox2 promoter15. In this model, Cre is expressed in most cells in the developing embryo but not in the placenta. We found that this procedure prevented embryonic lethality. The overall structure of the placenta and the number and ploidy of trophoblast giant cells were normal in Fzr1Δ/Δ; Sox2–Cre+/T (data not shown) and Fzr1Δ/−; Sox2–Cre+/T embryos (Fig. 2a,b). In addition, we recovered viable Fzr1Δ/Δ; Sox2–Cre+/T and Fzr1Δ/−; Sox2–Cre+/T embryos at all points of development, including E18.5 (Fig. 2c) and postnatal day (P) 3. In these embryos, the excision of exons 2 and 3 of Fzr1 by Cre was complete, as detected by specific PCR amplification and immunublot analysis of cultured cells from these embryos (Supplementary Information, Fig. S1).
Proliferative defects in Cdh1-deficient MEFs
Primary MEFs isolated from E10.5 Fzr1−/− embryos had no detectable Cdh1 expression (Fig. 1c). These mutant MEFs proliferated at a slightly slower rate, compared with control MEFs (Fig. 3a), and contained a slightly greater number of cells in S and G2/M phases of the cell cycle (Fig. 3b), suggesting a partial defect in these stages of the cell cycle. In addition, Fzr1−/− MEFs were significantly more susceptible to the senescence-like crisis period after 2–3 passages (versus 4–5 passages in wild-type cells) following a 3T3 protocol (Fig. 3c). Most of these cultures formed immortal clones, although Fzr1−/− MEFs required 13–15 passages to become immortalized (versus 8–9 in wild-type cultures). These immortalized Fzr1−/− cultures showed proliferation defects that were similar to the corresponding mutant primary cells (Supplementary Information, Fig. S2).

(a) Growth curves of primary (passage 2) Fzr1+/+ and Fzr1−/− MEFs in complete medium with 10% or 2% FBS. (b) DNA content profile of asynchronous Fzr1+/+ and Fzr1−/− primary MEFs. Percentage of cells in each phase of the cell cycle is shown. (c) These cells were maintained following a standard 3T3 protocol and cell numbers were scored in every passage. Although mutant MEFs enter the crisis period at earlier passages, all of these genotypes become immortalized after 13–15 passages (Supplementary Information, Fig. 2). (d) Western blot analysis of asynchronous Fzr1+/+, Fzr1+/− and Fzr1−/− MEFs with antibodies directed against the indicated cell-cycle proteins. (e) Western blot analysis of the indicated cell-cycle regulators in serum-starved Fzr1+/+ and Fzr1−/− MEFs at the indicated times after removal of growth factors. N and H indicate asynchronous NIH3T3 and Hela cells, respectively. β-actin was used as a loading control. Data are mean±s. d. (n = 3 for each genotype). Uncropped images of the scans are shown in Supplementary Information, Fig. S5.
The absence of Cdh1 correlates with increased levels of Cdc20 (Fig. 3d), consistent with a role for APC/CCdh1 in the degradation of Cdc20 (refs 16, 17). In addition, the protein levels of cyclin A, but not other cyclins (data not shown), were significantly increased in Cdh1-null cells. Cyclin B protein levels were decreased in asynchronous Cdh1-null cells. The 4N DNA content was increased in these cells, suggesting either accumulation of non-mitotic (4N-G1) cells or a defective mitotic progression (see below). To further analyse the degradation of cell-cycle regulators in non-proliferating Fzr1−/− MEFs, we removed growth factors from these cells. Many APC/CCdh1 targets were efficiently degraded in wild-type cells after serum removal, including Cdc20, geminin, Aurora A (Fig. 3e), cyclin A or Aurora B (data not shown). All of these known APC/CCdh1 targets were stabilized in serum-starved Fzr1−/− MEFs. In addition, the protein levels of the cell-cycle regulator p16INK4a were also stabilized in Cdh1-null cells (Fig. 3e), suggesting that deficient proliferation and earlier senescence in these mutant cells may be a consequence of an increase in the level of this cell-cycle inhibitor.
Serum-starved Fzr1−/− MEFs entered S phase after serum stimulation with different kinetics, compared with their wild-type counterparts (Fig. 4a). A greater number of Fzr1−/− cells than wild-type cells had entered S phase 18h after serum stimulation (60% versus 42%, respectively). However, DNA replication was less efficient in mutant cells, as shown by a decrease in the number of BrdU-positive Fzr1−/− cells after this stage (Fig. 4a). Cdc20, Aurora A and cyclin B levels were increased in Cdh1-null cells at all stages analysed (Fig. 4b). Some APC/CCdh1 substrates involved in DNA replication, such as Cdc6 and, more markedly, geminin, were upregulated in Cdh1-null cells. Cyclin E was slightly downregulated in mutant cells, whereas cyclin A was also upregulated during S phase. This upregulation of cyclin A, and perhaps cyclin B, may explain the slight increase in Cdk2 and Cdk1 kinase activities by 10–15h after serum stimulation (Fig. 4c).

(a) MEFs were serum-starved for 72h, forced to enter the cell cycle after stimulation with 15% FBS and pulsed with BrdU. The percentage of BrdU-positive cells was scored at different time-points. By 18h after stimulation with serum, Fzr1−/− MEFs showed increased incorporation of BrdU in S-phase (right panels). However, by 24h the percentage of cells that incorporated BrdU was reduced, compared with wild-type cells. (b) Immunodetection of the indicated cell-cycle proteins in total lysates from Fzr1+/+ and Fzr1−/− MEFs at the indicated time-points after serum stimulation. An additional sample from asynchronous HeLa cells was used to identify the human proteins. β-actin was used as a loading control. (c) Total lysates were isolated at the indicated points and Cdk2 or Cdk1 complexes were immunoprecipitated (IP). Cdk2 and Cdk1 kinase activity (KA) was assayed using histone H1 as a substrate (M, mock immunoprecipitation). (d) Immunodetection of soluble and chromatin-bound proteins in Fzr1+/+ and Fzr1−/− MEFs at the indicated time-points after serum stimulation. Mek2 (a cytosolic kinase) and histone H4 (a tightly-bound chromatin protein) are shown as controls of the fractionation protocol. Uncropped images of the scans are shown in Supplementary Information, Fig. S5.
These alterations in Cdh1-null cells were accompanied by slightly reduced levels of both Mcm4 and Mcm5 (minichromosome maintenance proteins) in the chromatin, suggesting a defect in the formation of pre-replicative complexes (Fig. 4d). Interestingly, 15h after serum stimulation, a stage when BrdU incorporation peaks in the mutant cells, cyclin A was bound to chromatin in Fzr1−/− but not Fzr1+/+ cells (Fig. 4d). Thus, in Cdh1-deficient cells, S phase proceeds with decreased loading of Mcm complexes to chromatin, an increased level of the DNA replication inhibitor geminin and premature Cdk1 activation, thus generating an inefficient environment for DNA replication.
Delayed mitosis and genomic instability in Cdh1-deficient cells
The anti-mitotic drug nocodazole, which disrupts microtubules, had similar effects in arresting Fzr1−/− and Fzr1+/+ cells, suggesting that there is no defect in the spindle assembly checkpoint (Fig. 5a). However, whereas most wild-type cells had exited mitosis and undergone cytokinesis 2h after drug removal, 60% of the Fzr1−/− MEFs remained with 4N content of DNA, as measured by flow cytometry. Microscopic observation revealed that 35% of Fzr1−/− cells were binucleated, whereas in Fzr1+/+ cultures, only 8.8% cells were binucleated (Fig. 5b). Most of the Fzr1−/− cells had lost the MPM2 phosphorylation signal, although with delayed kinetics (about 30min delay), compared with wild-type cells (Fig. 5c–d). These results indicate that Fzr1−/− MEFs remain as MPM2-negative, binucleated cells because of defective cytokinesis. This abnormal exit from mitosis may result from inefficient degradation of Cdc20, cyclin B and Plk1 (Fig. 5ef).

(a) DNA content of Fzr1+/+ and Fzr1−/− MEFs after release from nocodazole-induced prometaphase arrest. Most wild-type cells became diploid after 2h, whereas a significant fraction of Fzr1−/− cells remained as 4N cells. (b) This was accompanied by a significant increase of Fzr1−/− binucleated cells in the nocodazole-released cultures. The percentage of binucleated cells was scored in a total of 250 Fzr1+/+ and 250 Fzr1−/− cells (data are mean±s. d.). These cultures were stained with antibodies against α-tubulin (red), phospho-H3 (green) and with DAPI (blue). Scale bar, 10μm. (c) Quantification of phospho-MPM2 (a mitotic marker) levels after release from nocodazole showing a delay of about 30min in the exit from mitosis. (d) MPM2/propidium iodide double-staining of these cells showing a significant number of Fzr1−/− MPM2-positive mitotic cells 1h after nocodazole release. At this stage the MPM2 mark was lost or appears in diploid cells in the Fzr1+/+ cultures. Some Fzr1−/− but not Fzr1+/+ cells remained positive for MPM2 3h after nocodazole release. (e) Immunodetection of the indicated cell-cycle proteins in total lysates from Fzr1+/+ and Fzr1−/− MEFs at the indicated time-points after release from nocodazole. β-actin was used as a loading control. (f) Effect of Cdc20 or Cdc27 downregulation by RNA interference. Fzr1+/+ and Fzr1−/− MEFs were transfected with siRNAs against Cdc20 or Cdc27 and protein levels were analysed in asynchronous cells (0h) or 36h after serum removal. The indicated proteins were immunodetected using specific antibodies. Uncropped images of the scans are shown in Supplementary Information, Fig. S5.
Most Cdh1 substrates are stabilized in Cdh1-null cells but are degraded eventually. We therefore tested whether Cdc20 or other APC/C complexes may cooperate in degrading these proteins. Wild-type or Cdh1-null MEFs were transfected with specific small interference oligonucleotide RNAs (siRNAs) against Cdc20 or Cdc27, a structural subunit of APC/C. As shown in Fig. 5f, the specific siRNAs caused an incomplete but significant reduction in Cdc20 and Cdc27 protein levels. Most APC/C targets were stabilized after genetic deletion of Cdh1 in this assay, both in asynchronous (0h) and serum-starved (36h) cells (Fig. 5f). Downregulation of Cdc27 phenocopies the effect of Cdh1 deletion, suggesting that Cdh1 function is mediated by APC/C. Cyclin A2, Aurora B and geminin did not display significant stabilization after knocking down Cdc20 although Cdc20 may contribute to the degradation of cyclin B1, Aurora A and Plk1. This stabilization was not maintained in serum-starved cells, suggesting that Cdh1 is the major APC/C cofactor during G0.
The replicative defects and mitotic alterations in Cdh1-null cells did not prevent cell-cycle progression, as both primary (Fig. 3a) and immortalized (Supplementary Information, Fig. S2) cells proliferated in vitro. However, Fzr1−/− immortalized cultures showed a variety of aberrations, including binucleated or multinucleated cells, metaphase plates with mis-aligned chromosomes in diploid or polyploid cells and multipolar spindles (Fig. 6a). Although these aberrations were not frequent in stably proliferating immortalized cultures (6% of Fzr1−/− cells versus 1% Fzr1+/+ cells), they may contribute to the increased genomic instability. Indeed, most Fzr1−/− immortalized cultures had aberrant chromosome numbers (Fig. 6b), including highly aneuploid cells with 100–150 chromosomes. Chromosome breaks and non-disjunction figures were also observed in almost all metaphases analysed (n=80) in five out of six Fzr1−/− clones (Fig. 6c). Spectral karyotypic analysis of these cells showed a large variety of chromosomal aberrations including three, four or five copies of specific chromosomes, small chromosomal fragments and complex translocations affecting more than two chromosomes (Fig. 6d; Supplementary Information, Fig. S3). Interestingly, chromosome breaks were also observed in 14% of metaphases analysed in primary Fzr1−/− cultures. However, in Fzr1+/+ cells only 4.8% of metaphases contained such breaks (data not shown).

(a) Representative pictures of genomic and mitotic aberrations in immortal Fzr1−/− cultures. Cells were stained with antibodies against α-tubulin (red), and phospho-H3 (green). DAPI (blue) was also used in the top panels. (b) Quantification of chromosome numbers in Fzr1+/+ and Fzr1−/− immortalized cells. Both diploid and tetraploid cells were grouped in each histogram to indicate cells with complete sets of chromosomes (red bars) or carrying altered (aneuploid, other colours) genome composition. (c) Karyotype analysis of Fzr1−/− immortalized cells showing chromosome breaks and non-disjunction structures (arrowheads). (d) Spectral karyotypic (SKY) analysis of immortalized Fzr1−/− clones with genomic alterations including translocations and complex genomic compositions affecting the indicated chromosomes. Data are mean±s.d., n = 80 metaphases from 6 different Fzr1−/− clones and 50 metaphases from 4 different Fzr+/+ clones. Scale bars, 10 μm.
Cdh1 functions as a haploinsufficient tumour suppressor in vivo
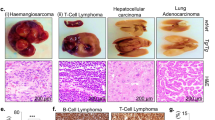
Histological analysis of several tissues in young (3-month) or adult (12-month) Fzr1+/− animals did not reveal any gross abnormalities (data not shown). In addition, when subjected to a classical skin carcinogenesis protocol, Fzr1+/− mice developed fewer and smaller tumours than their wild-type littermates (Supplementary Information, Fig. S4). These results indicate that tumour development is impaired in young Fzr1+/− mice, probably as a consequence of decreased proliferation rates, as observed in cultured cells. Survival of Fzr1+/− mice was decreased at later ages (Fig. 7a). Whereas 60% of Fzr1+/+ mice were alive at 25 months of age, only 22% of Fzr1+/− survived to this age. Moreover, 25% of aged Fzr1+/− mice developed epithelial neoplasias that were not present in their wild-type counterparts (Fig. 7b–h). These tumours include adenocarcinomas and fibroadenomas of the mammary gland (17% of females), as well as tumours in the lung, liver, kidney, testis and sebaceous gland (Supplementary Information, Table S2). Approximately 20% of Fzr1+/− mice developed B cell lymphomas, a common type of tumour observed at the same frequency in wild-type mice. These data indicate that with age, Fzr1+/− mice become significantly more susceptible to epithelial tumours. As shown in Fig. 7i, the wild-type allele of Fzr1 was retained in these epithelial tumours, suggesting that Cdh1 may function as a haploinsufficient tumour suppressor in vivo.

(a) Survival curve of Fzr1+/+ (n=19) and Fzr1+/− (n=51) mice. (b) Summary of tumours in these mice classified by cellular origin (c–h) Representative sections (H&E staining at the indicated original magnification) of major pathologies in Fzr1+/− mice: mammary gland adenocarcinoma (×25, c); mammary gland fibroadenoma (×25, d); lung adenoma (×200, e); sebaceous gland adenoma (×25, f); renal tubule adenoma (×100, g); plasmacytosis (asterisk) in a lymph node (×25, h). (i) Analysis of loss of heterozygosity in the following Fzr1+/− tumours (T): mammary gland adenocarcinoma (lane 1), sebaceous gland adenoma (2); lung adenocarcinoma (4), testis adenoma (5) and fibroadenoma (6). Lanes 3 and 7 correspond to control samples from healthy (N) lungs in Fzr1+/− and Fzr1+/+ mice, respectively. The migration of wild-type (9.0kbp) and mutant (7.5kbp) alleles is shown. Scale bars, 200 μm.
Unscheduled proliferation of brain progenitors and behavioural abnormalities in Fzr1+/− mice
Cdh1 is known to be highly expressed in the brain18 and controls axonal growth and patterning in the mammalian brain19. The nervous system of Fzr1+/− mice did not show any gross abnormality, either in the brain or peripheral tissues. However, we detected a significant increase in the proliferation rate of a specific fraction of cells in the subventricular zone of the brain (Fig. 8a). This region is the richest source of stem cells in the adult brain and contains a niche of Sox2- and Sox9-expressing stem cells that maintain neurogenesis20,21. The subventricular zone of Fzr1+/− mice had a high percentage of Ki67-positive cells and higher cellularity (Fig. 8a). However, the number of Sox2- or Sox9-positive cells was not significantly altered in this niche, suggesting an increase in asymmetric proliferation that generates transient amplifying or mature cells negative for these two stem-cell markers. We did not find differences in the structure of the Fzr1+/− hippocampus (data not shown), the other major neurogenic region in adult brain.

(a) Representative pictures of brain sections in Fzr1+/+ and Fzr1+/− mice stained with H&E, or antibodies against Ki67, Sox2 or Sox9. The area of the subventricular zone analysed is indicated by small squares. Quantification of Ki67-positive (grey bars) or total number (black bars) of cells in the subventricular zone niche close to the ependyma of these Fzr1+/+ and Fzr1+/− mice is shown (as number of cells per brain coronal section). Original magnification is ×50 (H&E images) or ×400 (immunostaining). Scale bars, 1mm (top panels) or 50μm (middle and bottom panels). (b) Performance of Fzr1+/+ and Fzr1+/− mice in the tightrope test. The percentage of mice that successfully passed the test after three trials is shown in the left panel. The percentage of mice that failed in the different trials is shown in the right panel. (c) Performance of Fzr1+/+ and Fzr1+/− mice in the object recognition test. The average discrimination ratio is shown for Fzr1+/+ and Fzr1+/− males and females (left panel). The percentage of mice with discrimination ratios greater than 0.5 is shown in the right panel. (data are mean±s. d., n = 11 males and 9 females from each of the genotypes). Asterisks indicate statistically significant differences at the confidence level shown.
To further understand whether the reduced levels of Cdh1 may have physiological consequences in Fzr1+/− mice, we performed two different behavioural tests in 1.5-year-old mice. Compared with wild-type mice, Fzr1+/− mice performed poorly in the tightrope test, a widely used marker of neuromuscular coordination. Wild-type mice were able to stay on a circular bar after three trials of 60s duration each, whereas approximately 55% of Fzr1+/− mice fell in the third trial (P <0.01) and about 10% fell even after 5 trials, suggesting defects in neuromuscular vigour and coordination. In an object recognition assay, recognition memory of Fzr1+/− mice was significantly reduced when compared with wild-type littermates, (P <0.015; Fig. 8c). Approximately 90% of Fzr1+/+ mice had a discrimination ratio higher than 0.5 (they dedicate more time to the 'new' object), whereas 60% of Fzr1+/− were unable to discriminate between new objects. These results suggest that reduced levels of Cdh1 contribute to neuromuscular and memory defects.
Discussion
Cdh1 is not essential in Saccharomyces cerevisiae, Schizosaccharomyces pombe22,23,24,25, Caenorhabditis elegans26 or Drosophila melanogaster27, suggesting that this protein is dispensable for the basic cell cycle. Genetic ablation of Fzr1, the mouse gene that encodes Cdh1, results in embryonic lethality. This is accompanied by reduced flux of fetal blood in the placental labyrinth and defective endoreduplication of giant trophoblast cells. These defects are rescued when Cdh1 is specifically ablated in the embryo but not in the placenta, indicating that placental abnormality is the cause of death in Fzr1−/− animals. At birth, no gross morphological abnormalities are found in Fzr1Δ/−; Sox2–Cre+/T neonates, although preliminary results indicate that these mutant mice die a few days after birth (data not shown).
Genetic elimination of Cdh1 in mammalian cells results in the accumulation of genomic aberrations linked to defects in the timing of DNA replication, exit from mitosis and cytokinesis. These defects correlate with increased levels of specific S phase and mitotic regulators. Cyclin A and cyclin B are thought to be targeted for degradation by both APC/CCdc20 and APC/CCdh1. Cdh1-null cells, however, have increased levels of cyclin A despite having increased levels of Cdc20, indicating that in cultured fibroblasts, cyclin A levels are mainly controlled by APC/CCdh1. In addition, Cdc20 downregulation by RNA interference does not significantly stabilize cyclin A (Fig. 5f). On the other hand, cyclin B levels seem to be modulated by both Cdh1 and Cdc20, at least in asynchronous cells. Both cyclin A and cyclin B have S-phase promoting activity and their upregulation28,29 and the concomitant increase in Cdk1 activity (Fig. 4) may result in early but inefficient DNA replication. In addition, unscheduled replication in Fzr1−/− cells occurs with reduced cyclin E, reduced Mcm loading to the chromatin and increased levels of inhibitors, such as p16INK4a and geminin, resulting in inefficient DNA replication. Some of these replicative defects, as well as the delay in exit from mitosis, have been observed using RNA interference and genetically modified chicken cell lines30,31.
Five of the Cdh1 targets upregulated in Fzr1−/− cells, namely Aurora A, Plk1, cyclin A, cyclin B and Cdc20, are part of the chromosomal instability signature of human tumours and show among the highest scores of correlation with poor prognosis in cancer13,32. Similarly, unbalanced expression of licensing DNA replication factors is also frequent in several tumour types33,34. These data suggest that loss of Cdh1 may induce gross genomic alterations during cell proliferation, which can lead to tumour development. Indeed, Fzr1+/− mice develop epithelial tumours, such as mammary gland adenocarcinomas and fibroadenomas, which are not observed in wild-type mice. Moreover, these tumours maintain the Fzr1 wild-type allele suggesting that Cdh1 is haploinsufficient for tumour suppression. Although the reduction in Cdh1 levels in young animals may result in inefficient proliferation of induced tumours (skin carcinogenesis protocol), the accumulation of genomic instability may facilitate tumour development in old mice, and may specifically promote the development of epithelial tumours that typically require a genomic instability component35. Similar results in CENP-E mutant mice have led to the hypothesis that genomic instability may act both as a tumour suppressor and an oncogene in vivo36. Consistent with these results, Cdh1 protein levels are decreased in some cancer cell lines31 and re-expression of Cdh1 reduced tumour development by a malignant B cell lymphoma cell line37. Although the human Fzr1 gene contains a consensus CpG island in its promoter, hypermethylation of this region is not frequent in human tumours (I.G.H, S.M. and M.M., unpublished observations). Thus, the mechanism for the reduction in Cdh1 protein levels in some human tumours remains unidentified.
Cdh1 is known to be highly expressed in neurons18 and Cdh1 silencing by RNA interference has specific effects on the morphology and growth of neuronal processes19. In addition, Cdh1 depletion in postmitotic neurons and human neuroblastoma cells in culture leads to apoptotic cell death38. This effect is accompanied by increased cyclin B protein levels and increased BrdU incorporation, suggesting that postmitotic neurons may be more susceptible to apoptotic cell death induced by unscheduled proliferation. Although Fzr1+/− mice do not show alterations in the structure of the nervous system, we have identified a specific set of cells in the subventricular zone that are particularly susceptible to reduced Cdh1 levels and show increased proliferation in mutant mice. These cells are found in a niche of cells positive for Sox2 and Sox9, two markers of neuronal stem cells21,39. These proliferative defects are accompanied by defects in neuromuscular coordination and learning in Fzr1+/− mice. Whether these behavioural defects are a consequence of the alteration in subventricular zone progenitors is currently unknown. A deeper characterization of the role of Cdh1 in these progenitor cells will require specific deletion of the Fzr1 gene to eliminate both alleles in these neuronal progenitors.
Methods
Generation of Fzr1-targeted mice and histological analysis.
The Fzr1 gene-targeting construct was assembled by flanking exons 2 and 3 of the murine Fzr1 locus with loxP sequences (Fig. 1a). A neomycin phosphotransferase (neo) cassette was used for positive selection of clones, whereas the thymidine kinase gene was used for negative selection with gancyclovir. Recombinant ES cells and clones were selected by Southern blot analysis using the probe 'a' as indicated in Fig. 1a. Four out of 386 ES cell clones analysed underwent homologous recombination at the Fzr1 locus. Two recombinant ES cell clones were used to generate Fzr1-deficient animals. Oligonucleotides and protocols for genotyping this colony of mice are available in the Supplementary Information.
TgpCAG–flpe40 or TgCMV–Cre41 transgenic mice were used for ubiquitous expression of Flp or Cre recombinases, respectively. To remove the neoR selection marker we crossed Fzr1+/loxfrt mice with pCAG–Flpe transgenic animals40 that express the Flp recombinase in a ubiquitous manner. To generate a null allele, we crossed Fzr1+/lox mice with transgenic mice that express Cre recombinase in a ubiquitous manner41. Cre-mediated recombination between the two loxP sites eliminates exons 2 and 3 (Fig. 1a). Splicing from exon 1 to exon 4 sequences causes a frameshift in the open reading frame that results in the synthesis of a putative small polypeptide unrelated to Cdh1. Sox2–Cre mice that express Cre in embryonic but not in extra-embryonic tissues were reported previously15. All animals were maintained in a mixed 129/Sv (25%) × CD1 (25%) × C57BL/6J (50%) background. Mice were housed in the pathogen-free animal facility of the Centro Nacional de Investigaciones Oncológicas (Madrid) following the animal care standards of the institution. These animals were observed on a daily basis and sick mice were killed humanely in accordance with the Guidelines for Humane End Points for Animals used in biomedical research. Tumour latency has been considered equivalent to lifespan. For histological observation, dissected organs were fixed in 10%-buffered formalin (Sigma) and embedded in paraffin wax. Sections of 3- or 5-μm thickness were stained with haematoxylin and eosin (H&E). Additional immunohistochemical examination of the tissues and pathologies analysed were performed using specific antibodies against Ki67 (Master Diagnostica), Sox2 or Sox9 (Chemicon) or active caspase 3 (RyD Systems).
For quantification of DNA ploidy in trophoblast giant cells, placentas were collected from E10.5 wild-type and mutant embryos. Sections of 5-μm thickness were cut and stained with Feulgen stain. Four wild-type and four Fzr1−/− placentas were analysed. Images were quantified using ImageJ software (National Institutes of Health, Bethesda, Maryland, USA, http://rsb.info.nih.gov/ij/). The ploidy of trophoblast giant cells was expressed relative to diploid decidual cells.
Cell culture and cytometry.
MEFs were prepared from E10.5 embryos and cultured using standard protocols42. The whole embryo was minced and dispersed in 0.1% trypsin (20min at 37°C). Cells were grown for two population doublings and then frozen. On reaching confluence, MEFs were subcultured at a ratio of 1:4; each passage was considered to be two population doubling levels (PDLs). All cultures were maintained in Dulbecco's modified Eagle's medium (DMEM; Gibco) supplemented with 2mM glutamine, 1% penicillin/streptomycin and 10% fetal bovine serum (FBS) or donor calf serum (CS).
Growth properties were analysed using the classical 3T3 protocol. Every 3 days, cells were detached using trypsin, counted and 106 cells were plated per 10-cm plate. The relative number of cells is considered as a measure of the number of cells per passage related to the initial number of cells seeded per plate. DNA content was analysed by flow cytometry (Becton-Dickinson). For growth-curve assays, cells were plated at a density of 1×105 cells per well in 6-well plates. For cell-cycle re-entry, cells were starved in serum-free DMEM for 72h before being re-stimulated with 15% FBS for the indicated times. To analyse mitotic progression, cells were pre-synchronized in G0 by serum starvation, released with 15% FBS in the presence of aphidicolin (0.2μgml−1) for 24h, and then treated with nocodazole (100mM, Sigma) for 12–14h.
For RNA interference, Fzr1+/+ and Fzr1−/− immortal MEFs were transfected with Cdc20 or Cdc27 siRNAs (oligonucleotide sequences are available from Dharmacon) using Hyperfect (Dharmacon) according to the manufacturer's instructions. Forty-eight hours after transfection, cells were either harvested (time 0) or serum starved for 36h before whole lysates were isolated for protein analysis.
Immunodetection of total protein levels or chromatin-associated DNA replication proteins.
Cells were washed twice with ice-cold PBS and lysed in NP-40 lysis buffer (150mM NaCl, 1% NP-40, 50mM Tris-HCl pH8.0, 1mM PMSF, 1μgml−1 leupeptin, 25μgml−1 aprotinin, 1mM EDTA). After 30min on ice, samples were vortexed (5min at 4°C) and cleared by centrifugation. In addition, cells were subjected to a biochemical fractionation protocol that separates soluble and chromatin-bound proteins43. Proteins were separated on SDS–PAGE, transferred to nitrocellulose membranes (BioRad), probed using specific antibodies and detected using donkey (Rockland) or goat (Invitrogen) anti rabbit fluorescent secondary antibodies followed by detection using the Odyssey Infrared Imaging System (Li-Cor Biosciences). After transfer of the protein lysates, we probed nitrocellulose membranes with antibodies against Cdh1 (Neomarkers), p27Kip1 (Transduction Laboratories), Cdc6 and histone H4 (Upstate Signaling), geminin, Cdc20 and cyclin A2 (Santa Cruz Biotechnology), cyclin B1 (Chemicon International), Aurora A, Aurora B, Cdc27 and Mek2 (Becton Dickinson), Plk1 (Zymed), Tpx2 and cyclin E (Abcam), phospho-MPM2 and phospho-histone H3 (Upstate Signaling), BrdU (Alexa Fluor 647 Conjugate; Molecular Probes) and β-Actin (Sigma). New anti-Mcm4 and anti-Mcm5 polyclonal sera were generated in rabbits using as antigens synthetic peptides corresponding to amino acids 23–36 in mouse Mcm4 and amino acids 593–606 in mouse Mcm5. Cdk2 and Cdk1 kinase activities were measured as described previously44 using histone H1 as a substrate.
Scoring of chromosomal abnormalities.
Metaphase preparations were made using 0.075M KCl hypotonic treatment and methanol/acetic acid fixation from MEFs after a 2h treatment with colcemid (0.01μgml−1). Slides were stained with 5% Giemsa in pH 6.8 buffer for 10min. A total of 50 metaphases were scored in each clone analysed. For spectral karyotyping (SKY), painting probes for each chromosome were generated from flow-sorted mouse chromosomes using sequence-independent DNA amplification. Labelling was performed by incorporating four different dyes in a combination sequence that allows unique and differential identification of each chromosome. Slides were prepared from fixative-stored material and hybridized and washed using the SKY method according to the manufacturer's protocol (Applied Spectral Imaging). Chromosomes were counterstained with DAPI.
Behavioural assays.
The tightrope test was used as described previously45. Briefly, mice were placed on a bar of circular section (60-cm long and 1.5-cm diameter) and the test was considered successful when a mouse did not fall during a period of 60s in at least one out of five consecutive trials. Animals underwent the object recognition test following a protocol described previously46 with minor modifications. Briefly, mice were placed in an experimental cage that consists of an open plastic box (73-cm long × 55-cm wide × 32-cm high) with opaque walls. The two similar objects (objects X and Y) were plastic parallelepipeds and the dissimilar object (object Z) was a plastic bottle. The objects were placed along the long axis of the experimental cage, each 7cm from each cage end. Mice were first exposed to the cage and to objects X and Y. After a familiarization period, each mouse was released in the experimental cage with objects X and Z. After each exposure, the objects and the cage were wiped with 70% ethanol to eliminate odour cues. The size and weight of the objects used did not allow their movement or displacement by the animals. Animals were considered to show recognition activity when the head of the animal was less than 2cm close to the object. The total time of recognition activity was scored manually using a stopwatch and recorded.
Statistical analysis.
Statistical analysis was performed using Student's t-test. All data are shown as mean±s. d.; probabilities of P <0.05 were considered significant.
Note: Supplementary Information is available on the Nature Cell Biology website.
References
Reed, S.I. Ratchets and clocks: the cell cycle, ubiquitylation and protein turnover. Nature Rev. Mol. Cell Biol. 4, 855–864 (2003).
Nakayama, K.I. & Nakayama, K. Ubiquitin ligases: cell-cycle control and cancer. Nature Rev. Cancer 6, 369–381 (2006).
Cardozo, T. & Pagano, M. The SCF ubiquitin ligase: insights into a molecular machine. Nature Rev. Mol. Cell Biol. 5, 739–751 (2004).
Harper, J.W., Burton, J.L. & Solomon, M.J. The anaphase-promoting complex: it's not just for mitosis any more. Genes Dev. 16, 2179–2206 (2002).
Peters, J.M. The anaphase promoting complex/cyclosome: a machine designed to destroy. Nature Rev. Mol. Cell Biol. 7, 644–656 (2006).
Acquaviva, C. & Pines, J. The anaphase-promoting complex/cyclosome: APC/C. J. Cell Sci. 119, 2401–2404 (2006).
Pines, J. Mitosis: a matter of getting rid of the right protein at the right time. Trends Cell Biol. 16, 55–63 (2006).
Sullivan, M. & Morgan, D.O. Finishing mitosis, one step at a time. Nature Rev. Mol. Cell Biol. 8, 894–903 (2007).
Bashir, T., Dorrello, N.V., Amador, V., Guardavaccaro, D. & Pagano, M. Control of the SCF(Skp2–Cks1) ubiquitin ligase by the APC/C(Cdh1) ubiquitin ligase. Nature 428, 190–193 (2004).
Wei, W. et al. Degradation of the SCF component Skp2 in cell-cycle phase G1 by the anaphase-promoting complex. Nature 428, 194–198 (2004).
Vodermaier, H.C. APC/C and SCF: controlling each other and the cell cycle. Curr. Biol. 14, R787–R796 (2004).
Ang, X.L. & Harper, J.W. Interwoven ubiquitination oscillators and control of cell cycle transitions. Sci. STKE 2004, pe31 (2004).
Carter, S.L., Eklund, A.C., Kohane, I.S., Harris, L.N. & Szallasi, Z. A signature of chromosomal instability inferred from gene expression profiles predicts clinical outcome in multiple human cancers. Nature Genet. 38, 1043–1048 (2006).
Cross, J.C. How to make a placenta: mechanisms of trophoblast cell differentiation in mice--a review. Placenta 26 (suppl. A), S3–S9 (2005).
Hayashi, S., Lewis, P., Pevny, L. & McMahon, A.P. Efficient gene modulation in mouse epiblast using a Sox2Cre transgenic mouse strain. Mech. Dev. 119 (suppl 1), S97–S101 (2002).
Prinz, S., Hwang, E.S., Visintin, R. & Amon, A. The regulation of Cdc20 proteolysis reveals a role for APC components Cdc23 and Cdc27 during S phase and early mitosis. Curr. Biol. 8, 750–760 (1998).
Shirayama, M., Zachariae, W., Ciosk, R. & Nasmyth, K. The Polo-like kinase Cdc5p and the WD-repeat protein Cdc20p/fizzy are regulators and substrates of the anaphase promoting complex in Saccharomyces cerevisiae. EMBO J. 17, 1336–1349 (1998).
Gieffers, C., Peters, B.H., Kramer, E.R., Dotti, C.G. & Peters, J.M. Expression of the CDH1-associated form of the anaphase-promoting complex in postmitotic neurons. Proc. Natl Acad. Sci. USA 96, 11317–11322 (1999).
Konishi, Y., Stegmuller, J., Matsuda, T., Bonni, S. & Bonni, A. Cdh1–APC controls axonal growth and patterning in the mammalian brain. Science 303, 1026–1030 (2004).
Alvarez-Buylla, A. & Garcia-Verdugo, J.M. Neurogenesis in adult subventricular zone. J. Neurosci. 22, 629–634 (2002).
Ferri, A.L. et al. Sox2 deficiency causes neurodegeneration and impaired neurogenesis in the adult mouse brain. Development 131, 3805–3819 (2004).
Visintin, R., Prinz, S. & Amon, A. CDC20 and CDH1: a family of substrate-specific activators of APC-dependent proteolysis. Science 278, 460–463 (1997).
Schwab, M., Lutum, A.S. & Seufert, W. Yeast Hct1 is a regulator of Clb2 cyclin proteolysis. Cell 90, 683–693 (1997).
Yamaguchi, S., Murakami, H. & Okayama, H. A WD repeat protein controls the cell cycle and differentiation by negatively regulating Cdc2/B-type cyclin complexes. Mol. Biol. Cell 8, 2475–2486 (1997).
Kitamura, K., Maekawa, H. & Shimoda, C. Fission yeast Ste9, a homolog of Hct1/Cdh1 and Fizzy-related, is a novel negative regulator of cell cycle progression during G1-phase. Mol. Biol. Cell 9, 1065–1080 (1998).
Fay, D.S., Keenan, S. & Han, M. fzr-1 and lin-35/Rb function redundantly to control cell proliferation in C. elegans as revealed by a nonbiased synthetic screen. Genes Dev. 16, 503–517 (2002).
Sigrist, S.J. & Lehner, C.F. Drosophila fizzy-related down-regulates mitotic cyclins and is required for cell proliferation arrest and entry into endocycles. Cell 90, 671–681 (1997).
Strausfeld, U.P. et al. Both cyclin A and cyclin E have S-phase promoting (SPF) activity in Xenopus egg extracts. J. Cell Sci. 109 (Pt 6), 1555–1563 (1996).
Moore, J.D., Kirk, J.A. & Hunt, T. Unmasking the S-phase-promoting potential of cyclin B1. Science 300, 987–990 (2003).
Engelbert, D., Schnerch, D., Baumgarten, A. & Wasch, R. The ubiquitin ligase APC(Cdh1) is required to maintain genome integrity in primary human cells. Oncogene 27, 907–917 (2008).
Sudo, T. et al. Activation of Cdh1-dependent APC is required for G1 cell cycle arrest and DNA damage-induced G2 checkpoint in vertebrate cells. EMBO J. 20, 6499–6508 (2001).
Perez de Castro, I., de Carcer, G. & Malumbres, M. A census of mitotic cancer genes: new insights into tumor cell biology and cancer therapy. Carcinogenesis 28, 899–912 (2007).
Karakaidos, P. et al. Overexpression of the replication licensing regulators hCdt1 and hCdc6 characterizes a subset of non-small-cell lung carcinomas: synergistic effect with mutant p53 on tumor growth and chromosomal instability-evidence of E2F-1 transcriptional control over hCdt1. Am. J. Pathol. 165, 1351–1365 (2004).
Pinyol, M. et al. Unbalanced expression of licensing DNA replication factors occurs in a subset of mantle cell lymphomas with genomic instability. Int. J. Cancer 119, 2768–2774 (2006).
DePinho, R.A. The age of cancer. Nature 408, 248–254 (2000).
Weaver, B.A., Silk, A.D., Montagna, C., Verdier-Pinard, P. & Cleveland, D.W. Aneuploidy acts both oncogenically and as a tumor suppressor. Cancer Cell 11, 25–36 (2007).
Wang, C.X., Fisk, B.C., Wadehra, M., Su, H. & Braun, J. Overexpression of murine fizzy-related (fzr) increases natural killer cell-mediated cell death and suppresses tumor growth. Blood 96, 259–263 (2000).
Almeida, A., Bolanos, J.P. & Moreno, S. Cdh1/Hct1–APC is essential for the survival of postmitotic neurons. J. Neurosci. 25, 8115–8121 (2005).
Nait-Oumesmar, B. et al. Activation of the subventricular zone in multiple sclerosis: evidence for early glial progenitors. Proc. Natl Acad. Sci. USA 104, 4694–4699 (2007).
Rodriguez, C.I. et al. High-efficiency deleter mice show that FLPe is an alternative to Cre–loxP. Nature Genet. 25, 139–140 (2000).
Schwenk, F., Baron, U. & Rajewsky, K. A cre-transgenic mouse strain for the ubiquitous deletion of loxP-flanked gene segments including deletion in germ cells. Nucleic Acids Res. 23, 5080–5081 (1995).
Malumbres, M. et al. Mammalian cells cycle without the D-type cyclin-dependent kinases Cdk4 and Cdk6. Cell 118, 493–504 (2004).
Mendez, J. & Stillman, B. Chromatin association of human origin recognition complex, cdc6, and minichromosome maintenance proteins during the cell cycle: assembly of prereplication complexes in late mitosis. Mol. Cell Biol. 20, 8602–8612 (2000).
Quereda, V., Martinalbo, J., Dubus, P., Carnero, A. & Malumbres, M. Genetic cooperation between p21Cip1 and INK4 inhibitors in cellular senescence and tumor suppression. Oncogene 26, 7665–7674 (2007).
Miquel, J. & Blasco, M. A simple technique for evaluation of vitality loss in aging mice, by testing their muscular coordination and vigor. Exp. Gerontol. 13, 389–396 (1978).
Bevins, R.A. & Besheer, J. Object recognition in rats and mice: a one-trial non-matching-to-sample learning task to study 'recognition memory'. Nature Protoc. 1, 1306–1311 (2006).
Acknowledgements
We thank Gardenia Fresneda and Manuel Eguren for help with carcinogenic assays and molecular analysis; and Sheila Rueda and Blanca Velasco for their valuable help in the management of the mouse colony. We also thank Sagrario Ortega and the CNIO Transgenic Unit for their expertise in ES cell manipulations, members of the CNIO Comparative Pathology Unit for histological and pathological processing and the CNIO Cytogenetics Unit for their help with karyotype analysis. I.G.H. is supported by a Ramón y Cajal contract (Ministerio de Educación y Ciencia). E.M. is supported by an FIS fellowship (Ministerio de Sanidad). This work was supported by grants from the Association pour la Recherche contre le Cancer and the Région Aquitaine (to P.D.), Ministerio de Educación y Ciencia (SAF2004-05611 to I.G.H.; BFU2004-04886 to J.M.; BFU2005-03195 and GEN2003-20243-C08-05 to S.M.; and SAF2006-05186 to M.M.), the Consolider-Ingenio 2010 Programme (CSD2007-00015 to J.M. and S.M.; and CSD2007-00017 to M.M.), Comunidad de Madrid (OncoCycle Programme; S-BIO-0283-2006), Fundación Ramón Areces, and Fundación Médica Mutua Madrileña Automovilística (to M.M.); and Fundación Científica de la Asociación Española contra el Cáncer (to S.M. and M.M.).
Author information
Authors and Affiliations
Contributions
I.G.H and E.M. performed most of experiments and J.M. carried out the analysis of chromatin-bound proteins. P.D. and M.C. performed the histological and pathological analysis of the samples. M.M. and S.M. designed and supervised the study and M.M. wrote the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Supplementary Information
Supplementary Figures S1, S2, S3, S4, S5, Supplementary Tables S1, S2 and Supplementary Methods (PDF 4416 kb)
Rights and permissions
About this article
Cite this article
García-Higuera, I., Manchado, E., Dubus, P. et al. Genomic stability and tumour suppression by the APC/C cofactor Cdh1. Nat Cell Biol 10, 802–811 (2008). https://doi.org/10.1038/ncb1742
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ncb1742
This article is cited by
-
SIRT1 ubiquitination is regulated by opposing activities of APC/C-Cdh1 and AROS during stress-induced premature senescence
Experimental & Molecular Medicine (2023)
-
SRC kinase-mediated signaling pathways and targeted therapies in breast cancer
Breast Cancer Research (2022)
-
Efficient terminal erythroid differentiation requires the APC/C cofactor Cdh1 to limit replicative stress in erythroblasts
Scientific Reports (2022)
-
Insufficiency of FZR1 disturbs HSC quiescence by inhibiting ubiquitin-dependent degradation of RUNX1 in aplastic anemia
Leukemia (2022)
-
Ubiquitin signaling in cell cycle control and tumorigenesis
Cell Death & Differentiation (2021)
