
Abstract
Despite a century of research on complex traits in humans, the relative importance and specific nature of the influences of genes and environment on human traits remain controversial. We report a meta-analysis of twin correlations and reported variance components for 17,804 traits from 2,748 publications including 14,558,903 partly dependent twin pairs, virtually all published twin studies of complex traits. Estimates of heritability cluster strongly within functional domains, and across all traits the reported heritability is 49%. For a majority (69%) of traits, the observed twin correlations are consistent with a simple and parsimonious model where twin resemblance is solely due to additive genetic variation. The data are inconsistent with substantial influences from shared environment or non-additive genetic variation. This study provides the most comprehensive analysis of the causes of individual differences in human traits thus far and will guide future gene-mapping efforts. All the results can be visualized using the MaTCH webtool.
Similar content being viewed by others

Main
Insight into the nature of observed variation in human traits is important in medicine, psychology, social sciences and evolutionary biology. It has gained new relevance with both the ability to map genes for human traits and the availability of large, collaborative data sets to do so on an extensive and comprehensive scale. Individual differences in human traits have been studied for more than a century, yet the causes of variation in human traits remain uncertain and controversial. Specifically, the partitioning of observed variability into underlying genetic and environmental sources and the relative importance of additive and non-additive genetic variation are continually debated1,2,3,4,5. Recent results from large-scale genome-wide association studies (GWAS) show that many genetic variants contribute to the variation in complex traits and that effect sizes are typically small6,7. However, the sum of the variance explained by the detected variants is much smaller than the reported heritability of the trait4,6,7,8,9,10. This 'missing heritability' has led some investigators to conclude that non-additive variation must be important4,11. Although the presence of gene-gene interaction has been demonstrated empirically5,12,13,14,15,16,17, little is known about its relative contribution to observed variation18.
In this study, our aim is twofold. First, we analyze empirical estimates of the relative contributions of genes and environment for virtually all human traits investigated in the past 50 years. Second, we assess empirical evidence for the presence and relative importance of non-additive genetic influences on all human traits studied. We rely on classical twin studies, as the twin design has been used widely to disentangle the relative contributions of genes and environment, across a variety of human traits. The classical twin design is based on contrasting the trait resemblance of monozygotic and dizygotic twin pairs. Monozygotic twins are genetically identical, and dizygotic twins are genetically full siblings. We show that, for a majority of traits (69%), the observed statistics are consistent with a simple and parsimonious model where the observed variation is solely due to additive genetic variation. The data are inconsistent with a substantial influence from shared environment or non-additive genetic variation. We also show that estimates of heritability cluster strongly within functional domains, and across all traits the reported heritability is 49%. Our results are based on a meta-analysis of twin correlations and reported variance components for 17,804 traits from 2,748 publications including 14,558,903 partly dependent twin pairs, virtually all twin studies of complex traits published between 1958 and 2012. This study provides the most comprehensive analysis of the causes of individual differences in human traits thus far and will guide future gene-mapping efforts. All results can be visualized with the accompanying MaTCH webtool.
Results
The distribution of studied traits is nonrandom
We systematically retrieved published classical twin studies in which observed variation in human traits was partitioned into genetic and environmental influences. For each study, we collected reported twin correlations for continuous traits and contingency tables for dichotomous traits, estimates from genetic model-fitting and study characteristics (sample size, population, age cohort and ascertainment scheme) (Supplementary Table 1 and Supplementary Note). We manually classified the investigated traits using the chapter and subchapter levels of the International Classification of Functioning, Disability and Health (ICF) or the International Statistical Classification of Diseases and Related Health Problems (ICD-10) (Online Methods). ICD-10 and ICF subchapter levels refer to actual diseases (for example, atopic dermatitis) and traits (for example, temperament and personality functions), respectively. We identified 2,748 relevant twin studies, published between 1958 and 2012. Half of these were published after 2004, with sample sizes per study in 2012 of around 1,000 twin pairs (Supplementary Table 2). Each study could report on multiple traits measured in one or several samples. These 2,748 studies reported on 17,804 traits. Twin subjects came from 39 different countries, with a large proportion of studies (34%) based on US twin samples. The continents of South America (0.5%), Africa (0.2%) and Asia (5%) were heavily under-represented (Fig. 1a,b and Supplementary Table 3). The total number of studied twins was 14,558,903 partly dependent pairs, or 2,247,128 when correcting for reporting on multiple traits per study. The majority of studies (59%) were based on the adult population (aged 18–64 years), although the sample sizes available for studies of the elderly population (aged 65 years or older) were the largest (Supplementary Table 4). Authorship network analyses showed that 61 communities of authors wrote the 2,748 published studies. The 11 largest authorship communities contained >65 authors and could be mapped back to the main international twin registries, such as the Vietnam Era Twin Registry, the Finnish Twin Cohort and the Swedish Twin Registry (Supplementary Fig. 1).

(a) The number of investigated traits in classical twin studies across all countries. (b) The average number of twin pairs included per study across countries. (c) The number of investigated traits according to functional trait domain and trait characteristic (inset). (d) Monozygotic and dizygotic twin correlations and reported estimates of h2 and c2 as a function of sample size. Contour lines indicate the density of the data in that region. The lines are colored by 'heat' from blue to red, indicating increasing data density.
The investigated traits fell into 28 general trait domains. The distribution of the traits evaluated in twin studies was highly skewed, with 51% of studies focusing on traits classified under the psychiatric, metabolic and cognitive domains, whereas traits classified under the developmental, connective tissue and infection domains together accounted for less than 1% of all investigated traits (Fig. 1c and Supplementary Tables 5–7). The ten most investigated traits were temperament and personality functions, weight maintenance functions, general metabolic functions, depressive episode, higher-level cognitive functions, conduct disorders, mental and behavioral disorders due to use of alcohol, anxiety disorders, height and mental and behavioral disorders due to use of tobacco. Collectively, these traits accounted for 59% of all investigated traits.
Equal contribution of genes and environment
We did not find evidence of systematic publication bias as a function of sample size (for example, where studies based on relatively small samples were only published when larger effects were reported) (Fig. 1d, Supplementary Figs. 2,3,4,5,6 and Supplementary Tables 8–11). We calculated the weighted averages of correlations for monozygotic (rMZ) and dizygotic (rDZ) twins and of the reported estimates of the relative contributions of genetic and environmental influences to the investigated traits using a random-effects meta-analytic model to allow for heterogeneity across different studies (Supplementary Tables 12–15). The meta-analyses of all traits yielded an average rMZ of 0.636 (s.e.m. = 0.002) and an average rDZ of 0.339 (s.e.m. = 0.003). The reported heritability (h2) across all traits was 0.488 (s.e.m. = 0.004), and the reported estimate of shared environmental effects (c2) was 0.174 (s.e.m. = 0.004) (Fig. 2a,b, Table 1 and Supplementary Fig. 7).

(a) Distribution of rMZ and rDZ estimates across the traits investigated in 2,748 twin studies published between 1958 and 2012. rMZ estimates are based on 9,568 traits and 2,563,628 partly dependent twin pairs; rDZ estimates are based on 5,220 traits and 2,606,252 partly dependent twin pairs (Table 1). (b) Relationship between rMZ and rDZ, using all 5,185 traits for which both were reported. (c) Random-effects meta-analytic estimates of twin correlations (top) and reported variance components (bottom) across all traits separately for four age cohorts. Error bars, standard errors. (d) Random-effects meta-analytic estimates of twin correlations (top) and reported variance components (bottom) across all traits, and within functional domains for which data on all correlations and variance components were available. Error bars, standard errors.
Estimates of h2 and c2 cluster in functional domains
We found that heritability estimates clustered in functional domains, with the largest heritability estimates for traits classified under the ophthalmological domain (h2 = 0.712, s.e.m. = 0.041), followed by the ear, nose and throat (h2 = 0.637, s.e.m. = 0.064), dermatological (h2 = 0.604, s.e.m. = 0.043) and skeletal (h2 = 0.591, s.e.m. = 0.018) domains. The lowest heritability estimates were for traits in the environment, reproduction and social values domains (Fig. 2d and Supplementary Table 16). All weighted averages of h2 across >500 distinct traits had a mean greater than zero (Supplementary Tables 17–24). The lowest reported heritability for a specific trait was for gene expression, with an estimated h2 = 0.055 (s.e.m. = 0.026) and an estimated c2 of 0.736 (s.e.m. = 0.033) (but note that these trait averages are based on reported estimates of variance components derived from only 20 data points reporting on the expression levels of 20 genes; Supplementary Table 21). We found the largest influence of c2 for traits in the cell domain (c2 = 0.674, s.e.m. = 0.048), followed by traits in the infection (c2 = 0.351, s.e.m. = 0.153), hematological (c2 = 0.324, s.e.m. = 0.090), endocrine (c2 = 0.322, s.e.m. = 0.050), reproduction (c2 = 0.320, s.e.m. = 0.061), social values (c2 = 0.271, s.e.m. = 0.032), environment (c2 = 0.269, s.e.m. = 0.020) and skeletal (c2 = 0.265, s.e.m. = 0.019) domains (Fig. 2d and Supplementary Table 16).
Heterogeneity of twin correlations across sex and age
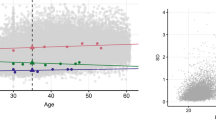
Across all traits, the weighted averages of twin correlations and reported h2 and c2 values did not show evidence of heterogeneity by sex, although there was some evidence for lower correlation in opposite-sex twin pairs in comparison to same-sex dizygotic twin pairs (Table 1 and Supplementary Note). The data showed a decrease in monozygotic and dizygotic twin resemblance after adolescence and an accompanying decrease in the estimates of both h2 and c2 (Fig. 2c and Supplementary Table 15).
In the top 20 most investigated traits for twin correlations, the weighted estimates did not show consistent evidence for heterogeneity by sex, with rMZM (monozygotic male twin correlation) and rMZF (monozygotic female twin correlation) as well as rDZM (dizygotic male twin correlation) and rDZF (dizygotic female twin correlation) remarkably similar across the majority of the top 20 specific traits investigated (Fig. 3), although for several traits the correlations for opposite-sex twin pairs were lower than the estimates for same-sex twin pairs, mostly after age 11 years (for example, for weight maintenance functions, functions of brain, mental and behavioral disorders due to the use of alcohol, and mental and behavioral disorders due to the use of tobacco). Heterogeneity of weighted twin correlations by age was more prominent than heterogeneity by sex (Fig. 3). For example, when considering rMZ, we note that for most of the top 20 investigated traits the estimate tended to decrease with age, especially after adolescence, a trend that was generally mirrored in the rDZ estimates (Fig. 3). Meta-analysis results across all traits across different countries are provided in Supplementary Table 25.

Alc., alcohol; dis., disorders; depr., depressive; endocr., endocrine; imm., immunological; funct., functions; maint., maintenance; metab., metabolic; ment. beh., mental and behavioral; spec. personal., specific personality; temp. pers., temperament and personality; tob., tobacco; r, correlation; MZ, monozygotic twins; DZ, dizygotic twins; M, males; F, females; SS, same-sex pairs only; DOS, dizygotic opposite-sex pairs. Inclusion for the top 20 most investigated traits was conditional on the reporting of rMZ and rDZ. Empty cells denote insufficient information available to calculate weighted estimates; error bars, standard errors. We note that estimates and graphs for all specific traits are available from the online MaTCH webtool.
Model-fitting and selection leads to underestimation of h2
Falconer's equations can be used to calculate ĥ2 and ĉ2 on the basis of twin correlations18. In these equations, ĥ2 = 2 × (rMZ − rDZ) and ĉ2 = 2 × rDZ − rMZ. When these are applied using the weighted averages of rMZ and rDZ, we find an ĥ2 estimate of 2 × (0.636 − 0.339) = 0.593 and a ĉ2 estimate of 2 × 0.339 − 0.636 = 0.042 (Table 1 and Supplementary Fig. 8). We note that the ĥ2 estimate based on twin correlations is larger than the weighted average of reported h2 values (Supplementary Figs. 9 and 10). As a consequence, the ĉ2 estimate based on twin correlations is lower than the weighted average of the reported c2 component. To test whether this discrepancy was due to a bias in studies reporting only twin correlations or only variance components, we conducted the meta-analysis only on studies reporting both. This analysis yielded similar estimates with a similar discrepancy (Supplementary Table 13), ruling out the explanation that twin correlations may have been reported on traits that happened to be more heritable than traits for which the estimates of variance components were reported. Through theory, we show that such a discrepancy can arise when individual studies represent a mixture of traits that follow a pattern of rMZ > 2rDZ and rMZ < 2rDZ and where the choice of fitting a model that includes shared environment or non-additive genetic influences is based on the observed pattern of twin correlations (Supplementary Note). More specifically, because c2 and non-additive genetic influences cannot be estimated simultaneously from twin correlations, an ACE model (for additive genetic (A), common environmental (C), and error or non-shared environmental (E) influences) is fitted to the data if 2rDZ − rMZ > 0. In contrast, if 2rDZ − rMZ < 0, an ADE model, including non-additive genetic (D) instead of common environmental (C) influences, is selected. This leads to sampling bias in estimating h2 from the full model. We show (Supplementary Table 26) that such per-study choices cause bias and can lead to a 10% downward bias in the reported estimates of h2 in comparison to those based on twin correlations, consistent with the observed discrepancy between our meta-analysis of variance component estimates calculated from twin correlations and the reported variance components.
Overall twin correlations imply a simple additive model
There may be many causes of similarities and differences within monozygotic and dizygotic twin pairs, and these are typically interpreted in terms of additive or non-additive genetic influences and shared or non-shared environmental influences19. Yet, there are essentially only two estimable and testable variance components of interest in the twin design. Therefore, inference from classical twin studies on all underlying, unobserved sources of variation that lead to the resemblance between relatives is limited. However, there are two simple and parsimonious hypotheses that can be tested across traits from estimated correlation coefficients for monozygotic twin pairs (rMZ) and dizygotic twin pairs (rDZ). The first is that the correlations for the monozygotic and dizygotic twin populations (ρMZ and ρDZ) are the same, consistent with twin resemblance being solely due to non-genetic factors. The second hypothesis involves a twofold ratio of ρMZ to ρDZ, consistent with twin resemblance being solely due to additive genetic variation. Across-trait consistency with either of these hypotheses is not a proof of these simple models but would provide an extremely parsimonious model against which other experimental designs (for example, DNA sequence–based studies) should be tested. For the vast majority of traits (84%), we found that monozygotic twin correlations were larger than dizygotic twin correlations. Using the weighted estimates of rMZ and rDZ across all traits, we showed that, on average, 2rDZ − rMZ = 0.042 (s.e.m. = 0.007) (Table 1), which is very close to a twofold difference in the correlation of monozygotic twins relative to dizygotic twins (Supplementary Figs. 11 and 12). The proportion of single studies in which the pattern of twin correlations was consistent with the null hypothesis that 2rDZ = rMZ was 69%. This observed pattern of twin correlations is consistent with a simple and parsimonious underlying model of the absence of environmental effects shared by twin pairs and the presence of genetic effects that are entirely due to additive genetic variation (Table 2). This remarkable fitting of the data with a simple mode of family resemblance is inconsistent with the hypothesis that a substantial part of variation in human traits is due to shared environmental variation or to substantial non-additive genetic variation.
Most specific traits follow an additive genetic model
Although across all traits 69% of studies showed a pattern of monozygotic and dizygotic twin correlations consistent with an rMZ that was exactly twice the rDZ, this finding is not necessarily representative of the majority of studies in functional domains or for every specific trait (that is, at the ICD-10 or ICF subchapter level). We thus calculated the proportion of studies consistent with 2rDZ = rMZ within functional domains and for each specific trait and found that traits consistent with this hypothesis tended to cluster in specific functional domains (Supplementary Tables 27–29). A pattern of twin correlations consistent with 2rDZ = rMZ was most prominent for traits included in the neurological, ear, nose and throat, cardiovascular and ophthalmological domains, with 99.5%, 97%, 95% and 87% of studies, respectively, being consistent with a model where all resemblance was entirely due to additive genetic variance. In only 3 of 28 general trait domains were most studies inconsistent with this model. These domains were activities (35%), reproduction (44%) and dermatological (45%) (Table 2 and Supplementary Table 27). Of the 59 specific traits (ICD-10 or ICF subchapter classifications) for which we had sufficient information to calculate the proportion of studies consistent with 2rDZ = rMZ, 21 traits showed a proportion less than 0.50, whereas for the remaining 38 traits the majority of individual studies were consistent with 2rDZ = rMZ (Supplementary Table 29). Of the top 20 most investigated specific traits, we found that for 12 traits the majority of individual studies were consistent with a model where variance was solely due to additive genetic variance and non-shared environmental variance, whereas the pattern of monozygotic and dizygotic twin correlations was inconsistent with this model for 8 traits, suggesting that, apart from additive genetic influences and non-shared environmental influences, either or both non-additive genetic influences and shared environmental influences are needed to explain the observed pattern of twin correlations (Table 2). These eight traits were conduct disorders, height, higher-level cognitive functions, hyperkinetic disorders, mental and behavioral disorders due to the use of alcohol, mental and behavioral disorders due to the use of tobacco, other anxiety disorders and weight maintenance functions. For all eight traits, meta-analyses on reported variance components resulted in a weighted estimate of reported shared environmental influences that was statistically different from zero (Supplementary Table 21). Comparison of weighted twin correlations for these specific traits resulted in positive estimates of 2rDZ − rMZ, except for hyperkinetic disorders, where 2rDZ − rMZ was −0.130 (s.e.m. = 0.034) on the basis of 144 individual reports and 207,589 twin pairs, which suggests the influence of non-additive genetic variation for this trait or any other source of variation that leads to a disproportionate similarity among monozygotic twin pairs.
Discussion
We have conducted a meta-analysis of virtually all twin studies published in the past 50 years, on a wide range of traits and reporting on more than 14 million twin pairs across 39 different countries. Our results provide compelling evidence that all human traits are heritable: not one trait had a weighted heritability estimate of zero. The relative influences of genes and environment are not randomly distributed across all traits but cluster in functional domains. In general, we showed that reported estimates of variance components from model-fitting can underestimate the true trait heritability, when compared with heritability based on twin correlations. Roughly two-thirds of traits show a pattern of monozygotic and dizygotic twin correlations that is consistent with a simple model whereby trait resemblance is solely due to additive genetic variation. This implies that, for the majority of complex traits, causal genetic variants can be detected using a simple additive genetic model.
Approximately one-third of traits did not follow the simple pattern of a twofold ratio of monozygotic to dizygotic correlations. For these traits, a simple additive genetic model does not sufficiently describe the population variance. An incorrect assumption about narrow-sense heritability (the proportion of total phenotypic variation due to additive genetic variation) can lead to a mismatch between the results from gene-finding studies and previous expectations. If the pattern of twin correlations is consistent with a substantial contribution from shared environmental factors, as we find for conduct disorders, religion and spirituality, and education, then gene-mapping studies may yield disappointing results. If the cause of departure from a simple additive genetic model is the existence of non-additive genetic variation, as is, for example, suggested by the average twin correlations for recurrent depressive disorder, hyperkinetic disorders and atopic dermatitis, then it may be tempting to fit non-additive models in gene-mapping studies (for example, GWAS or sequencing studies). However, the statistical power of such scans is extremely low owing to the many non-additive models that can be fitted (for example, within-locus dominance versus between-locus additive-by-additive effects) and the penalty incurred by multiple testing. Our current results signal traits for which an additive model cannot be assumed. For most of these traits, dizygotic twin correlations are higher than half the monozygotic twin correlations, suggesting that shared environmental effects are causing the deviation from a simple additive genetic model. Yet, data from twin pairs only do not provide sufficient information to resolve the actual causes of deviation from a simple additive genetic model. More detailed studies may identify the likely causes of such deviation and may as such uncover epidemiological or biological factors that drive family resemblance. To make stronger inferences about the causes underlying resemblance between relatives for traits that deviate from the additive genetic model, additional data are required, for example, from large population samples with extensive phenotypic and DNA sequence information, detailed measures of environmental exposures and larger pedigrees including non-twin relationships.
We note that our inference is based on twin studies published between 1958 and 2012 and that it generally applies to complex traits but does not necessarily generalize to mendelian subtypes of traits. Most mendelian traits are rare in the population and are therefore not studied by researchers of twins because they cannot ascertain enough affected twin pairs to reliably estimate genetic parameters. In the rare case where sufficient numbers of affected twin pairs were available, the mendelian subtypes were analyzed together with the subtypes of the same trait that were due to common causes, as it was unknown whether the studied trait was a mendelian subtype. If the traits we studied were in fact a mix of mendelian and complex subtypes, our inference would be biased away from our main result because mendelian diseases tend to be dominant or recessive, not additive. In addition, there may be heterogeneity in measurement errors between studies for the same trait and between traits. A test-retest correlation would quantify measurement error when contrasted with a correlation between monozygotic twins, but few twin studies report such correlations in the same papers that estimate heritability.
Our results provide the most comprehensive empirical overview of the relative contributions of genes and environment to all human traits that have been studied in twins thus far, which can guide and serve as a reference for future gene-mapping efforts.
Methods
Identifying relevant studies.
We searched PubMed for studies published between 1 January 1900 and 31 December 2012 that provided twin correlations, concordance rates, and a heritability estimate (h2) or an estimate of shared environmental influences (c2), using monozygotic and dizygotic twins. The following search term was used
(“English”[Language] AND (“1900/01/01”[Date - Publication]: “2012/12/31”[Date - Publication]) AND twin AND “journal article”[Publication Type] AND “humans”[Filter] AND (heritability[Title/Abstract] OR “genetic influence”[Title/Abstract] OR “environmental influence”[Title/Abstract] OR “genetic factors”[Title/Abstract] OR “environmental factors”[Title/Abstract]) AND “journal article”[Publication Type]) NOT review[Title] NOT review[Publication Type])
The search was run on 31 January 2013 and again on 29 April 2013, which yielded an additional 44 publications, with the difference likely due to keywords or tags that had been added to publications in the intermittent period.
The last PubMed search yielded 4,388 unique studies. From these, we deleted studies that were not relevant for the current purpose using the following exclusion criteria: (i) studies with only monozygotic twins available; (ii) studies with no heritability estimates, twin correlations or concordances available; (iii) review studies; (iv) meta-analyses; and (v) multivariate studies that provided information on completely overlapping traits and samples with previously published univariate studies. Some studies investigating h2 for the brain (for example, voxel-based brain measures) were not included for practical purposes. These studies typically presented their results in graphs with color-coded point estimates of heritability mapped onto the brain. Such estimates could not be quantified, and these studies were thus not included.
From the remaining 2,748 studies, we were able to retrieve the full text from all but 5 papers (99.8%). Of the studies without full-text availability, we included relevant information based on the abstract. An overview of authors and journals and a full reference list of all 2,748 studies are provided in Supplementary Tables 30–32.
Primary information obtained from each study.
From every study, we retrieved basic information on the PubMed ID, the authors, the trait as named in the study and the year of publication. In addition, the following information was retrieved:
-
Country of origin of the study population. We used standard ISO country names, and where possible data entry was done separately for each country investigated in the study.
-
Age group of the study population. The study population was classified into four age cohorts on the basis of the average age of the included sample: age >0 and <12; age ≥12 and <18; age ≥18 and <65; and age ≥65.
-
Monozygotic and dizygotic twin correlations. Twin correlations were entered as provided in the study and could be calculated as intraclass, Pearson, polychoric or tetrachoric correlations or on the basis of least-squares or maximum-likelihood estimates. When available, we entered the twin correlations separately for males and females (i.e., monozygotic male (MZM), monozygotic female (MZF), dizygotic male (DZM), dizygotic female (DZF) and dizygotic opposite-sex (DOS) pairs). If correlations were not available for males and females separately, we entered the MZ and DZ correlations, i.e., the correlations based on both sexes. In cases where it was clear that the dizygotic correlation was based on same-sex twins only, we entered the dizygotic same-sex (DZSS) correlation.
-
Estimates of heritability (h2) and shared environmental component (c2), under the full ACE (or ADE) model. We entered 'h2_FULL' and 'c2_FULL', on the basis of estimates under the full ACE (including additive genetic and shared and non-shared environmental influences) or ADE (including additive and non-additive genetic and non-shared environmental influences) model. When an ACE model was fitted, the estimate for A was entered in 'h2_FULL' and the estimate for C was entered in 'c2_FULL'. When an ADE model was fitted, the estimates of A and D were summed and entered for 'h2_FULL' and zero was entered for 'c2_FULL'. When estimates were provided separately for males and females, they were entered separately. In the case of multivariate analyses, univariate estimates were always preferred to allow comparison across studies.
-
Estimates of heritability (h2) and shared environmental component (c2), under the best-fitting ACE (or ADE) model. We entered 'h2_BEST' and 'c2_BEST', on the basis of estimates under the best-fitting ACE or ADE model as provided in the study. When an ACE model was the best-fitting model, the estimate for A was entered in 'h2_BEST' and the estimate for C was entered in 'c2_ BEST'. When an ADE model was the best-fitting model, the estimates of A and D were summed and entered for 'h2_ BEST' and zero was entered for 'c2_ BEST'. When estimates were provided separately for males and females, they were entered separately. In the case of multivariate analyses, univariate estimates were always preferred to allow comparison across studies. In cases where estimates for the best-fitting model were not directly provided but information available in the paper indicated that the best-fitting model was AE (or CE or E), we entered zero for 'c2_ BEST' and missing for 'h2_ BEST' (when the best-fitting model was an AE model), missing for 'c2_ BEST' and zero for 'h2_ BEST' (when the best-fitting model was a CE model), and zero for 'c2_ BEST' and zero for 'h2_ BEST' (when the best-fitting model was described to be an E model).
-
The total number of twin pairs as used for each entered estimate.
-
Whether the study was a classical twin study. All 2,748 studies in the database included monozygotic and dizygotic twins. However, a classical twin study was defined as a study that involved only reared-together monozygotic and dizygotic twins. From studies that included siblings, extended families, adoptees or reared-apart twins, only estimates based on the reared-together twin sample were used for the meta-analyses. Most of the non-classical twin studies did provide twin correlations for the classical twin design and were thus included in the meta-analysis for twin correlations. When A and C estimates were based on extended twin designs, they were excluded from the meta-analyses.
-
The method used for estimating the variance components. We entered the statistical method used for estimating the variance components, which included, for example, ANOVA, Bayesian, maximum-likelihood (ML), DeFries-Fulker regression, least-squares (LS) or intrapair differences. We also listed a dichotomized version of this indicating whether the method used was 'ML or LS' or 'not ML or LS', for all other methods. In the meta-analyses for h2 and c2 estimates, only those based on maximum likelihood or least squares were included.
-
Whether the trait was dichotomous or continuous. Traits measured as 0 or 1, as well as traits measured on a quantitative scale but dichotomized before analysis, were listed as dichotomous. All other traits, including ordinal traits, were listed as 'not dichotomous' and treated as continuous.
-
Whether the study involved ascertainment for the trait. When the trait under investigation was the same trait that was used to select probands, the study was listed as 'ascertained'.
-
Number of concordant and discordant pairs. In cases of dichotomous traits, the total numbers of pairs for discordant and concordant affected pairs were entered separately for each zygosity. In cases of dichotomous traits that were not ascertained, the number of concordant unaffected pairs was also entered.
-
Prevalence. In cases of dichotomous traits, the population prevalence, separately for monozygotic and dizygotic twins when available, was entered. Prevalence was based on what was provided in the study or was calculated using (2c + d)/2n, where c is the number of concordant affected pairs, d is the number of discordant pairs and n is the total number of pairs in non-ascertained traits.
Thus, provided that there was availability, the statistics in Supplementary Table 1 were obtained for each trait reported on in every study. When the five basic twin correlations were available (rMZM, rMZF, rDZM, rDZF and rDOS), we calculated rMZ, rDZSS and rDZ, using the weighted average via Fisher z transformation and using sample size as weights. In situations where rMZM (or rMZF) was exactly 1 (or −1), we subtracted (or added in the case of −1) 0.00001 to the correlation to ensure non-problematic Fisher z transformation. Sample sizes of MZ, DZSS and DZ were obtained by summing the sample sizes of MZM and MZF, of DZM and DZF, and of DZM, DZF and DOS, respectively. Estimates of h2 and c2 were calculated across sex as the n-weighted average across the separate male and female estimates, when available. For the number of concordant and discordant pairs, MZ, DZ and DZSS were calculated on the basis of the numbers available for MZM, MZF, DZM, DZF and DOS. Prevalences for pooled entries were calculated as an n-weighted average.
Data entry checks.
Studies were entered and cross-checked for obvious typos by T.J.C.P. and D.P. After initial data entry and initial cross-checking, all data points were manually checked (D.P.) by looking up the entered values in the original paper. In addition, automatic checks were run (D.P. and B.B.) to identify typos, strange outliers or obvious mistakes. These checks included: (i) identifying highly unlikely values (clear typos, for example, correlation of 120); (ii) testing whether the sum of h2 and c2 was <100; (iii) testing for strange discrepancies between estimates from the full and best-fitting models; and (iv) checking for outliers on the basis of extreme sample size and extreme χ2 values for rejecting the null hypothesis that either 2 × (rMZ − rDZ) or 2 × rDZ − rMZ is equal to zero.
Classification of traits.
After data entry, all traits were manually classified using the ICF. The ICF is the framework of the World Health Organization (WHO) for health and disability and provides the conceptual basis for the definition, measurement and policy formulations for health and disability. It is a universal classification of disability and health for use in health and health-related sectors. ICF belongs to the WHO family of international classifications, the best-known member of which is the ICD-10. ICD-10 provides an etiological framework for the classification of diseases, disorders and other health conditions, whereas ICF classifies functioning and disability associated with health conditions. The ICD-10 and ICF are therefore complementary (see URLs).
Most traits investigated in twin studies concern healthy functioning (for example, cognitive function, social attitudes, body height and personality) and were classified according to ICF. In cases where the studied traits were diseases or symptoms of disease, ICD-10 was used. Traits were given two hierarchical classifications corresponding to the ICF or ICD-10 hierarchical structure, using the chapter structure (for example, b1) and the level directly under the chapter (for example, b110), which corresponds to the code for the actual disease (ICD-10) or trait (ICF).
Six new classes at the chapter level and 17 new classes at the subchapter level were created to accommodate traits that could not be classified under either ICF or ICD-10. For the chapter level, the created classes were cell, function of DNA, functions of the nervous system, medication effects, mortality and structure of DNA. For the subchapter level, the classes created were all-cause mortality, cell cycle, cell growth, diazepam effects, expression, function of brain, gene expression, height, methylation, mortality from heart disease, mtDNA, physical appearance, receptor binding, sister chromatid exchange, structure of DNA, telomeres and X inactivation.
In addition to the two standard ICF or ICD-10 classification levels, we added a general classification of functional trait domains. We thus classified all traits using a 3-level scheme that included 28 broad, functional domains, 54 ICF or ICD-10 chapter-level classes and 313 subchapter-level classes. A small proportion of studied traits (<0.1%) could not be classified meaningfully on the chapter level (two traits) or the subchapter level (three traits). There were 326 unique combinations across the 3 levels of trait categorization (Supplementary Table 33). All analyses were conducted on all entries of each of the three levels of classification. In addition, we analyzed all traits together. Although this is unspecific in terms of diseases or traits, it provides a general overview of the relationship between monozygotic and dizygotic twin correlations and shows general patterns of, for example, sex and age differences. The most specific level was the subchapter level, which was the actual ICD-10 diagnosis or a similar ICF classification for normal functioning, reflecting specific traits such as cleft lip, hyperkinetic disorders or higher-level cognitive function. As researchers do not necessarily adhere to the ICD-10 or ICF trait nomenclature, traits with the same subchapter classification could have different trait names in the original study: for example, for higher-level cognitive function, the original studies included the trait names total IQ score, cognitive ability, intelligence or 'g'.
Tests for publication bias.
Publication bias can occur when studies that report relatively large heritability estimates or high twin correlations are more likely to be submitted and/or accepted for publication than studies that report more modest effects. Such a publication bias would lead to an overestimation of the true twin correlations or the true heritability and environmental estimates. We used several standard statistical tools to aid in identifying and quantifying possible publication bias, including inspection of funnel plots, Begg and Mazumdar's test20, Egger's regression test21 and Rosenthal's fail-safe N22.
Meta-analysis methods of twin correlations and variance components.
We used the DerSimonian-Laird (DSL) random-effects meta-analytical approach with correlation coefficients as effect sizes, as described by Schulze23 and implemented in the R package metacor. This function transforms a correlation to its Fisher z value with corresponding standard error before the meta-analysis. This method is preferred over conducting a meta-analysis directly on the correlations because the standard error of a twin correlation is a function of not only sample size but also the correlation itself, with larger correlations having a smaller standard error. This can cause problems in a meta-analysis, as it would lead to the larger correlations appearing more precise and being assigned more weight in the analysis, irrespective of sample size. To avoid this problem, the DSL method transforms correlations to the Fisher's z metric, whose standard error is determined solely by sample size. All n-weighted computations were thus performed using Fisher's z metric, and the results were converted back to correlations for interpretation.
The random-effects approach allows for heterogeneity of the true twin correlations across different studies. That is, rather than assuming that there is one true level of twin correlation, the random-effects model allows a distribution of true correlations. The combined effect of the random-effects model represents the mean of the population of true correlations. For computational reasons, correlations of −1 and 1 were converted to −0.99999 and 0.99999 before meta-analysis. We set a threshold of at least five pairs of twins available per estimate and at least two studies available per category. Meta-analyses were conducted for each category of all three levels of classification.
We note that twin samples used in different publications were not independent. For example, studies using Australian twins are predominantly based on twins from the Australian Twin Registry. These studies sometimes include different subsamples but may also include completely overlapping samples used to investigate different traits. As participants are anonymous, it is not possible to determine the extent of overlap in the studies included in our analyses. We thus assumed independency of samples in the meta-analyses. This assumption leads to an underestimation of the variance of weighted estimates and an overestimation of their precision. We expect that the dependency of study samples is lowest at the specific level of the ICD-10 or ICF subchapters and highest for the general functional domains.
Meta-analysis for dichotomous, non-ascertained traits.
In the DSL random-effects model, the standard error of a correlation is calculated on the basis of the provided n (pairs). The estimated standard errors for continuous traits are correct, but for dichotomous traits the resulting standard error is incorrect. That is because twin correlations for non-ascertained, dichotomous traits are typically based on three categories of pairs: concordant, unaffected (CON−), discordant (DIS) and concordant, affected (CON+) pairs. Whereas the total number of participating pairs is the sum of these, the information that determines the twin correlation and its significance is mostly derived from the latter two categories. For non-ascertained, dichotomous traits, we calculated the study-specific tetrachoric twin correlation on the basis of the contingency table (i.e., CON−, DIS and CON+ pairs), under the assumption that the dichotomous traits represent latent variables that follow a bivariate normal distribution24. We used a maximum-likelihood estimator described by Olsson25, implemented in the R function polycor, to calculate the study-specific twin correlation and its standard error. As our meta-analysis required twin correlation and sample size (not standard error) as input and we wanted to be able to pool across continuous and dichotomous traits, we calculated the 'effective' number of pairs on the basis of the obtained standard error. The effective number of pairs was defined as the number of pairs that produced the exact same standard error within the DSL meta-analyses as the standard error obtained from the contingency table.
Meta-analysis for dichotomous, ascertained traits.
For ascertained traits, it was not possible to calculate the twin correlations and standard errors on the basis of the contingency table, as the traits included only pairs with at least one proband. Without information on the number of concordant, unaffected pairs, the prevalence of the affected status would be required to calculate a twin correlation. We used the algorithms derived from Falconer26 and Smith27. Again, for practical purposes, calculated standard errors were transformed to an effective number of pairs for use in the DSL meta-analysis.
Proportion of studies consistent with specific hypotheses.
We estimated the proportion of studies that were consistent with H0: 2 × (rMZ − rDZ) = 0 (π0(h)) and the proportion of observations consistent with H0: 2 × rDZ − rMZ = 0 (π0(c)), using the Jiang and Doerge method28, as well as the q-value method29.
Authorship network analysis.
We used the approach more fully described previously30. Briefly, we retrieved from PubMed the full Medline listing for all twin studies included in this meta-analysis using NCBI eutils. The output was parsed to capture the names of all authors. The twin study author list was manually reviewed to resolve clear inconsistencies in the spelling of the names of authors between publications. Gephi was used to construct a network to understand twin study publication patterns. For clarity, we removed individuals who had published only one paper (i.e., we required authorship on ≥2 papers). The substructure of the network was investigated by estimating community membership modules using the Louvain method31 implemented in Gephi.
URLs.
ICF classification, http://apps.who.int/classifications/icfbrowser/; ICD-10 classification, http://apps.who.int/classifications/icd10/browse/2010/en. The data used for this manuscript have been integrated in a web application, where user-specified selections of traits can be made to apply the analyses presented in this work. The web application is called MaTCH (Meta-analysis of Twin Correlations and Heritability) and is accessible via http://match.ctglab.nl/; Gephi, http://gephi.github.io/.
References
Moore, J.H. Analysis of gene-gene interactions. Curr. Protoc. Hum. Genet. Chapter 1, Unit 1.14 (2004).
Hill, W.G., Goddard, M.E. & Visscher, P.M. Data and theory point to mainly additive genetic variance for complex traits. PLoS Genet. 4, e1000008 (2008).
Traynor, B.J. & Singleton, A.B. Nature versus nurture: death of a dogma, and the road ahead. Neuron 68, 196–200 (2010).
Zuk, O., Hechter, E., Sunyaev, S.R. & Lander, E.S. The mystery of missing heritability: genetic interactions create phantom heritability. Proc. Natl. Acad. Sci. USA 109, 1193–1198 (2012).
Phillips, P.C. Epistasis—the essential role of gene interactions in the structure and evolution of genetic systems. Nat. Rev. Genet. 9, 855–867 (2008).
Visscher, P.M., Brown, M.A., McCarthy, M.I. & Yang, J. Five years of GWAS discovery. Am. J. Hum. Genet. 90, 7–24 (2012).
Manolio, T.A. et al. Finding the missing heritability of complex diseases. Nature 461, 747–753 (2009).
Stranger, B.E., Stahl, E.A. & Raj, T. Progress and promise of genome-wide association studies for human complex trait genetics. Genetics 187, 367–383 (2011).
Maher, B. Personal genomes: the case of the missing heritability. Nature 456, 18–21 (2008).
Eichler, E.E. et al. Missing heritability and strategies for finding the underlying causes of complex disease. Nat. Rev. Genet. 11, 446–450 (2010).
Nelson, R.M., Pettersson, M.E. & Carlborg, Ö. A century after Fisher: time for a new paradigm in quantitative genetics. Trends Genet. 29, 669–676 (2013).
Barker, J.S. Inter-locus interactions: a review of experimental evidence. Theor. Popul. Biol. 16, 323–346 (1979).
Cockerham, C.C. An extension of the concept of partitioning hereditary variance for analysis of covariances among relatives when epistasis is present. Genetics 39, 859–882 (1954).
Cockerham, C.C. in Statistical Genetics and Plant Breeding 53–94 (Nat. Acad. Sci. Nat. Res. Council Publ., 1963).
Kempthorne, O. On the covariances between relatives under selfing with general epistacy. Proc. R. Soc. Lond. B Biol. Sci. 145, 100–108 (1956).
Crow, J.F. & Kimura, M. An Introduction To Population Genetics Theory (Harper and Row, 1970).
Carlborg, O. & Haley, C.S. Epistasis: too often neglected in complex trait studies? Nat. Rev. Genet. 5, 618–625 (2004).
Falconer, D.S. & Mackay, T.F.C. Quantitative Genetics (Longman Group, 1996).
Lynch, M. & Walsch, B. Genetics and Analysis of Quantitative Traits (Sinauer Associates, 1998).
Begg, C.B. & Mazumdar, M. Operating characteristics of a rank correlation test for publication bias. Biometrics 50, 1088–1101 (1994).
Egger, M., Davey Smith, G., Schneider, M. & Minder, C. Bias in meta-analysis detected by a simple, graphical test. Br. Med. J. 315, 629–634 (1997).
Rosenthal, R. The file drawer problem and tolerance for null results. Psychol. Bull. 86, 91–106 (1979).
Schulze, R. Meta-Analysis: A Comparison Of Approaches (Hogrefe & Huber, 2004).
Drasgow, F. in Encyclopedia of Statistical Sciences (eds. Kotz, S., Read, C.B., Balakrishnan, N. & Vidakovic, B.) Vol. 7, 68–74 (John Wiley & Sons, 2006).
Olsson, U. Maximum likelihood estimation of the polychoric correlation coefficient. Psychometrika 44, 443–460 (1979).
Falconer, D.S. The inheritance of liability to certain diseases, estimated from the incidence among relatives. Ann. Hum. Genet. 29, 51–76 (1965).
Smith, C. Concordance in twins: methods and interpretation. Am. J. Hum. Genet. 26, 454–466 (1974).
Jiang, H. & Doerge, R.W. Estimating the proportion of true null hypotheses for multiple comparisons. Cancer Inform. 6, 25–32 (2008).
Storey, J.D. & Tibshirani, R. Statistical significance for genomewide studies. Proc. Natl. Acad. Sci. USA 100, 9440–9445 (2003).
Bulik-Sullivan, B.K. & Sullivan, P.F. The authorship network of genome-wide association studies. Nat. Genet. 44, 113 (2012).
Blondel, V., Guillaume, J., Lambiotte, R. & Lefebvre, E. Fast unfolding of community hierarchies in large networks. J. Stat. Mech. 10, P10008 (2008).
Acknowledgements
We would like to thank M. Frantsen, M.P. Roeling, R. Lee and D.M. DeCristo for their contribution to collecting the full texts of selected twin studies and data entry. This work was funded by the Netherlands Organization for Scientific Research (NWO VICI 453-14-005, NWO Complexity 645-000-003), by the Australian Research Council (DP130102666) and by the Australian National Health and Medical Research Council (APP613601).
Author information
Authors and Affiliations
Contributions
D.P., B.B., P.F.S. and P.M.V. performed the analyses. D.P. conceived the study. D.P., T.J.C.P. and P.M.V. designed the study. T.J.C.P. and D.P. collected and entered the data. D.P. and P.F.S. categorized traits according to standard classifications. A.v.B. and C.A.d.L. checked data entries, and checked and wrote statistical scripts. A.v.B. designed and programmed the webtool. D.P., T.J.C.P. and P.M.V. wrote the manuscript. All authors discussed the results and commented on the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Integrated supplementary information
Supplementary Figure 1 Authorship co-occurrence matrix on 2,748 twin studies published between 1958 and 2012.
Each colored cell represents two authors who appeared on the same paper; darker cells indicate authors that co-published more frequently. The filter of at least 25 papers per author was set for readability. The web application MaTCH has an interactive version of this matrix.
Supplementary Figure 2 Funnel plots across all traits for twin correlations and variance components.
Z, Z-converted correlation; MZ, monozygotic twins; DZ, dizygotic twins; DZSS, DZ same-sex twins; MZM, MZ male twins; MZF, MZ female twins; DZM, DZ male twins; DZF, DZ female twins; DOS, DZ opposite-sex twins; h2, heritability; c2, shared environment; h2 same sex; c2 same sex; h2 males; c2 males; h2 female; c2 females.
Supplementary Figure 3 Funnel plots for rMZ across the major trait domains.
The plots denote the relationship between the Z-transformed rMZ and its standard error. SE, standard error.
Supplementary Figure 4 Funnel plots for rDZ across the major trait domains.
The plots denote the relationship between the Z-transformed rDZ and its standard error. SE, standard error.
Supplementary Figure 5 Funnel plots for h2 across the major trait domains.
The plots denote the relationship between the Z-transformed h2 and its standard error. SE, standard error.
Supplementary Figure 6 Funnel plots for c2 across the major trait domains.
The plots denote the relationship between the Z-transformed c2 and its standard error. SE, standard error.
Supplementary Figure 7 Distribution of twin correlations and variance components in full and best models across all traits from 2,748 studies.
rMZ, monozygotic twin correlation; rDZ, dizygotic twin correlation; rDZSS, DZ same-sex twin correlation; rMZM, MZ male twin correlation; rMZF, MZ female twin correlation; rDZM, DZ male twin correlation; rDZF, DZ female twin correlation; rDOS, DZ opposite-sex twin correlation; h2, heritability; c2, shared environment; h2 same sex;c2 same sex; h2 males; c2 males; h2 females; c2 females. “BEST” denotes estimates from the most parsimonious models per study. All other estimates are from “FULL” models.
Supplementary Figure 8 Distribution of differences between MZ and DZ correlations.
rMZ, monozygotic twin correlation; rDZ, dizygotic twin correlation; rDZSS, DZ same-sex twin correlation; rMZM, MZ male twin correlation; rMZF, MZ female twin correlation; rDZM, DZ male twin correlation; rDZF, DZ female twin correlation; rDOS, DZ opposite-sex twin correlation.
Supplementary Figure 11 Scatterplots of all MZ versus DZ correlations.
Contour lines indicate the density of the data in that region. The lines are ‘heat’ colored from blue to red, indicating increasing data density.
Supplementary Figure 12 QQ plots of the χ2 test statistics for testing the null hypothesis that 2(rMZ – rDZ) = 0 and 2rDZ – rMZ = 0 and relationship with sample size.
(a) The deviation from the null hypotheses is quantified with the inflation λ in the QQ plots. (b) Effects as a function of sample size.
Supplementary information
Supplementary Text and Figures
Supplementary Figures 1–12, Supplementary Note and Supplementary Tables 1–19, 22–24 and 26–31. (PDF 4786 kb)
Supplementary Tables 20, 21, 25, 32 and 33.
Supplementary Tables 20, 21, 25, 32 and 33. (XLSX 609 kb)
Rights and permissions
About this article

Cite this article
Polderman, T., Benyamin, B., de Leeuw, C. et al. Meta-analysis of the heritability of human traits based on fifty years of twin studies. Nat Genet 47, 702–709 (2015). https://doi.org/10.1038/ng.3285
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ng.3285
This article is cited by
-
The genetic architecture of youth anxiety: a study protocol
BMC Psychiatry (2024)
-
Attention-deficit/hyperactivity disorder
Nature Reviews Disease Primers (2024)
-
Genetic effects on variability in visual aesthetic evaluations are partially shared across visual domains
Communications Biology (2024)
-
Examining intergenerational risk factors for conduct problems using polygenic scores in the Norwegian Mother, Father and Child Cohort Study
Molecular Psychiatry (2024)
-
Can flow proneness be protective against mental and cardiovascular health problems? A genetically informed prospective cohort study
Translational Psychiatry (2024)
