
Abstract
To identify susceptibility loci for bipolar disorder, we tested 1.8 million variants in 4,387 cases and 6,209 controls and identified a region of strong association (rs10994336, P = 9.1 × 10−9) in ANK3 (ankyrin G). We also found further support for the previously reported CACNA1C (alpha 1C subunit of the L-type voltage-gated calcium channel; combined P = 7.0 × 10−8, rs1006737). Our results suggest that ion channelopathies may be involved in the pathogenesis of bipolar disorder.
Similar content being viewed by others

Main
Recent genome-wide association studies (GWAS) have identified genetic variants that show consistent association with common, complex diseases, such as type 2 diabetes1, prostate cancer2 and Crohn's disease3. In many cases, a critical component for success involved combining results and data across multiple, smaller studies to provide adequate power to detect common variants of modest relative risk. Given that a single study will often not be sufficient, here we present the results of combining two previously published4,5 and one new (N = 2,365) GWAS of bipolar disorder.
Three groups have performed independent GWAS of bipolar disorder, with little agreement among the most associated regions. Baum et al.6, using DNA pooling, identified a SNP located in DGKH (diacylglycerol kinase eta) that was associated with bipolar disorder in 1,233 cases and 1,439 controls with a P value of 1.5 × 10−8. The Wellcome Trust Case Control Consortium (WTCCC)4 analyzed 1,868 cases and 2,938 controls and identified a locus in a gene-rich region of high linkage disequilibrium (LD) on chromosome 16p12 with P = 6.3 × 10−8. Recently, Sklar et al.5 performed a GWAS of 1,461 cases and 2,008 controls (the STEP-UCL study), finding the strongest single SNP result in MYO5B (myosin 5B), with P = 1.7 × 10−7. Although there was no obvious correspondence among the studies' top few hits, a broader comparison of the WTCCC and STEP-UCL studies identified CACNA1C as showing the strongest consistent signal5.
We genotyped a new sample of 1,098 individuals with bipolar disorder and 1,267 controls on a similar platform as used by the WTCCC and STEP-UCL studies (Supplementary Table 1 online), with 331,786 SNPs passing quality control (Supplementary Methods online). This dataset, referred to as ED-DUB-STEP2, included additional samples from the STEP-BD study5 as well as cases and controls from the University of Edinburgh and Trinity College Dublin. Approval for each study was obtained from the appropriate review boards of the participating institutions and informed consent was obtained from all subjects. SNPs were tested for association using logistic regression co-varying for sample collection site and quantitative indices of ancestry based on a multi-dimensional scaling analysis; the genomic inflation factor (see Supplementary Methods) was λ1,000 = 1.056 (Supplementary Fig. 1 online).
Analysis of the ED-DUB-STEP2 samples identified 14 chromosomal regions associated at P < 5 × 10−5 (Supplementary Table 2 online). No region exceeded our threshold for genome-wide significance of 5 × 10−8, correcting for an effective number of one million independent, common SNPs in the genome1,7. However, one of these regions spanned the CACNA1C association identified previously in the WTCCC and STEP-UCL studies5 (Supplementary Fig. 2 online), thus providing further independent support that this gene is associated with risk of bipolar disorder. In this region, the best single SNP across the WTCCC and STEP-UCL studies showed a consistent association in ED-DUB-STEP2 (rs1006737, P = 0.011, allele A, OR = 1.21). Furthermore, the SNP with strongest association in ED-DUB-STEP2 (rs10774037, P = 4.6 × 10−5, allele G, OR = 1.43) also had a consistent effect (P = 0.0008, OR = 1.14) in the WTCCC and STEP-UCL samples and was in LD with rs1006737 (r2 = 0.39).
Next, we combined individual genotyping data from the WTCCC, STEP-UCL and ED-DUB-STEP2 studies, resulting in an overall sample of 4,387 cases and 6,209 controls genotyped on 325,690 overlapping SNPs, which substantially improves power to detect risk alleles of modest effect (Supplementary Table 1). The case sample comprised the following diagnoses: 81% bipolar 1, 16% bipolar 2, 2% schizoaffective manic and 1% bipolar NOS (Supplementary Table 3 online).
Genomic coverage was increased by imputing the genotypes for 1,444,258 additional HapMap SNPs using PLINK8 (Supplementary Methods), resulting in a total set of 1,769,948 SNPs. Applying a leave-one-out procedure for every genotyped SNP, we estimated concordance between imputed and true genotypes as 0.987 (Supplementary Table 4 online). Similar results were obtained using MACH1 (ref. 9; data not shown).
Our primary analysis was a logistic regression of disease state on single SNP allelic dosage, either directly genotyped or imputed, controlling for study site and two quantitative indices of ancestry (Supplementary Methods). The genomic inflation factor was λ1,000 = 1.024 when considering all SNPs, and λ1,000 = 1.021 for directly genotyped SNPs (Supplementary Fig. 1 and Supplementary Fig. 3 online).
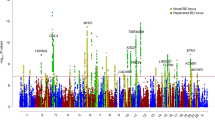
Results for the primary analysis are shown in Figure 1a and available for download (http://pngu.mgh.harvard.edu/purcell/bpwgas2/). Thirty-nine SNPs showed a P < 10−6 and were located in three distinct chromosomal regions (Table 1). An additional 18 regions had at least one SNP with a P < 10−5 (Supplementary Fig. 4 and Supplementary Table 5 online). Cluster plots for these SNPs are shown in Supplementary Figure 5 online.

(a) Genome-wide results (−log10P) are shown in chromosomal order for directly genotyped (N = 325,690) and imputed (N = 1,444,258) SNPs that were tested for association in the overall sample of 4,387 bipolar cases and 6,209 controls. Horizontal lines indicate a P value of 5 × 10−8 (dashed) and 1 × 10−6 (dotted). (b) Plots for the three regions of strongest association. Results (−log10P) are shown for directly genotyped (diamonds) and imputed (circles) SNPs. The most associated SNP for each region is shown in blue, and the color of the remaining markers reflects the linkage disequilibrium (r2) with the top SNP in each panel (increasing red hue associated with increasing r2). The recombination rate (second y axis) is plotted in light blue and is based on the CEU HapMap population. The dashed horizontal line indicates a P = 5 × 10−8. Exons for each gene are represented by vertical bars, based on all isoforms available from the Mar 2006 UCSC genome browser assembly.
The strongest association was in ANK3 on chromosome 10q21 for the imputed SNP rs10994336, with P = 9.1 × 10−9 (Fig. 1b). This result was supported by the following observations. First, multiple SNPs were associated across a 195-kb region. Second, three genotyped SNPs showed P values similar to rs10994336 (for example, rs1938526, P = 1.3 × 10−8, r2 = 0.40 with rs10994336). Third, haplotype analysis of genotyped SNPs supported the single SNP results (most associated haplotype P = 2.0 × 10−9, Supplementary Table 6 online). Fourth, the association remained (rs10994336, P = 5.2 × 10−9) after including the six additional WTCCC disease panels as extra controls for the WTCCC dataset ('expanded reference group' analysis). Finally, individual Sequenom genotyping confirmed that rs10994336 was imputed with high accuracy (concordance = 0.983, N = 3,293).
The second strongest region of association was located in the third intron of CACNA1C on chromosome 12p13 (rs1006737, P = 7.0 × 10−8). The third region was 3.3 kb away from an uncharacterized gene (C15orf53) on chromosome 15q14 (rs12899449, P = 3.5 × 10−7), in the same LD block with the brain-expressed RASGRP1 (RAS guanyl releasing protein 1) gene. The association with both regions remained in the expanded reference group analysis (rs1006737, P = 4.0 × 10−8; rs12899449, P = 9.3 × 10−6).
Genotypic and heterogeneity analyses for these three regions suggested that the effect at each locus was consistent with an additive model (Supplementary Table 7 online) and of similar magnitude across studies (Supplementary Table 8 online). There was no clear indication of differential association across bipolar subtypes, psychosis, age-at-onset and sex (Supplementary Table 9 online). No significant treatment response effects were observed for these three loci (data not shown).
Finally, we carried out a two-locus genome-wide search to identify epistatically acting loci of moderate-to-large effect that single-locus analysis may have missed10,11. From over 4 × 1010 pairwise tests, we identified 61 interactions with a P < 5 × 10−10, representing seven distinct pairs of regions (Supplementary Table 10 online). No single interaction exceeded a Bonferroni corrected–threshold of 1.2 × 10−12, in spite of reasonable power to detect large interaction effects (Supplementary Methods).
In summary, we present evidence that variation in ANK3 confers risk of bipolar disorder in three independent datasets. ANK3 is an adaptor protein found at axon initial segments that has been shown to regulate the assembly of voltage-gated sodium channels12,13. We also provide independent support that CACNA1C is associated with bipolar disorder. In addition, we have recently shown that both ANK3 and subunits of the calcium channel are downregulated in the mouse brain in response to lithium14, one the most effective bipolar pharmacotherapies. Taken together, these results point to the possibility that bipolar disorder is in part an ion channelopathy15.
Note: Supplementary information is available on the Nature Genetics website.
References
Diabetes Genetics Initiative et al. Science 316, 1331–1336 (2007).
Gudmundsson, J. et al. Nat. Genet. 39, 631–637 (2007).
Rioux, J.D. et al. Nat. Genet. 39, 596–604 (2007).
Wellcome Trust Case Control Consortium. Nature 447, 661–678 (2007).
Sklar, P. et al. Mol. Psychiatry 13, 558–569 (2008).
Baum, A.E. et al. Mol. Psychiatry 13, 197–207 (2008).
The International HapMap Consortium. Nature 437, 1299–1320 (2005).
Purcell, S. et al. Am. J. Hum. Genet. 81, 559–575 (2007).
Li, Y. & Abecasis, G.R. Am. J. Hum. Genet. S79, 2290 (2006).
Evans, D.M., Marchini, J., Morris, A.P. & Cardon, L.R. PLoS Genet. 2, e157 (2006).
Marchini, J., Donnelly, P. & Cardon, L.R. Nat. Genet. 37, 413–417 (2005).
Kordeli, E., Lambert, S. & Bennett, V. J. Biol. Chem. 270, 2352–2359 (1995).
Poliak, S. & Peles, E. Nat. Rev. Neurosci. 4, 968–980 (2003).
McQuillin, A., Rizig, M. & Gurling, H.M. Pharmacogenet. Genomics 17, 605–617 (2007).
Gargus, J.J. Biol. Psychiatry 60, 177–185 (2006).
Acknowledgements
Study supported by US National Institute of Mental Health (MH062137, MH067288, P.S.; MH063445, J.W.S.; MH63420, V.L.N.; N01MH80001, G.S.S., M.E.T. and M.S. Bauer), NARSAD (R.H.P., S.P., J.F., P.S.); Johnson & Johnson Pharmaceutical Research & Development, Sylvan C. Herman Foundation, Stanley Medical Research Institute, Dauten Family, Merck Genome Research Institute (E.M.S.); and National Health Medical Research Council (MRC), Australia (M.A.R.F.). Broad Institute Center for Genotyping and Analysis supported by U54 RR020278, National Center for Research Resources. UCL sample collection supported by MDF–The Bipolar Organization, Neuroscience Research Charitable Trust, central London NHS Blood Transfusion Service, Priory Hospitals and UK MRC (G9623693N, G0500791). Dublin sample collection supported by the Health Research Board and Science Foundation Ireland; controls supplied by J. McPartlin, Trinity College Biobank. Edinburgh cohort collection supported by the Wellcome Trust, London, Chief Scientist Office of the Scottish Executive and MRC, London. WTCCC genotype analyses were funded by the Wellcome Trust and undertaken within the context of the WTCCC. WTCCC members are listed in Supplementary Note.
Author information
Authors and Affiliations
Consortia
Contributions
Writing group: M.A.R.F., P.S., N.C., S.M.P. Analytic group: S.M.P., M.A.R.F., Y.A.M., D.M.R., J. Fan, M.J.D., P.A.H., M.C.O. N.C., P.S. Project management: P.S., E.M.S., A.P.C., D.H.B., H.M.G., M.C.O., N.C. Clinical characterization for STEP-BD: J.W.S., V.L.N., R.H.P., M.E.T., G.S.S. Clinical characterization for Trinity College Dublin: D.W.M., M.G., A.P.C. Clinical characterization for University of Edinburgh: W.J.M., K.A.M., D.J.M., A.W.M., M.V.B., D.H.B. Clinical characterization for University College London: A. McQuillin, N.J.B., M.R., J.L., A.C.P.P., R.K, A.A., D.C., H.M.G. Clinical characterization, phenotype assessment and sample management and curation for WTCCC: G.B., D.S. (Aberdeen); S.C., K.G.-S. L.J. (Birmingham); C.F., E.K.G., D.G., M.L.H., P.A.H., I.R.J., G.K., V.M., I.N., M.C.O., M.J.O., N.C. (Cardiff); D.A.C., A.E., A.F., R.W., P.M. (London); A.H.Y., I.N.F. (Newcastle). DNA sample QC and genotyping for ED-DUB-STEP2: K.C., J.S., J. Fan, J. Franklin, K.G.A., S.B.G., B.B., M.D.
Corresponding authors
Additional information
A full list of members is provided in the Supplementary Note.
Supplementary information
Supplementary Text and Figures
Supplementary Methods, Supplementary Figures 1–5, Supplementary Tables 1–10, Supplementary Note (PDF 8946 kb)
Rights and permissions
About this article
Cite this article
Ferreira, M., O'Donovan, M., Meng, Y. et al. Collaborative genome-wide association analysis supports a role for ANK3 and CACNA1C in bipolar disorder. Nat Genet 40, 1056–1058 (2008). https://doi.org/10.1038/ng.209
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ng.209
This article is cited by
-
Progress and Implications from Genetic Studies of Bipolar Disorder
Neuroscience Bulletin (2024)
-
The association of genetic variation in CACNA1C with resting-state functional connectivity in youth bipolar disorder
International Journal of Bipolar Disorders (2023)
-
Abnormal patterns of sleep and waking behaviors are accompanied by neocortical oscillation disturbances in an Ank3 mouse model of epilepsy-bipolar disorder comorbidity
Translational Psychiatry (2023)
-
Expression of ANK3 moderates the association between childhood trauma and affective traits in severe mental disorders
Scientific Reports (2023)
-
Calciopathies and Neuropsychiatric Disorders: Physiological and Genetic Aspects
Neuroscience and Behavioral Physiology (2023)
