
Abstract
Tumours recruit mesenchymal stem cells to facilitate healing, which induces their conversion into cancer-associated fibroblasts that facilitate metastasis. However, this process is poorly understood on the molecular level. Here we show that CXCL16, a ligand for CXCR6, facilitates mesenchymal stem cell or very small embryonic-like cells recruitment into prostate tumours. CXCR6 signalling stimulates the conversion of mesenchymal stem cells into cancer-associated fibroblasts, which secrete stromal-derived factor-1, also known as CXCL12. CXCL12 expressed by cancer-associated fibroblasts then binds to CXCR4 on tumour cells and induces an epithelial-to-mesenchymal transition, which ultimately promotes metastasis to secondary tumour sites. Our results provide the molecular basis for mesenchymal stem cell recruitment into tumours and how this process leads to tumour metastasis.
Similar content being viewed by others


Introduction
Tumours have long been considered as wounds that do not heal1. Wound healing normally requires the participation of many different cell types as well as the activation of a vast number of cellular processes including matrix degradation, proliferation and recruitment of inflammatory cells. In addition, cells such as fibroblasts, epithelial and endothelial cells are also recruited and they too must coordinate their activities with inflammatory cells to pattern regeneration of normal tissues. As in normal wound healing, tumours also activate the recruitment of host cells into tumour beds to regulate survival and proliferation2. In this context, recent attention has focused on the roles of dendritic, tumour-associated macrophages and other early hematopoietic lineage populations that establish niches within tumours that foster and protect cancer stem cells from cytotoxic and metabolic stresses3. Moreover, many of these same cell populations are thought to promote and establish premetastatic niches at distant sites, which ultimately facilitate the ability of disseminated tumour cells to establish metastatic foci4,5.
Mesenchymal stem cells (MSCs) are multipotent cells that contribute to tissue homoeostasis and regeneration. Normally, MSCs are rapidly recruited into sites of injury and inflammation where they differentiate into a variety of connective tissue cell types6,7. Recently, marrow-derived MSCs were shown to participate in tumour progression by establishing a favourable tumour microenvironment, differentiating into cancer-associated fibroblasts (CAFs), which establish cytokine networks that promote progression and migration8,9,10,11,12,13. Precisely how MSCs are recruited into primary tumour sites, how they contribute to the development of tumour niches for cancer stem cells, what regulates the conversion of MSCs into CAFs and how CAFs promote metastasis is not entirely understood.
Skeletal metastases are one of the most serious complications of prostate cancer14. Growing evidence suggests that the CXC chemokine ligand 16 (CXCL16) and its receptor CXCR6 have important roles in tumour progression and bone metastasis15,16,17,18. CXCL16 is one of a small number of chemokines expressed as both soluble and cell surface molecules and it functions as a chemoattractant for many cell types19. CXCL16 is secreted by cells in response to IFN-γ, TNF-α and IL-1β (refs 18, 20, 21, 22, 23, 24, 25, 26). CXCL16 is the sole ligand for CXCR6, a member of the seven transmembrane G protein-coupled receptor family, which signals through the AKT/mTOR pathways16. Our group has shown that in primary and metastatic prostate cancer, CXCL16 is highly expressed compared with normal prostate epithelial cells16,27. In addition, CXCL16/CXCR6 is involved in prostate cancer migration and invasion16,19,24,27.
In the present study we demonstrate that tumour growth is dependent on the recruitment of MSCs into human and mouse prostate cancer in response to CXCL16. Once in the tumour, CXCL16 binding to CXCR6 expressed by MSCs, stimulates their conversion into CAFs, which subsequently secrete enhanced levels of CXCL12. CXCL12 expression by CAFs promotes an epithelial-to-mesenchymal transition (EMT) of the cancer cells, which supports metastasis to secondary sites. Together, these studies provide the molecular basis for MSC recruitment into primary tumours, and the conversion of MSCs into CAFs that ultimately lay the foundations for the EMT required for establishing distant metastasis.
Results
CXCL16 secreted by prostate cancer recruits MSCs
We reasoned that cells with stem cell-like properties must rapidly migrate into wounds to initiate tissue regeneration. We hypothesized that CXCR6-expressing MSCs from the bone marrow are likely rapidly recruited into tumours in response to CXCL16. Therefore, human and mouse bone marrow MSCs (Supplementary Fig. S1a) were evaluated for CXCR6 expression. Human (Fig. 1a) and freshly isolated non-passaged (P0) murine MSCs (Lin−Sca-1+CD45− or very small embryonic-like (VSEL) stem cells)7,28,29 and second passage MSCs (P2) expressed CXCR6, while MSCs isolated from CXCR6−/− (MSCCXCR6−/−) mice did not (Fig. 1c). Tissue microarrays from prostate cancer patients demonstrated that CXCL16 expression correlated with tumour aggressiveness (Fig. 1e; Supplementary Fig. S1b). Prostate cancer and breast cancer cell lines expressed significant levels of CXCL16 (Fig. 1f; Supplementary Fig. S1c–g). In vitro, P0 or P2 MSCs isolated from CXCR6 wild-type mice (MSCCXCR6+/+) migrated towards CXCL16, while MSCCXCR6−/− did not (Fig. 1h). To determine what role CXCL16 has in recruiting MSCs into tumours, prostate cancer was implanted subcutaneously (s.c.) into CXCR6+/+ or CXCR6−/− mice (Supplementary Fig. S1h). Significantly, greater tumour volume was observed when the tumours were grown in CXCR6+/+ versus CXCR6−/− mice, suggesting that CXCR6-expressing host cells modulate tumour growth (Fig. 1i). Surprisingly, more MSCs were found in the tumours grown in the CXCR6+/+ mice than in the tumours grown in CXCR6−/− mice (Fig. 1j; Supplementary Table S1), though there were no differences in MSC numbers in the marrow of the CXCR6+/+ versus CXCR6−/− mice (Supplementary Fig. S1i), suggesting a specific recruitment of MSCs into tumours facilitates growth.

(a) CXCR6 mRNA expression by human MSCs (P0 and P2). (b) Expression of CXCR6 protein by human MSCs. Controls included isotype-matched antibodies and fibroblast-specific protein 1 (FSP1) for MSCs. Scale bars, 100 μm. (c) CXCR6 mRNA by mMSCs. CXCR6 expression was determined in freshly isolated, non-cultured (P0) or P2 murine MSCs from CXCR6+/+ or CXCR6−/− mice. Human and murine osteoblasts (HOB and MC3T3-E1) were used as a negative control. (d) Expression of CXCR6 by murine P2 CXCR6+/+ or CXCR6−/− MSCs by immunohistochemistry (IHC) staining. Scale bar, 100 μm. Data in a–c are representative of mean with s.d. for triplicates in each of the three independent experiments (Student’s t-test). (e) CXCL16 expression in human prostate cancer tissue microarray in Supplementary Fig. S1b. Differences noted between normal prostate (n=30), Gleason 4+5 (n=9), Gleason 6+7 (n=18), and Gleason 8+9 (n=15) (mean±s.d. Student’s t-test). Secretion of CXCL16 by human prostate cancer cell lines (f) and murine prostate cancer cell lines (g) as determined by ELISA (mean±s.d., n=3 independent experiments, Student’s t-test). (h) Migration of freshly isolated, non-cultured (P0) or P2 murine MSCs from CXCR6+/+ or CXCR6−/− mice in response to CXCL16. The % migrated MSC was determined by hemocytometer counting (mean±s.d., n=3 independent experiments, Student’s t-test). (i) CXCR6+/+ or CXCR6−/− mice were implanted s.c. with RM1 cells and caliper measurements of tumour growth performed over 25 days. *Significant differences between tumours grown CXCR6+/+ and CXCR6−/− mice (mean±s.d., for 7 animals per group, n=3 independent experiments, P<0.05; Student’s t-test). (j) % MSCs (P0) present in RM1 tumours grown in CXCR6+/+ or CXCR6−/− mice at day 25 (mean±s.d. for 7 animals per group, n=3 independent experiments, Student’s t-test). (k) SCID mice were implanted s.c. with PC3 cells mixed with MSCP0CXCR6+/+ or MSCP0CXCR6−/− cells and tumour growth was evaluated by caliper measurements over 42 days. *Significant differences between tumours grown with PC3 cells mixed with MSCP0CXCR6+/+ and MSCP0CXCR6−/− cells (mean±s.d. for n=5 animals per group, n=1 independent experiment, P<0.05, Student’s t-test).
To validate that these results were representative of other tumours and not specific to subcutaneous tumour growth, the studies were repeated with human prostate cancer and breast cancer cell lines in an orthotopic setting. As seen previously, robust MSC recruitment into the tumours occurred when prostate cancer or breast cancer cell lines were implanted in an orthotopic setting (Supplementary Fig. S1j–r; Supplementary Table S1). To confirm that MSCs signalling through CXCR6 has a critical role in tumour growth, prostate cancer cells were mixed with MSCP0CXCR6+/+ or MSCP0CXCR6−/− and tumour growth was monitored. As predicted, significantly larger tumour growth occurred when the tumour cells were mixed with MSCs expressing CXCR6 (MSCP0CXCR6+/+) compared with tumours established with MSCs not in which CXCR6 expression is knocked out (MSCP0CXCR6−/−) (Fig. 1k). Together these findings suggest a key role for CXCL16/CXCR6 in recruiting MSCs into tumours, and supporting tumour growth.
CXCL16/CXCR6 signalling induces CAF formation and CXCL12
Local and recruited MSCs are known to convert into tumour-associated fibroblasts (TAFs) or CAFs in close proximity to tumour cells30,31. To test whether prostate cancer-derived CXCL16 facilitates the conversion of MSCs into CAFs, MSCs were treated with CXCL16 and examined for expression of α-SMA and vimentin. MSCsCXCR6+/+ converted to α-SMA+ and vimentin+ expressing cells after CXCL16 stimulation, while MSCsCXCR6−/− did not (Fig. 2a–d). To further investigate the role that CXCL16/CXCR6 signalling has in tumour growth, MSCs isolated from CXCR6+/+ or CXCR6−/− mice were treated with conditioned media derived from human and murine prostate cancer cell lines and examined for expression of α-SMA and vimentin. MSCCXCR6+/+ cells expressed significant levels of α-SMA and vimentin after treatment with conditioned media derived from prostate cancer cell lines, while MSCCXCR6−/− cells did not (Fig. 2e; Supplementary Fig. S2a,b). To validate these observations, prostate tumours grown in CXCR6+/+ or CXCR6−/− mice were probed for the CAF phenotype (Supplementary Fig. S2c). Paralleling the in vitro findings, fewer α-SMA+ and vimentin+ cells were identified in tumours grown in the CXCR6−/− mice compared with CXCR6+/+ mice (Fig. 2g). Previously, we demonstrated that CXCL16 expression in human tumours corresponds with increasing Gleason grade27. Therefore, to validate the murine observations in a human setting, tumour tissue microarrays derived from human prostate cancer samples were stained for vimentin. The data demonstrate that more CAFs expressing vimentin were detected in the Gleason 4+5 prostate cancer than in the benign prostate cancer tissues (Fig. 2h; Supplementary Fig. S2d). A second critical feature of the CAF phenotype is the expression of stromal-derived factor-1 (SDF-1 or CXCL12), which facilitates metastases32,33. Colocalization studies identified that more α-SMA+/CXCL12+ and vimentin+/CXCL12+-expressing cells were observed in tumours isolated from CXCR6+/+ versus CXCR6−/− mice (Fig. 2j) and greater levels of CXCL12 were identified in the extracellular milieu of tumours grown in CXCR6+/+ versus CXCR6−/− mice (Fig. 2l) or when mMSCs are treated with CXCL16 (Supplementary Fig. S2e). Next, CXCL12 secretion by MSCs was examined in response to CXCL16. MSCCXCR6+/+ but not MSCsCXCR6−/− secreted CXCL12 in response to CXCL16 (Fig. 2m), which was regulated through Erk and NF-κB signalling (Supplementary Fig. S2f–h).

MSCs isolated from CXCR6+/+ or CXCR6−/− mice (P2) were exposed to vehicle or CXCL16 (100 ng ml−1) for 7 days. The expression of α-SMA (a) mRNA by qRT–PCR or (b) protein by immunohistochemistry (IHC) (red, α-SMA, white arrows; blue, DAPI nuclear stain), or for vimentin (c) mRNA or (d) protein (red, vimentin, white arrows; blue, DAPI nuclear stain) were evaluated. Scale bars, 100 μm. MSCs isolated from CXCR6+/+ or CXCR6−/− mice were exposed to human prostate cancer cell conditioned media for 7 days. The expression of α-SMA (e) and vimentin (f) mRNA were evaluated by qRT–PCR. (g) IHC of localization of α-SMA and vimentin-positive cells within tumours grown in CXCR6+/+ or CXCR6−/− mice (red, α-SMA or vimentin, white arrow; blue, DAPI nuclear stain). Scale bars, 100μm. Data in a, c, e and f are representative of mean with s.d. for triplicates in each of the three independent experiments (Student’s t-test). (h) IHC of vimentin or FSP1-positive cells within benign or Gleason 4+5 prostate cancers in human tissue microarrays (TMAs) (red, vimentin, white arrows; green, FSP1,white arrows; blue, DAPI nuclear stain). Staining for FSP1 served as a positive control of MSCs. Scale bars, 100 μm. (i) Quantification of Fig. 2h. Mean expression scores were multiplied by percent of positive cells in the field. Significant differences were noted between benign (n=30) or Gleason 4+5 prostate (n=6) (mean±s.d., Student’s t-test). Colocalization of CXCL12 expression with (j) α-SMA- and (k) vimentin-positive cells (white arrows) within tumours grown in CXCR6+/+ or CXCR6−/− mice. Scale bars, 100 μm. (l) CXCL12 protein expression in the extracellular milieu within tumours grown in CXCR6+/+ or CXCR6−/− mice (mean±s.d., for triplicates in each of three independent experiments, Student’s t-test). (m) Secretion of CXCL12 from MSCP2CXCR6+/+ cells or MSCP2CXCR6−/− cells were observed following exogenous CXCL16 treatment by ELISA (mean±s.d. for triplicates in each of three independent experiments, Student’s t-test, analysis of variance). (n) Colocalization of CXCL12 with vimentin following exposure of MSCP2CXCR6+/+ cells or MSCP2CXCR6−/− cells to CXCL16 in vitro. Scale bars, 100 μm.
Knockdown of CXCL16 reduces MSC recruitment and CAF formation
To further explore the role of CXCL16 secreted by tumour cells and MSC cell recruitment, lentiviral vectors were used to silence CXCL16 expression in RM1 cells (RM1shCXCL16). After clonal selection, individual clones were pooled and assayed by qRT–PCR and ELISA for the reduction of CXCL16 expression (Fig. 3a). We then tested whether RM1shCXCL16 cells have the same capabilities to stimulate migration of MSCs compared with control (RM1Control) cells. As expected, the knockdown of CXCL16 expression in RM1 cells inhibited the migration of MSC cells in vitro (Fig. 3c). In conjunction with these studies, tumour growth over time was evaluated in CXCR6+/+ mice (Fig. 3d). As shown in Fig. 3e, tumours generated from the RM1Control cells rapidly developed, while tumour growth of the RM1shCXCL16 cells was dramatically suppressed. Importantly, more P0 MSCs were identified in the tumours grown with RM1Control cells than in tumours grown with RM1shCXCL16 cells (Fig. 3f; Supplementary Table S1). Taken together, these data suggest that CXCL16 expression by prostate tumours is critical for tumour growth and MSC cell recruitment.

(a) Expression of CXCL16 mRNA in the RM1Control or RM1shCXCL16 cells by qRT–PCR. (b) Secretion of CXCL16 by RM1Control or RM1shCXCL16 cells was determined by ELISA. (c) Migration of MSCCXCR6+/+ cells was determined towards RM1Control cells or RM1shCXCL16 cells. Data in a–c are representative of mean with s.d. for triplicates in each of three independent experiments (Student’s t-test). Significance was determined using a Student’s t-test. (d) Experimental scheme of RM1Control or RM1shCXCL16 cell implantation to CXCR6+/+ mice for examining tumour growth and MSC cell recruitment to tumours. (e) The tumour growth of RM1Control or RM1shCXCL16 cells on CXCR6+/+ mice was evaluated by caliper measurements over 23 days. *Significant differences between tumours grown with RM1Control and RM1shCXCL16 cells (mean±s.d., for n=5 animals per group, n=2 independent experiments, P<0.05; analysis of variance). (f) % MSCs present in RM1Control or RM1shCXCL16 tumours grown in CXCR6+/+ mice (mean±s.d., for n=5 animals per group, n=2 independent experiments, Student’s t-test).
Further studies examined the generation of the CAF phenotype in response to prostate cancer expressing CXCL16. In in vitro studies, MSCCXCR6+/+ cells expressed high levels of α-SMA and vimentin after treatment with conditioned media from RM1Control cells but not after treatment with conditioned media isolated from RM1shCXCL16 cells (Supplementary Fig. S3a,b). In in vivo studies, the tumours were generated from RM1Control or RM1shCXCL16 cells in CXCR6+/+ mice (Supplementary Fig. S3c). Fewer α-SMA+ and vimentin+ cells were identified in tumours generated from RM1shCXCL16 cells compared with tumours generated from RM1Control cells (Supplementary Fig. S3d). Colocalization studies identified that more α-SMA+/CXCL12+ and vimentin+/CXCL12+ cells were observed in tumours from RM1Control versus RM1shCXCL16 cells (Supplementary Fig. S3e,f).
CAF CXCL12 promotes prostate cancer cell EMT
To explore the extent to which CXCL16 drives metastasis, we determined whether CAF-derived CXCL12 activates an EMT in prostate cancer cells (Supplementary Fig. S4a). Loss of cell–cell contacts and the emergence of a spindle-shaped morphology was observed following CXCL12 treatments of prostate cancer cells or when they were cocultured with MSCsCXCR6+/+, but not when cocultured with MSCCXCR6−/− (Fig. 4a). In fact, when prostate cancer cells were treated with CXCL12 or cocultured with MSCsCXCR6+/+, but not MSCsCXCR6−/− cells, a near complete loss of the epithelial transcriptome occurred including E-cadherin, reduced cytokeratin, enhanced expression of N-cadherin, vimentin, α-SMA, β-catenin, snail and slug were observed (Fig. 4a–c). When tumour microarrays were stained for E-cadherin or N-cadherin, more E-cadherin-expressing prostate cancer cells were detected in the benign prostate tissues, whereas more N-cadherin-expressing prostate cancer cells were detected in the Gleason 4+5 prostate cancers (Fig. 4d; Supplementary Fig. S4b)34,35,36. Enhanced expression of the CXCL12 receptor CXCR4 is known to facilitate migration and metastasis in vivo37,38. We observed that CXCR4 expression by prostate cancer was enhanced following induction of an EMT phenotype in vitro and was associated with enhanced tumour growth in vivo (Fig. 4f). Studies with anti-CXCR4 antibody and the CXCR4 inhibitor AMD3100 showed that CXCL12 induces prostate cancer towards an EMT phenotype (Fig. 4h; Supplementary Fig. S4c–e). In fact, prostate cancer cells that had undergone an EMT were significantly more responsive than their parental counterparts to CXCL12 or serum (Fig. 5a), such that CXCR4 blockade prevented prostate cancer migration in vitro (Fig. 5b).

(a) Vehicle-or CXCL12-treated RM1 cells, or RM1 cells cocultured with MSCs from CXCR6+/+ or CXCR6−/− mice were examined by phase contrast microscopy and immunohistochemistry (IHC) staining for cytokeratin, E-cadherin, N-cadherin, vimentin and α-SMA. Scale bars, 100 μm. Representative images from two independent studies. (b) Western blots analysis for epithelial (E-cadherin) and mesenchymal (N-cadherin, β-catenin, snail, slug) markers. Representative images from two independent studies. (c) EMT markers in the primary tumour were examined by IHC. Colocalization of E-cadherin or N-cadherin with FSP1 was observed. More E-cadherin by prostate cancer cells (red; white arrows) was detected in close proximity to FSP1-expressing MSC cells (green; orange arrows) in tumours grown in CXCR6−/− mice compared with tumours grown in CXCR6+/+ mice. In contrast, more N-cadherin-expressing prostate cancer cells (red; white arrows) were detected in close proximity to N-cadherin and FSP1 co-expressing CAF cells (yellow; yellow arrows) when the tumours were grown in CXCR6+/+ mice compared with tumours grown in CXCR6−/− mice. Blue, DAPI nuclear stain. Scale bars, 100 μm. Representative images derived from n=7 mice per group. (d) IHC of E-cadherin- or N-cadherin-positive cells within benign or Gleason 4+5 prostate cancers in human prostate tissue microarrays (TMAs) (red, E-cadherin or N-cadherin, white arrows; blue, DAPI nuclear stain). Scale bars, 100 μm. (e) Quantification of panel d. Mean expression scores were multiplied by percent of positive cells in the field. Significant differences were noted between benign (n=30) or Gleason 4+5 prostate (n=6) (mean±s.d., analysis of variance). (f) CXCR4 mRNA was determined for EMT-induced RM1 cells following CXCL12 treatment or co-culture with MSCs derived from CXCR6+/+ or CXCR6−/− mice (mean±s.d., n=3 independent experiments). (g) More CXCR4-expressing RM1 cells (red; white arrows) were detected in close proximity to CXCR4 and FSP1 (green; orange arrows) co-expressing CAF cells (yellow; yellow arrows) when the tumours were grown in CXCR6+/+ mice compared with tumours grown in CXCR6−/− mice. Scale bars, 100 μm. Representative images from an experiment with n=7 animals per group. (h) AMD3100 or anti-CXCR4 antibody prevents the development of EMT by RM1 cells following CXCL12 exposure. Scale bars, 100 μm. Representative images from two independent studies.

(a) Migration assays were performed in Transwell plates using 10% serum or CXCL12 as chemoattractants. Migration towards 0.5% serum was used as a negative control. (b) Blockade of CXCR4 by AMD3100 or anti-CXCR4 antibody prevents prostate cancer migration towards CXCL12 or MSCs isolated from CXCR6+/+, but not CXCR6−/− animals. Data in a and b are representative data from two independent studies (mean±s.d., analysis of variance). Significance was determined using a Student’s t-test. RFP-labelled RM1WT or RM1EMT cells (Supplementary Fig. S5a) were incubated with vehicle or AMD3100 in vitro, and then inoculated by intra-cardiac (i.c.) injection into CXCR6+/+ or CXCR6−/− (n=7). Metastasis was assessed by qPCR for RFP in a number of tissues. (c,d) Number of metastatic RM1 cells following i.c. injection. *Significance between RM1WT treated with vehicle and RM1WT treated with AMD3100 (P<0.05). #Significance between RM1WT treated with vehicle and RM1EMT cells treated with vehicle (P<0.05). †Significance between RM1EMT treated with vehicle and RM1EMT treated with AMD3100 (P<0.05). Error bars represents mean±s.d., n=2 independent experiments, P<0.05; Student’s t-test. (e–h) RM1 cells expressing RFP were identified in the femur of CXCR6+/+ or CXCR6−/− mice following i.c. injection. Red arrows identify RM1 cells. White arrows identify osteoblast on the bone surface staining positive for CXCL12 expression. Scale bars, 100 μm. (f,h) Quantification of panels e and g, respectively. The numbers of RM1 cells were quantified on the endosteal region of the seven long bones. Endosteal regions were defined as 12 cell diameters from bone surfaces. (mean±s.d. (n=3), analysis of variance).
EMT-induced CXCR4 expression promotes metastasis
In an animal model of bone metastasis, red fluorescent protein (RFP)-expressing wild-type (RM1WT) or EMT-induced RM1 cells (RM1EMT) (Supplementary Fig. S5a) were incubated with AMD3100 or vehicle in vitro, and then injected by an intra-cardiac route into CXCR6+/+ or CXCR6−/− mice to establish prostate cancer bone metastases (Supplementary Fig. S5b). First, we examined CXCL12 levels in a number of osseous sites and in blood (Supplementary Fig. S5c). All the animals from CXCR6+/+ or CXCR6−/− mice had significant numbers of disseminated tumour cells (DTCs) in their bones 10 days following injection of RM1WT or RM1EMT cells (Fig. 5c–h). In contrast, RM1WT cells pretreated with AMD3100 had a reduced number of DTCs in their calvaria, spine and femur from CXCR6+/+ mice (Fig. 5c). Strikingly, animals receiving RM1EMT cells showed a significant increase of in the total DTC load in most osseous tissues compared with animals injected with the RM1WT cells alone. Critically, the number of DTCs were significantly reduced following CXCR4 blockade (Fig. 5c). However, fewer DTCs were identified in the bones of the CXCR6−/− versus CXCR6+/+ mice (Fig. 5d). Together, these data suggest that CXCL16 initiated induction of an EMT, and CXCR4 expression via MSC activation has an important and critical role in prostate cancer cell dissemination and metastasis.
Discussion
Tumours arise from cells that have sustained and multiple genetic mutations resulting in deregulation of normal growth-control mechanisms39. Recent evidence also suggests that the microenvironment itself regulates crucial neoplastic progression steps in haematological tumours40. Cancer cells not only interact with each other, their extracellular matrix and inflammatory cells, but also with recruited and resident cells of mesenchymal origin. The characteristic transformation of stromal cells that accompanies, or precedes the malignant conversion of epithelial cells has been linked to CAFs41,42,43. Several cancer types demonstrate the concept that these fibroblasts can determine the fate of the epithelial cells, promote malignant progression either through soluble factors, and cell–cell interactions and/or alterations of the extracellular matrix43. The complexity of these interactions has been amplified by studies showing alterations in resident cells may be drivers in cancer progression44.
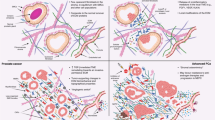
MSCs are multipotent cells that contribute to tissue homoeostasis and regeneration. Tumours recruit MSCs to facilitate tumour progression and metastasis. Here, we provide evidence that the recruitment of MSCs into prostate cancer is dependent on the expression of the CXCR6 ligand, CXCL16 by tumour cells (Fig. 6). CXCR6 signalling supports the recruitment, conversion and activation of MSC into CXCL12-secreting CAFs. Moreover, enhanced CXCL12 secretion supports an EMT conversion of the prostate cancer cells and an increase in the expression of the CXCL12 receptor, CXCR4. These events result in enhanced tumour progression and ultimately extravasation and metastasis. Targeting MSCs and the CXCL16/CXCR6 axis may prevent tumour progression and metastasis of prostate cancer and provide a more effective therapeutic strategy for prostate cancer.

Model showing potential mechanisms underlying primary prostate cancer progression by the recruitment of MSCs and bone metastasis. Secretion of CXCL16 by cancer cells recruits MSCs into tumour sites. Tumour-derived CXCL16 interacts with its receptor, CXCR6 on MSCs and activates signal transduction, leading MSCs to convert into CAFs, which secrete high levels of CXCL12. CXCL12 promotes the malignant transformation of proliferating cancer cells to an EMT. EMT enhances CXCR4 expression in prostate cancer cells. CXCR4 expression facilitates metastasis.
Several lines of evidence demonstrate that cells with stem cell-like properties must be able to migrate into wound sites rapidly for regeneration to occur45,46,47,48. In the context of neoplasia, bone-marrow-derived MSCs have been shown to increase the metastatic potential of weakly metastatic human ovarian cancer cells, in part through the conversion to a CAF phenotype49. We demonstrate that primary small Lin−Sca-1+CD45− cells (VSEL stem cells) isolated from marrow and MSCs passaged in vitro express high mRNA levels of CXCR6 and migrate towards CXCL16. CXCL16 exists both in soluble and transmembrane forms50. CXCL16 is expressed on monocytes, macrophages, B cells and dendritic cells51,52, and both forms of CXCL16 are expressed by human tumour cells15,16,22,53. The precise role of soluble versus transmembrane CXCL16 in tumour progression remains unclear. Transmembrane CXCL16 may function to suppress tumour proliferation, while soluble CXCL16 induces proliferation and migration of cancer cells19. But which form of CXCL16 regulates prostate cancer growth within tumours is still unclear.
Previous work by our group demonstrated that CXCR6 expression in tumours correlated with Gleason score. Using lentiviral vectors to overexpress CXCR6 in PC3, LNCaP C4-2B and LNCaP cells, or by reducing CXCR6 expression by siRNA, we also found that modifying CXCR6 expression altered the ability of prostate cancer cell lines to invade and grow both in vitro and in vivo16. In addition, CXCR6 regulates the expression of several proangiogenic factors including IL-8 and vascular endothelial growth factor, both of which are likely to participate in the regulation of tumour angiogenesis16. In part, binding of CXCL16 to CXCR6 induces activation of Akt, p70S6K, and eukaryotic initiation factor 4E-binding protein 1 in prostate cancer cells in addition to mammalian target of rapamycin (mTOR) pathways16. Moreover, rapamycin not only drastically inhibited CXCL16-induced prostate cancer cell invasion and growth but also reduced secretion of IL-8 or vascular endothelial growth factor levels and inhibited expression of other CXCR6 targets including CD44 and matrix metalloproteinase16.
The present study further adds to the complexity of CXCL16/CXCR6 signalling and the role that paracrine/autocrine loops have in tumour progression. These findings are similar to previous observations showing that CXCL12/CXCR4 signalling54,55 supports and cross-talk between CAFs and prostate cancer33. Our results illustrate the molecular basis for MSC recruitment into tumours and how this process leads to tumour metastasis by coupling activities of the CXCL12/CXCR4 and CXCL16/CXCR6 axes. By demonstrating that MSCs have an active role in establishing an EMT in cancer, which ultimately facilitates metastasis, MSCs are critical components of the host-response network in tumours and represent viable entities for the design of targeting therapies to prevent the establishment of distant metastasis.
Methods
Animals
Male CXCR6+/+ or CXCR6−/− mice (5–7 weeks; Jackson Laboratory, Bar Harbor, ME) and SCID mice (CB.17. SCID; Taconic, Germantown, NY) were used as transplant recipients. All animal procedures were performed in compliance with the institutional ethical requirements and approved by the University of Michigan Committee for the Use and Care of Animals (UCUCA).
Cell lines
The human prostate cancer cell lines PC3, LNCaP and DU145, and murine prostate cancer cell lines RM1 and Tramp were used (American Type Culture Collection, Rockville, MD). LNCaP were originally isolated from a lymph node of a patient with disseminated bony and lymph node involvement. Prostate cancer cell lines were cultured in RPMI 1640 (Invitrogen, Grand Island, NY) with 10% fetal bovine serum (FBS, Invitrogen), 1% penicillin-streptomycin (P/S, Invitrogen). The human prostate epithelial cell line RWPE-1 (ATCC) was cultured in Keratinocyte-SFM with supplements (Invitrogen). The mouse prostate epithelial cell line NMPE (CHI Scientific, Maynard, MA) was cultured in Mouse Prostate PrimaCell medium (CHI Scientific). The human breast epithelial cell line MCF-10A and breast cancer cell line MCF-7 were kindly provided by Dr Max Wicha (University of Michigan). MCF-10A cell line was cultured in DMEM/F12 with supplements (Invitrogen) and MCF-7 cell line was cultured in DMEM with supplements (Invitrogen). Conditioned media were collected and frozen after filtration through a 0.22-μm filter. For induction of EMT, RM1 cells were cultured to confluence, and serum-starved for 24 h. The cells were cultured in RPMI with 0.1% FBS supplemented with 200 ng ml−1 CXCL12 (cat. 350-NS, R&D Systems, Minneapolis, MN) for 48 h, and termed RM1EMT versus RM1WT. RM1 cells used in metastasis assays were labelled with RFP by lentiviral transfection (Supplementary Fig. S5a) and selected by FACS.
MSCs
Human MSCs (hMSCs, Lonza, Walkersville, MD), and freshly isolated mouse MSCs (non-passaged (P0)) (Lin−Sca-1+CD45− cells or very small embryonic-like (VSEL) stem cells) and primary bone-marrow-derived MSCs (passaged once (P1) or twice (P2)) from CXCR6+/+ or CXCR6−/− mice were used for this study. The hMSCs were cultured in DMEM supplemented with 10% FBS, and 1% P/S. For mouse MSCs, after killing, marrow was flushed from femurs and tibias of both CXCR6+/+ and CXCR6−/− mice into α-MEM (Invitrogen) with 2% FBS to generate primary MSCs and cultured in α-MEM containing 10% FBS and 1% P/S. Once confluent, the cells were passaged 2–3 times to minimize macrophage contamination. Subsequently, repopulated bone-marrow-derived MSCs were obtained, and cells were cultured in DMEM containing 10% FBS and 1% P/S. For MSC differentiation assays, mouse primary bone-marrow-derived MSCs were cultured in adipogenic, osteogenic or chondrogenic conditions for 2 weeks, and cells were stained with Alizarin Red S, Oil Red O and Alcian Blue, respectively.
Isolation of MSCP0 (VSEL) cells
Small Lin−Sca-1+CD45− (or <8μm VSEL stem cells), referred to as MSCP0 herein, were isolated from mononuclear bone marrow cells (1 × 108 cells per ml) from both CXCR6+/+ and CXCR6−/− mice, and resuspended in PBS containing 2% heat-inactivated FBS and 1% P/S. The cells were incubated with the following antibodies: biotin-conjugated rat anti-mouse Ly-6A/E (Sca-1) (cat. 553334, 1:50 dilution, BD Pharmingen, San Diego, CA), streptavidin-PE-Cy5 (cat. 554062, 1:50 dilution, BD Pharmingen), anti-CD45-APC (cat. 557659, 1:30 dilution, BD Pharmingen), anti-CD45R/B220-PE (cat. 553089, 1:200 dilution, BD Pharmingen), anti-Gr-1-PE (cat. 553128, 1:200 dilution, BD Pharmingen), anti-TCRαβ PE (cat. 553172, 1:200 dilution, BD Pharmingen), anti-TCRγζ PE (cat. 553178, 1:200 dilution, BD Pharmingen), anti-CD11b PE (cat. 557397, 1:200 dilution, BD Pharmingen) and anti-Ter-119 PE (cat. 553673, 1:200 dilution, BD Pharmingen). MSCP0 cells were freshly isolated by cell sorting (FACSAria II, Becton Dickinson, Mountain View, CA).
Western blot analyses
Cells were lysed in RIPA buffer with protease inhibitors and lysates separated on 10% SDS-polyacrylamide gel and transferred to PVDF membranes. The membranes were incubated with 5% milk for 1 h and probed overnight at 4 °C with antibodies targeting N-cadherin (cat. 610921, 1:2,500 dilution, BD Transduction laboratory, Lexington, KY), E-cadherin (cat 610181, 1:25 dilution, BD Transduction laboratory, Lexington, KY), β-catenin (cat. 9582, 1:1,000 dilution, Cell Signaling Technology, Danvers, MA), snail (cat. 3879, 1:1000 dilution, Cell Signaling), slug (cat. 9585, 1:1,000 dilution, Cell Signaling) and β-actin (cat. 4970, 1:1,000 dilution, Cell Signaling). After washing, blots were incubated with peroxidase-conjugated anti-rabbit IgG HRP secondary antibodies (cat. W401B, 1:7,500 dilution, Promega, Madison, WI) for 1 h. Protein expression was detected with SuperSignal West Pico Chemiluminescent Substrate (cat. 34080, Thermo Scientific, Rockford, IL).
Immunohistochemistry
Cells were fixed and tissue sections were de-waxed and re-hydrated, then blocked with Image-iT FX signal enhancer for 30 min and incubated 2 h at room temperature in dark with 10 μg ml−1 primary antibodies combined with reagents of Zenon Alexa Fluor 488 (green) or 555 (red) labelling kit. CXCR6 (cat. NLS-1102, 1:100 dilution, Novus Biologicals, Littleton, CO), fibroblast-specific protein 1 (FSP1, cat. 07-2274, 1:100 dilution, Millipore, Temecula, CA), CXCL12 (cat. ab25117, 1:100 dilution, Abcam, Cambridge, MA), cytokeratin (cat. ab9377, 1:200 dilution, Abcam), E-cadherin (cat. 610181, 1:25 dilution, BD Transduction Laboratory), N-cadherin (cat. 610921, 1:25 dilution, BD Transduction Laboratory), vimentin (cat. ab8978, 1:100 dilution, Abcam), anti-mouse α-SMA (cat. ab5694, 1:20 dilution, Abcam), CXCR4 (cat. ab2074, 1:100 dilution, Abcam) and RFP (cat. ab62341, 1:50 dilution, Abcam) antibodies were diluted in PBST (PBS plus 0.2% Triton X-100). The cells and tissue sections were post-fixed with 10% formalin for 10 min followed by processing with ProLong Gold antifade reagent with DAPI medium. Images were acquired with an Olympus FV500 confocal microscope.
Human prostate tissue microarrays were purchased from US Biomax, Inc. (Rockville, MD). Anti-human CXCL16 (ab17537, 1:20 dilution, Abcam), FSP1 (cat. 07-2274, 1:100 dilution, Millipore), vimentin (cat. ab8978, 1:100 dilution, Abcam), E-cadherin (cat. 610181, 1:25 dilution, BD Transduction Laboratory) and N-cadherin (cat. 610921, 1:25 dilution, BD Transduction Laboratory) antibodies were used. Staining intensity was ranked from 1 to 4 (1, negative; 2, weak; 3, moderate; 4, strong intensity staining).
Subcutaneous tumour growth
Tumours were established by injecting RM1 (1 × 104) cells in growth factor-reduced Matrigel (cat. 354236, BD Bioscience, Bedford, MA) s.c. into 5- to 7-week-old male CXCR6+/+ or CXCR6−/− mice (Supplementary Fig. S1h; Supplementary Table S1). Tumours were also established by injecting RM1Control or RM1shCXCL16 (1 × 104) cells in growth factor-reduced Matrigel s.c. into 5- to 7-week-old male CXCR6+/+ mice (Fig. 3d; Supplementary Table S1). In some cases, tumours were also established by injecting human PC3 cells (2 × 105) mixed with MSCP0CXCR6+/+ or MSCP0CXCR6−/− cells (2 × 103) in growth factor-reduced Matrigel s.c. into 5- to 7-week-old male SCID mice. The animals were monitored daily and tumour volumes were evaluated every 3–7 days. Tumour volumes were calculated using the formula V=(the shortest diameter) × (the longest diameter) × height. After 23–42 days the animals were killed. The tumours were measured and prepared for Lin−Sca-1+CD45− cell analyses and histology.
Orthotopic tumour growth
After anaesthesia with 2–4% isoflurane inhalation, a low midline incision was made in the lower abdomen. The human and murine prostate cancer cells (5 × 104 to 5 × 105) in 20 μl of PBS were injected into the right or left dorsolateral lobe of the prostate of 5- to 7-week-old male SCID or CXCR6+/+ mice, and the wound was closed with surgical clips. The human breast cell lines (1 × 106) in growth factor-reduced Matrigel were injected abdominal fat pad of 5–7 week-old female SCID mice (Supplementary Fig. S1j; Supplementary Table S1).
In vivo metastasis assays
Cells (2 × 105 RFP-labelled RM1 cells/mouse) were incubated with 10 μM AMD3100 (cat. A5602, Sigma) or vehicle for 30 min at 4 °C, and then introduced into 5- to 7-week-old male CXCR6+/+ or CXCR6−/− mice by intra-cardiac injection. qPCR and immunohistochemistry were used to identify the location of the cells. Metastasis was first assessed in osseous tissues and blood samples at day 10 by qPCR using a probe for the red fluorescent protein gene (AICSVE0-F 5′-AGAGCATCTACATGGCCAAGAAG-3′ (forward), AICSVE0-R 5′-TCGTTGTGGCTGGTGATGTC-3′ (reverse) and FAM 5′-CTTGCTGTCCACGTAGTAGT-3′ (TaqMan probe; Applied Biosystems)). The data were normalized to mouse tissue β-actin (Mm00607939_s1). Immunohistochemistry for prostate cancer cells in the marrow was also used for metastasis assays. The numbers of RM1 cells were quantified on the endosteal region of the seven long bones defined as 10 cell diameters from the bone surfaces.
Statistical analyses
All in vitro experiments were performed at least three times with similar results and representative assays are shown. Statistical analysis was performed by analysis of variance or Student’s t-test using GraphPad Instat (GraphPad, San Diego, CA) with significance at P<0.05.
Additional information
How to cite this article: Jung, Y. et al. Recruitment of mesenchymal stem cells into prostate tumours promotes metastasis. Nat. Commun. 4:1795 doi: 10.1038/ncomms2766 (2013).
References
Dvorak, H. F. . Tumors: wounds that do not heal. Similarities between tumor stroma generation and wound healing. N. Engl. J. Med. 315, 1650–1659 (1986).
Wels, J., Kaplan, R. N., Rafii, S. & Lyden, D. . Migratory neighbors and distant invaders: tumor-associated niche cells. Genes Dev. 22, 559–574 (2008).
Yang, Z. J. & Wechsler-Reya, R. J. . Hit 'em where they live: targeting the cancer stem cell niche. Cancer Cell 11, 3–5 (2007).
Kaplan, R. N. et al. VEGFR1-positive haematopoietic bone marrow progenitors initiate the pre-metastatic niche. Nature 438, 820–827 (2005).
Rafii, S. & Lyden, D. . S100 chemokines mediate bookmarking of premetastatic niches. Nat. Cell Biol. 8, 1321–1323 (2006).
Quante, M. et al. Bone marrow-derived myofibroblasts contribute to the mesenchymal stem cell niche and promote tumor growth. Cancer Cell 19, 257–272 (2011).
Taichman, R. S. et al. Prospective identification and skeletal localization of cells capable of multilineage differentiation in vivo. Stem Cells Dev. 19, 1557–1570 (2010).
Whiteside, T. L. . The tumor microenvironment and its role in promoting tumor growth. Oncogene 27, 5904–5912 (2008).
Bergfeld, S. A. & DeClerck, Y. A. . Bone marrow-derived mesenchymal stem cells and the tumor microenvironment. Cancer Metastasis Rev. 29, 249–261 (2010).
Mi, Z. et al. Osteopontin promotes CCL5-mesenchymal stromal cell-mediated breast cancer metastasis. Carcinogenesis 32, 477–487 (2011).
Liu, Y. et al. Effects of inflammatory factors on mesenchymal stem cells and their role in the promotion of tumor angiogenesis in colon cancer. J. Biol. Chem. 286, 25007–25015 (2011).
Abarrategi, A., Marinas-Pardo, L., Mirones, I., Rincon, E. & Garcia-Castro, J. . Mesenchymal niches of bone marrow in cancer. Clin. Transl. Oncol. 13, 611–616 (2011).
Mishra, P. J., Mishra, P. J., Glod, J. W. & Banerjee, D. . Mesenchymal stem cells: flip side of the coin. Cancer Res. 69, 1255–1258 (2009).
Siegel, R., Naishadham, D. & Jemal, A. . Cancer statistics, 2012. CA Cancer J. Clin. 62, 10–29 (2012).
Lu, Y. et al. CXCL16 functions as a novel chemotactic factor for prostate cancer cells in vitro. Mol. Cancer Res. 6, 546–554 (2008).
Wang, J. et al. CXCR6 induces prostate cancer progression by the AKT/mammalian target of rapamycin signaling pathway. Cancer Res. 68, 10367–10376 (2008).
Chandrasekar, B., Bysani, S. & Mummidi, S. . CXCL16 signals via Gi, phosphatidylinositol 3-kinase, Akt, I kappa B kinase, and nuclear factor-kappa B and induces cell-cell adhesion and aortic smooth muscle cell proliferation. J. Biol. Chem. 279, 3188–3196 (2004).
Ha, H. K. et al. Clinical significance of CXCL16/CXCR6 expression in patients with prostate cancer. Mol. Med. Report 4, 419–424 (2011).
Deng, L., Chen, N., Li, Y., Zheng, H. & Lei, Q. . CXCR6/CXCL16 functions as a regulator in metastasis and progression of cancer. Biochim. Biophys. Acta 1806, 42–49 (2010).
van, V. et al. An alternatively spliced CXCL16 isoform expressed by dendritic cells is a secreted chemoattractant for CXCR6+ cells. J. Leukoc. Biol. 87, 1029–1039 (2010).
Nakayama, T. et al. Cutting edge: profile of chemokine receptor expression on human plasma cells accounts for their efficient recruitment to target tissues. J. Immunol. 170, 1136–1140 (2003).
Meijer, J. et al. The chemokine receptor CXCR6 and its ligand CXCL16 are expressed in carcinomas and inhibit proliferation. Cancer Res. 68, 4701–4708 (2008).
Matsushita, K. et al. Soluble CXCL16 in Preoperative Serum is a Novel Prognostic Marker and Predicts Recurrence of Liver Metastases in Colorectal Cancer Patients. Ann. Surg. Oncol. 19, (Suppl 3): S518–S527 (2011).
Hu, W. et al. CXCR6 is expressed in human prostate cancer in vivo and is involved in the in vitro invasion of PC3 and LNCap cells. Cancer Sci. 99, 1362–1369 (2008).
Held-Feindt, J. et al. Overexpression of CXCL16 and its receptor CXCR6/Bonzo promotes growth of human schwannomas. Glia 56, 764–774 (2008).
Darash-Yahana, M. et al. The chemokine CXCL16 and its receptor, CXCR6, as markers and promoters of inflammation-associated cancers. PLoS One 4, e6695 (2009).
Lu, Y. et al. CXCL16 Functions as a novel chemotactic factor for prostate cancer cells In vitro. Mol. Cancer Res. 6, 546–554 (2008).
Ratajczak, M. Z., Zuba-Surma, E. K., Machalinski, B., Ratajczak, J. & Kucia, M. . Very small embryonic-like (VSEL) stem cells: purification from adult organs, characterization, and biological significance. Stem Cell Rev. 4, 89–99 (2008).
Wang, Z., Song, J., Taichman, R. S. & Krebsbach, P. H. . Ablation of proliferating marrow with 5-fluorouracil allows partial purification of mesenchymal stem cells. Stem Cells 24, 1573–1582 (2006).
Orimo, A. & Weinberg, R. A. . Stromal fibroblasts in cancer: a novel tumor-promoting cell type. Cell Cycle 5, 1597–1601 (2006).
Karnoub, A. E. et al. Mesenchymal stem cells within tumour stroma promote breast cancer metastasis. Nature 449, 557–563 (2007).
Begley, L., Monteleon, C., Shah, R. B., Macdonald, J. W. & Macoska, J. A. . CXCL12 overexpression and secretion by aging fibroblasts enhance human prostate epithelial proliferation in vitro. Aging Cell 4, 291–298 (2005).
Wang, J. et al. Characterization of phosphoglycerate kinase-1 expression of stromal cells derived from tumor microenvironment in prostate cancer progression. Cancer Res. 70, 471–480 (2010).
Tran, N. L., Nagle, R. B., Cress, A. E. & Heimark, R. L. . N-Cadherin expression in human prostate carcinoma cell lines. An epithelial-mesenchymal transformation mediating adhesion with stromal cells. Am. J. Pathol. 155, 787–798 (1999).
Algaba, F., Arce, Y., Fernandez, S., Oliver, A. & Alcaraz, A. . Adhesion molecules expression as a potential marker of prostate cancer aggressivity. A TMA study of radical prostatectomy specimens. Arch. Ital. Urol. Androl. 78, 130–134 (2006).
Rubin, M. A. et al. E-cadherin expression in prostate cancer: a broad survey using high-density tissue microarray technology. Hum. Pathol. 32, 690–697 (2001).
Taichman, R. S. et al. Use of the stromal cell-derived factor-1/CXCR4 pathway in prostate cancer metastasis to bone. Cancer Res. 62, 1832–1837 (2002).
Muller, C. A. et al. Involvement of chemokine receptors in breast cancer metastasis. Nature 410, 50–56 (2001).
Knudson, A. G. Jr . Mutation and cancer: statistical study of retinoblastoma. Proc. Natl Acad. Sci. USA 68, 820–823 (1971).
Raaijmakers, M. H. et al. Bone progenitor dysfunction induces myelodysplasia and secondary leukaemia. Nature 464, 852–857 (2010).
Elenbaas, B. & Weinberg, R. A. . Heterotypic signaling between epithelial tumor cells and fibroblasts in carcinoma formation. Exp. Cell Res. 264, 169–184 (2001).
Nakagawa, H. et al. Role of cancer-associated stromal fibroblasts in metastatic colon cancer to the liver and their expression profiles. Oncogene 23, 7366–7377 (2004).
Micke, P. & Ostman, A. . Tumour-stroma interaction: cancer-associated fibroblasts as novel targets in anti-cancer therapy? Lung Cancer 45, (Suppl 2): S163–S175 (2004).
Hill, R., Song, Y., Cardiff, R. D. & Van Dyke, T. . Selective evolution of stromal mesenchyme with p53 loss in response to epithelial tumorigenesis. Cell 123, 1001–1011 (2005).
Kollet, O. et al. Rapid and efficient homing of human CD34(+)CD38(-/low)CXCR4(+) stem and progenitor cells to the bone marrow and spleen of NOD/SCID and NOD/SCID/B2m(null) mice. Blood 97, 3283–3291 (2001).
Lapidot, T., Dar, A. & Kollet, O. . How do stem cells find their way home? Blood 106, 1901–1910 (2005).
Peled, A. et al. Dependence of human stem cell engraftment and repopulation of NOD/SCID mice on CXCR4. Science 283, 845–848 (1999).
Sackstein, R. et al. Ex vivo glycan engineering of CD44 programs human multipotent mesenchymal stromal cell trafficking to bone. Nat. Med. 14, 181–187 (2008).
Coffelt, S. B. et al. The pro-inflammatory peptide LL-37 promotes ovarian tumor progression through recruitment of multipotent mesenchymal stromal cells. Proc. Natl Acad. Sci. USA 106, 3806–3811 (2009).
Wilbanks, A. et al. Expression cloning of the STRL33/BONZO/TYMSTRligand reveals elements of CC, CXC, and CX3C chemokines. J. Immunol. 166, 5145–5154 (2001).
Matloubian, M., David, A., Engel, S., Ryan, J. E. & Cyster, J. G. . A transmembrane CXC chemokine is a ligand for HIV-coreceptor Bonzo. Nat. Immunol. 1, 298–304 (2000).
Shimaoka, T. et al. Critical role for CXC chemokine ligand 16 (SR-PSOX) in Th1 response mediated by NKT cells. J. Immunol. 179, 8172–8179 (2007).
Wente, M. N. et al. Expression and potential function of the CXC chemokine CXCL16 in pancreatic ductal adenocarcinoma. Int. J. Oncol. 33, 297–308 (2008).
Wang, J., Loberg, R. & Taichman, R. S. . The pivotal role of CXCL12 (SDF-1)/CXCR4 axis in bone metastasis. Cancer Metastasis Rev. 25, 573–587 (2006).
Wang, J. et al. The role of CXCR7/RDC1 as a chemokine receptor for CXCL12/SDF-1 in prostate cancer. J. Biol. Chem. 283, 4283–4294 (2008).
Acknowledgements
This work is directly supported by the National Institutes of Health (CA163124, CA093900, CA166307, CA069568, DK082481, MH095589, Y.S., E.T.K. P.H.K, K.J.P. and R.S.T.), the Fund for Cancer Research (R.S.T.), the Department of Defense (E.T.K., Y.S. and R.S.T.), the Prostate Cancer Foundation (Y.S, K.J.P. and R.S.T.), National Natural Funding of China (81071747) (Jianhua Wang), National Key Program (973) for Basic Research of China (2011CB510106) (Jianhua Wang) and Shanghai Pujiang Program (10PJ1406400) (Jianhua Wang). Logistical support was also received from the University of Michigan FACS and Imaging Cores, and the Dental School’s Molecular Biology Core.
Author information
Authors and Affiliations
Contributions
Y.J., J.K.K., Y.S. and R.S.T. designed experiments. Y.J., J.K.K., Y.S., Jingcheng Wang, A.M., J.J., J.E.B., S.M., E.L., H.S., T.J., H.Z. and J.D. performed experiments and analysed the data. Jianhua Wang, P.H.K., E.T.K. and K.J.P. discussed the results and gave valuable critique on the paper. Y.J., Y.S. and R.S.T. wrote the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Supplementary Information
Supplementary Figures S1-S5, Supplementary Table S1 and Supplementary Methods (PDF 9024 kb)
Rights and permissions
About this article
Cite this article
Jung, Y., Kim, J., Shiozawa, Y. et al. Recruitment of mesenchymal stem cells into prostate tumours promotes metastasis. Nat Commun 4, 1795 (2013). https://doi.org/10.1038/ncomms2766
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/ncomms2766
This article is cited by
-
Claudin-18.2 mediated interaction of gastric Cancer cells and Cancer-associated fibroblasts drives tumor progression
Cell Communication and Signaling (2024)
-
Targeting IL-17A enhances imatinib efficacy in Philadelphia chromosome-positive B-cell acute lymphoblastic leukemia
Nature Communications (2024)
-
WNT Signaling in Stem Cells: A Look into the Non-Canonical Pathway
Stem Cell Reviews and Reports (2024)
-
Define cancer-associated fibroblasts (CAFs) in the tumor microenvironment: new opportunities in cancer immunotherapy and advances in clinical trials
Molecular Cancer (2023)
-
Mesenchymal stromal cells promote the drug resistance of gastrointestinal stromal tumors by activating the PI3K-AKT pathway via TGF-β2
Journal of Translational Medicine (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
