
Abstract
The eukaryotic genome consists of DNA molecules far longer than the cells that contain them. They reach their greatest compaction during chromosome condensation in mitosis. This process is aided by condensin, a structural maintenance of chromosomes (SMC) family member1,2. The spatial organization of mitotic chromosomes and how condensin shapes chromatin architecture are not yet fully understood. Here we use chromosome conformation capture (Hi-C)3,4 to study mitotic chromosome condensation in the fission yeast Schizosaccharomyces pombe5,6,7. This showed that the interphase landscape characterized by small chromatin domains is replaced by fewer but larger domains in mitosis. Condensin achieves this by setting up longer-range, intrachromosomal DNA interactions, which compact and individualize chromosomes. At the same time, local chromatin contacts are constrained by condensin, with profound implications for local chromatin function during mitosis. Our results highlight condensin as a major determinant that changes the chromatin landscape as cells prepare their genomes for cell division.
Similar content being viewed by others


Main
Mitotic chromosomes are one of the most recognizable structures in eukaryotic cells. While their microscopic description goes back to the nineteenth century8, knowledge of their molecular architecture remains scarce. Mitotic chromosome condensation is promoted by the chromosomal condensin complex, a ring-shaped multisubunit protein assembly that topologically entraps DNA and that is thought to establish linkages between more than one DNA1,2. How condensin promotes chromosome condensation remains a topic of intense current interest. Chromosome conformation capture is a powerful tool to visualize patterns of chromosomal DNA contacts, yielding insight into how the DNA is arranged inside chromosomes3,4,9,10. This technique identified large-scale homogenous folding of the chromatin chain within human mitotic chromosomes11. How local chromatin behavior changes as a result of mitotic chromosome compaction and how the condensin complex contributes to this process remain important open questions.
To understand the changes of mitotic chromatin organization at high resolution, we applied a high-throughput chromosome conformation capture variant, Hi-C, to fission yeast. The small size of the fission yeast genome, distributed among three chromosomes, and its use of a fundamentally conserved chromosome condensation machinery make this organism an attractive choice. We compared chromosomal DNA contact maps between asynchronously growing cells, >90% of which are in the G2 stage of interphase12, and cells arrested in mitosis by transcriptional repression of the Slp1Cdc20 activator of the anaphase-promoting complex (Supplementary Fig. 1a)13. In our Hi-C analysis, mapped paired-end reads indicative of a DNA interaction were assigned to the nearest restriction site used for DNA fragmentation (DpnII), and these sites were then grouped into 2-kb bins for further analysis (Supplementary Fig. 2a). This resulted in high-resolution DNA interaction frequency maps of the fission yeast genome (Fig. 1a). High reproducibility of the interaction maps under these and each of the following biological conditions was confirmed by clustering of the Euclidean distance between the directionality plots of three independent repeats of each experiment (Supplementary Fig. 2b,c).

(a) Normalized Hi-C contact probability maps in wild-type cells in interphase and mitosis (lower left and upper right triangles, respectively). The three fission yeast chromosomes are shown with centromere positions indicated (black dots). See Supplementary Figure 3 for an annotated schematic of the interactions that are recorded in this map. (b) Distribution of normalized contact probabilities between chromosomes (Inter-chr), within chromosome arms (Intra-arm) or between the two arms of the same chromosome (Inter-arm).
In addition to the expected richness of local DNA interactions along the three fission yeast chromosomes, seen in dark colors along the diagonal of the interphase contact map, prominent inter-centromeric and inter-telomeric interactions between chromosomes were apparent (Fig. 1a, lower left triangle; compare to Supplementary Fig. 3 for an annotated map). This is expected from the Rabl orientation of fission yeast chromosomes in interphase9,12,14. In mitosis, interactions appeared to spread to greater distances from the diagonal, seen by the broadening of the dark region (Fig. 1a, upper right triangle). In contrast, interactions between chromosomes and between the two arms of each chromosome were reduced, as indicated by the lighter orange color of these regions. Contact probability distribution plots confirmed a quantitative enrichment of interactions within chromosome arms in mitosis (Fig. 1b), at the expense of interactions between arms and between chromosomes. This provides a molecular correlate of the cytologically observed chromosome individualization and chromosome arm stiffening in mitosis (Supplementary Fig. 1a)7,8,15.
The changes in contact probabilities between interphase and mitosis were seen more clearly on Hi-C difference maps, obtained by simple subtraction of the normalized contact probabilities (Fig. 2a, upper right triangle). This confirmed an increased frequency of interactions within chromosome arms in mitosis, as well as the accompanying reduction in inter-arm and inter-chromosomal interactions. To elucidate the contribution of condensin to the altered contact probabilities in mitosis, we established a conditional shutoff allele for the condensin subunit Cut14Smc2 by combining promoter repression with an auxin-inducible degron (cut14SO; Supplementary Fig. 1a,b)5,16. The efficiency of Cut14 shutoff was confirmed by loss of mitotic chromosome compaction, measured by the distance between two fluorescent marks on chromosome I (Supplementary Fig. 1c–e)13. A Hi-C difference map between cells in interphase and condensin-depleted mitotic cells showed hardly any changes in contact probabilities (Fig. 2a, lower left triangle). Contact probability distribution plots confirmed that the previously seen changes in mitosis, namely increased intra-arm interactions, as well as reduced inter-chromosomal and inter-arm interactions, all depended on condensin (Fig. 2b; compare to Fig. 1b). Similar results were obtained after depletion of the condensin subunit Cnd3CAP-G or following inhibition of Aurora B kinase, a positive regulator of mitotic condensation6,17,18,19,20, although Cnd3CAP-G was depleted less efficiently than Cut14Smc2 and Aurora B inhibition only partly blocked chromosome condensation (Supplementary Figs. 1b,f and 4a–c). A Hi-C difference map of mitotic cells containing or depleted of Cut14Smc2 further illustrated that condensin is responsible for most of the observed contact probability changes in mitosis (Fig. 2c).

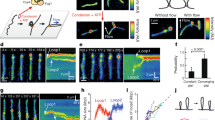
(a) Hi-C difference maps between interphase and mitosis of wild-type and cut14SO cells (upper right and lower left triangles, respectively). (b) Distribution of normalized contact probabilities. As in Figure 1b, but comparing interphase and cut14SO mitosis. (c) Hi-C difference map comparing wild-type and cut14SO cells in mitosis. (d) Quantification of inter-centromeric interaction changes between interphase and mitosis. Box plots of the Hi-C contact probability changes between bins within central cores + 10 kb are compared to the medians and range of 3C–qPCR between centromere II and centromeres I and III of two technical replicates each of three independent experiments. The box shows the 25th, 50th and 75th percentiles; whiskers indicate 1.58 times the interquantile distance divided by the square root of total interaction number. (e) Localization of centromeres is shown in fixed mitotic cells. Mitotic spindles were also visualized and DNA was counterstained using DAPI. Scale bar, 1 μm (f) The number of distinct centromere signals was counted. n = 100 cells were scored in three independent experiments each. The mean ± s.d. is shown together with individual results.
Hi-C differences indicate relative, but not necessarily absolute, changes of contact probabilities. We therefore used 3C followed by quantitative real-time PCR (3C–qPCR) to calibrate our Hi-C difference maps. We chose inter-centromeric interactions as a benchmark, which are among the strongest inter-chromosomal contacts and are reliably detectable. 3C–qPCR showed that the interaction frequency between centromeres dropped by just over twofold in mitosis, consistent with the almost twofold reduction in inter-centromeric interactions seen in the Hi-C difference maps (Fig. 2d). Both the Hi-C difference map and 3C–qPCR suggested that inter-centromeric contacts were retained in mitotic cells lacking condensin (Fig. 2d). To confirm this by an independent technique, we microscopically visualized centromere positioning in wild-type and cut14SO cells arrested in mitosis. Centromeres were separated and distributed along the short metaphase spindle in wild-type cells, as previously described (Fig. 2e,f)12. Centromeres remained clustered close to the spindle poles in Cut14-depleted cells, often displaying stretched signals. This pattern is consistent with persistent inter-centromere contacts and reminiscent of mitotic defects that have previously been reported in the absence of condensin19,21,22,23. Thus, the contact frequency changes reported in our Hi-C difference maps are a good reflection of absolute changes that occur during mitotic chromosome condensation.
We next focused our analysis on the most frequent DNA contacts, those within chromosome arms, as these are the most likely drivers of mitotic chromosome architecture changes. Figure 3a shows a magnified view of the Hi-C difference map along the right arm of chromosome II, demonstrating increased longer-range interactions within the arm. Unexpectedly, we also noticed a marked reduction in short-range contacts, seen as a blue line along the base. Both changes depended on the condensin complex (Fig. 3b), as well as Aurora B kinase activity (Supplementary Fig. 4). Plotting the median contact probability as a function of genomic distance showed fewer intra-arm interactions shorter than 84 kb in mitosis but increased interaction frequencies for distances between 84 and 940 kb (Fig. 3c). Interactions beyond a distance of 940 kb were again depleted in mitosis (the transition points lie around 90 kb and 900 kb, respectively, on all chromosome arms). Again, these rearrangements of contact probabilities depended on condensin and Aurora B kinase activity (Fig. 3c and Supplementary Fig. 4). The reduction of megabase-scale contacts is reminiscent of reduced longest-range interactions as human chromosomes condense11, a probable reflection of chromosome-arm stiffening as mitotic chromosomes gain shape.

(a,b) Hi-C difference maps of the chromosome II right arm comparing interphase and mitosis in wild-type (a) and cut14SO (b) cells. (c) Median contact probabilities in interphase and mitosis as a function of distance along the chromosome II right arm are shown in wild-type (top) and cut14SO (bottom) cells. (d) 4C-like plot of Hi-C contact probability from a viewpoint on the chromosome II right arm. The positions of 3C–qPCR primer-binding sites are shown (dotted and solid lines). (e) Interaction changes between interphase and mitosis determined by 3C–qPCR (mean ± s.e.m. of three biological repeats) are compared to 4C-like contact probability changes based on the Hi-C data (dots). Lines represent smoothed 4C-like contact probability changes. (f) Box plots of the distribution of contact frequencies within all chromosome arms under the indicated conditions: I, interphase; M, mitosis. Box plots show the median and 25th and 75th percentiles; whiskers indicate the 95% confidence interval. Outliers are also shown. (g) Moving intra-arm median interacting distances along chromosome II (solid lines) are shown together with shaded areas representing the 25th and 75th percentiles.
The chromosome volume decreases during mitotic condensation, resulting in chromatin compaction. A decrease in local chromatin contacts within mitotic chromosomes came therefore as a surprise. To further investigate this, we plotted interaction frequencies from one selected viewpoint in the Hi-C data to obtain a 4C-like plot4. Figure 3d shows such a plot from a viewpoint close to the middle of the chromosome II right arm. We selected seven distances from this viewpoint and compared contact frequency in interphase and mitosis by 3C–qPCR (Fig. 3e). This provided quantitative confirmation of the 4C-like plot. Intra-arm contacts closer than 90 kb were reduced up to twofold in mitosis, while longer-range interactions increased to a similar extent, consistent with the changes seen in the Hi-C difference map. Both effects again depended on condensin. Thus, an unanticipated consequence of chromosome condensation is a reduction in local chromatin contacts.
4C-like analysis also allowed us to address whether chromosomes condense homogeneously or whether compaction varies along chromosome arms. We used a sliding viewpoint and recorded the contact distributions at each position along chromosome II. The median length of all interactions was around 50 kb in interphase, which increased to around 150 kb in mitosis (Fig. 3f). Plotting the sliding median along chromosome II showed somewhat longer interactions in the middle of both the left and right chromosome arms and shorter interactions toward the centromeres and chromosome ends (Fig. 3g). This trend was not an artifact of the iteratively corrected normalization used to generate our Hi-C maps and was similarly seen when using an alternative normalization strategy, as well as in the raw read counts (Supplementary Fig. 5). This implies that fission yeast chromosomes are slightly more compact in the middle of their arms than in centromere-proximal regions or chromosome ends.
Chromosomes are thought to consist of a series of topologically associating domains (TADs)9,24,25,26, within which chromatin interactions are enriched. We analyzed TADs along fission yeast chromosomes in interphase and mitosis by plotting the directionality of interactions along the chromosome (Supplementary Fig. 2b), from which we determined TAD boundary positions (Fig. 4a). These boundaries correlated with local minima of insulation scores, thus validating the boundary assignments (Supplementary Fig. 2d,e). This analysis confirmed the existence of numerous TADs in interphase with a median size of 267 kb. The median TAD size nearly doubled in mitosis to 481 kb, dependent on condensin as well as Aurora B kinase activity (Fig. 4a,b and Supplementary Fig. 6). The condensin-mediated expansion of TAD size in mitosis is compatible with the size range of condensin-mediated chromosome interactions previously determined by ChIA-PET27. Notably, 69% (25 of 36) of the boundary positions in mitosis coincided with those observed in interphase, suggesting that interphase TADs fuse to give rise to larger chromosome domains in mitosis. We reached the same conclusions when we used an alternative algorithm (TopDom)28 to assign domain boundaries (Supplementary Fig. 7).

(a) Normalized Hi-C maps are shown with domain boundaries along a section of the chromosome I left arm under the indicated conditions. For domain visualization, a bin size of 5-kb was used. Black triangles indicate domain boundaries. (b) Density plots of domain size distributions. (c) Distribution of normalized contact probabilities of 5-kb bins containing or not containing a condensin-binding site. A Wilcoxon Mann–Whitney test was used to test the null hypothesis that contact probabilities between condensin-binding and non-binding sites are the same. (d) Contact probability changes between interphase and mitosis are plotted as a function of distance, separated into bins containing or not containing a condensin-binding site.
We started to address how condensin promotes longer-range interactions by performing ChIP to determine condensin-binding sites. We divided the chromosome into 5-kb bins that were categorized into those that did or did not contain a condensin-enriched site (Supplementary Table 1). The contact probability plot of these 5-kb bins showed that condensin-binding sites have a small but significantly greater probability of engaging in intra-arm interactions, as compared to non-binding sites (Fig. 4c). This suggests that condensin acts by engaging in contacts between its binding sites. Next, we analyzed whether condensin-binding sites show a preference for longer-range DNA interactions. The mitotic interaction frequency change of condensin-binding sites as a function of genomic distance confirmed that these sites engage in mitotic longer-range interactions in a condensin-dependent manner. However, sites not binding condensin showed a qualitatively similar behavior (Fig. 4d). Therefore, condensin-binding sites are preferential targets for intra-arm interactions that facilitate contacts between neighboring DNA sequences. While site-specific looping between condensin-binding sites has been seen by ChIA-PET27, our Hi-C approach finds that intra-arm interactions are widely spread. Condensin binding to chromosomes with limited sequence preference could be a reason for this.
To further clarify the role of condensin-binding sites in chromatin organization, we analyzed their relationship with chromatin boundary positions. Most interphase and mitotic boundaries contained a condensin-binding site (Supplementary Fig. 8), consistent with a role for condensin in domain formation and their fusion in mitosis.
An unexpected consequence of mitotic chromosome condensation was the reduction in local chromatin contacts. To address whether this is due to restricted chromatin motility in mitosis, we monitored local movement of two loci on chromosomes I and II, visualized using LacO repeats bound by LacI-GFP and TetO repeats bound by TetR-tdTomato, respectively, by high-speed confocal microscopy. Kymographs of the LacI-GFP movements on chromosome I showed greater motility in interphase than mitosis (Fig. 5a). To quantify this difference, we tracked the GFP locus at 20-ms intervals and derived mean square displacement plots. This confirmed that chromatin motility is constrained in mitosis (Fig. 5b). The same was observed on chromosome II (Supplementary Fig. 9). Constrained mitotic chromatin motility depended on condensin. Motility markedly increased following condensin depletion, exceeding even that observed in interphase (Fig. 5b). We could not directly measure condensin's impact on interphase motility, as the condensin depletion that we achieve during mitotic arrest requires longer than cells usually spend in interphase.

(a) Kymographs of the LacO locus on chromosome I, marked by LacI-GFP, in wild-type cells in interphase and mitosis. The schematic illustrates the experiment. Scale bar, 1 μm. (b) Mean square displacement (MSD) of the LacO locus under the indicated conditions. Mean ± s.e.m. is plotted (n = 61–72). (c) A model for how chromosome compaction by condensin is achieved by longer-range interactions, accompanied by chromatin domain enlargement as well as reduced local chromatin motility.
Our study describes the conformational changes of eukaryotic chromatin during mitotic chromosome condensation. Condensin replaces predominantly short-range local contacts in interphase with longer-range interactions in mitosis. Our results do not distinguish whether condensin establishes longer-range interactions by extruding or expanding DNA loops, by stabilizing stochastic chromatin interactions, or both29,30. In either scenario, a slower dissociation rate of condensin from chromosomes, which has been observed in mitosis23, could promote longer-range interactions. It will be interesting to computationally study predictions from the two models and compare them to our high-resolution experimental data sets. A notable consequence of mitotic longer-range interactions is restricted chromatin motility, accompanied by reduced local contacts (Fig. 5c). Establishment of gene silencing in budding yeast requires passage through mitosis, while transcriptional reprogramming in mammalian nuclei is promoted by mitotic factors, in ways that are not fully understood31,32,33. Future studies will explore whether reduced local chromatin motility facilitates either process. In line with the central role played by condensin during biochemically reconstituted chromosome assembly34, our findings illustrate condensin's principal contribution to chromatin reorganization during mitotic chromosome condensation.
Methods
S. pombe strains and culture.
All the strains used in this study are listed in Supplementary Table 2. PCR-based gene targeting35 and standard genetic manipulation36 were used during strain construction. To obtain the cut14so and cnd3so strains, the corresponding gene sequence for the C terminus was fused to the sequence encoding the auxin-inducible degron module IAA17 (ref. 37), and the endogenous gene promoter was replaced by the nmt81 promoter38 in cells harboring Skp1–Tir1 (ref. 16). For the Z2-Cnd3-GFP strain, a plasmid containing the cnd3 gene fused with GFP under the regulation of the endogenous cnd3 promoter was constructed by the module-based Golden Gate method for chromosomal integration plasmids in fission yeast39. The resultant plasmid was linearized by enzymatic digestion (FseI) and integrated at the Z2 locus. To replace the endogenous slp1 promoter, a kanR–nmt41 promoter–slp1 fragment was amplified from a previously described slp1 shutoff strain13 and integrated.
All strains were cultured in Edinburgh minimal medium (EMM) supplemented with L-glutamic acid as a nitrogen source. To shut off slp1 transcription and induce mitotic arrest, cells were cultured at 25 °C for 3 h after addition of 5 μg/ml thiamine. After this, 60–75% of cells were arrested in mitosis, displaying a short mitotic spindle. The efficiency of this arrest was unaffected by condensin depletion or Aurora B kinase inactivation. To deplete Cut14 or Cnd3, cells were cultured at 25 °C for 3 h with 5 μg/ml thiamine as well as 0.5 mM of the auxin 1-naphthaleneacetic acid (NAA). To inhibit Aurora B kinase (Ark1), ark1-as3 cells18 were cultured with 5 μg/ml thiamine at 25 °C for 1.5 h and then with 5 μM 1NM-PP1 or the equivalent amount of DMSO as a control for a further 1.5 h.
Fluorescence microscopy.
Cells were fixed with 70% ethanol and stained with 4′,6-diamidino-2-phenylindole (DAPI). Images were acquired as serial sections along the z axis on a DeltaVision microscope system (Applied Precision) and combined using the quick projection algorithm in SoftWoRx. To measure the distance between genomic loci marked with LacO repeats bound by LacI-GFP and TetO repeats bound by TetR-tdTomato, z stacks were merged and the distance was measured in 2D using Fiji40. To track chromatin motility, live-cell imaging was performed using an α Plan Apochromat 100×/1.46 NA oil objective (Carl Zeiss) in a temperature-controlled chamber. Images were captured on an Evolve 512 EMCCD camera (Photometrics), operated in streaming mode and mounted on an inverted microscope (AxioObserver.Z1, Carl Zeiss), the constituents of a custom-built spinning-disc confocal system (Intelligent Imaging Innovations). All hardware was controlled with SlideBook software (Intelligent Imaging Innovations). Images were collected at 20 Hz (TetR-tdTomato) or 50 Hz (LacI-GFP), following excitation with either a 488- or 568-nm laser. Fluorescent spot movement was automatically tracked, and MSD was calculated using the ParticleTracker41 plugin in Fiji (see URLs).
Immunoblotting.
Cell extracts were prepared by glass bead cell breakage as detailed below. Protein levels were analyzed by SDS–PAGE followed by immunoblotting using antibody to AID (Cosmo Bio, CAC-APC004AM) or Tat1 (a mouse monoclonal antibody generated in house against α-tubulin) as primary antibody.
Hi-C and 3C sample preparation.
The Hi-C experiment was performed as previously described, with modifications3. Approximately 7.5 × 108 fission yeast cells were fixed with 0.5% formaldehyde at 25 °C for 30 min. Excess formaldehyde was quenched with 0.125 M glycine at 25 °C for 15 min. Fixed cells were washed three times with wash buffer (50 mM HEPES-KOH pH 7.5, 140 mM NaCl, 1 mM EDTA, 1% Triton X-100 and 1 mg/ml NaN3) and resuspended in FA buffer (50 mM HEPES-KOH pH 7.5, 140 mM NaCl, 1 mM EDTA, 1% Triton X-100 and 0.1% sodium deoxycholate) containing cOmplete ULTRA protease inhibitor cocktail (Roche). Cells were ruptured using glass bead breakage in a Multi Beads Shocker (Yasui Kikai). Crude extract was centrifuged to retrieve the chromatin fraction at 20,800g at 4 °C for 15 min. Precipitated chromatin was washed once with DpnII restriction enzyme incubation buffer (New England BioLabs) and then resuspended in DpnII buffer and incubated at 65 °C for 10 min with 0.1% SDS. After addition of 1% Triton X-100, chromatin was digested overnight with DpnII (New England BioLabs) and RNase A (Sigma) at 37 °C. After heat inactivation of DpnII at 65 °C for 20 min, the digested chromatin was divided into three parts, one for 3C library preparation and the other two for Hi-C library preparation. For Hi-C library preparation, DpnII overhangs were filled in with biotin-14-dATP, dCTP, dGTP and dTTP using DNA polymerase I Klenow fragment (New England BioLabs) at 37 °C for 45 min. To capture proximity, chromatin was diluted to 8 ml in T4 DNA ligase buffer followed by incubation at 16 °C for 8 h with T4 DNA ligase (New England BioLabs). For 3C library preparation, digested chromatin was similarly ligated, but without the fill-in step. DNA was in both cases recovered by proteinase K digestion and reversal of cross-linking overnight at 65 °C, followed by phenol-chloroform extraction and isopropanol precipitation. Hi-C library preparation progressed by removing biotin-14-dATP at unligated ends by incubation at 20 °C for 4 h in the presence of 300 U of T4 DNA polymerase (New England BioLabs). After inactivating T4 DNA polymerase by addition of EDTA, DNA was fragmented using a focused ultrasonicator (Covaris, S220) and purified on AMPure XP beads (Beckman Coulter). The ends of the fragmented DNA were repaired, and sequencing adaptors were ligated using NEBNext Ultra DNA Library Prep kit reagents and NEBNext Multiplex Oligos for Illumina (Index Primers Set 1, New England BioLabs). Junction-containing biotinylated DNA fragments were adsorbed to MyOne Streptavidin C1 DynaBeads (Thermo Fisher) at room temperature for 15 min. Bead-bound DNA was amplified using NEBNext High-Fidelity 2× PCR Master Mix with index primer sets. DNA fragments in the 200- to 700-bp range were purified by double selection on SPRI selection beads (Beckman Coulter), followed by sequencing on the Illumina HiSeq platform (100-bp paired-end reads).
3C–qPCR.
3C libraries were prepared as described above. The efficiency of proximal ligation was quantified using primer pairs corresponding to interactions of interest (listed in Supplementary Table 3). Real-time qPCR was performed with PowerUp SYBR Green Master Mix using a QuantStudio 12K Flex Real-Time PCR System (Applied Biosystems). Ct values were normalized using internal controls (YK377–YK378 for inter-centromeric interactions, YK717–YK718 for intra-arm interactions), and ΔCt values comparing interphase and mitosis were then calculated. For quantification of inter-centromeric interactions, ΔCt values were converted to a linear scale to derive fold-change values. Two technical replicates were averaged in each of three biological repeats of the experiment. The median and range of values from the three repeat experiments are reported. To quantify intra-arm interactions, again two technical replicates were averaged in each of three biological repeats. The mean of ΔCt values between interphase and mitosis in the three biological repeats was plotted together with the standard error.
ChIP–seq sample preparation.
Cells were fixed with 0.5% formaldehyde, and crude cell extracts were then prepared by glass bead breakage as described above. Extracts were sonicated using a Bioruptor (Diagenode). After removing debris by centrifugation, extracts were split into input and ChIP samples. The ChIP sample was incubated with antibody to HA (F-7; Santa Cruz Biotechnology, sc-7392) overnight at 4 °C. Precipitated DNA–protein complexes were washed twice with FA buffer, twice with FA buffer containing 500 mM NaCl, twice with ChIP wash buffer (10 mM Tris-HCl pH 8, 250 mM LiCl, 0.5% NP-40, 0.5% sodium deoxycholate and 1 mM EDTA) and once with TE. DNA ends were repaired by incubation with Klenow enzyme (New England BioLabs) in the presence of dNTPs at 25 °C for 30 min. End-repaired DNA–protein complex was eluted with TE containing 1% SDS by incubating at 25 °C for 30 min. Eluted DNA–protein complexes were incubated with T4 DNA ligase and then incubated at 65 °C overnight, followed by incubation with RNase A at 37 °C for 60 min. DNA was recovered by proteinase K digestion and reversal of cross-linking at 55 °C for 2 h, followed by phenol-chloroform extraction and ethanol precipitation. Sequencing adaptors were ligated to the ends of purified DNA using the NEBNext Ultra DNA Library Prep kit and NEBNext Multiplex Oligos for Illumina. The adaptor-ligated DNA was amplified using NEBNext High-Fidelity 2× PCR Master Mix with index primer sets. Amplified fragments were purified by double selection on SPRI selection beads (Beckman Coulter), followed by sequencing on the Illumina HiSeq platform.
Hi-C data analysis.
Identification of ligation events. Reads containing DpnII recognition motifs were trimmed to immediately after the first occurrence of the motif. If either read of the resultant read pair was shorter than 20 bp, the pair was discarded. Each read of the read pair was aligned individually to the S. pombe genome (build ASM294v2.21) using the BWA-mem algorithm (version 0.7.12)42. Read pairs with unmapped reads or read alignments with a mapping quality of <10 were discarded. To remove reads derived from unligated genomic fragments, concordantly mapped read pairs within 2 kb of each other on the genome were also discarded. The sonication fragmentation step within the library preparation protocol should generate unique read pairs from the ligation of any two DpnII sites within the genome. Identically mapped read pairs were therefore assumed to be PCR duplicates. A single read from these putative duplicate reads was randomly selected, and the other reads were discarded. The remaining reads were extended to the nearest DpnII recognition site downstream of the read start. Read pairs where either read start site was >1 kb upstream of a DpnII site were presumed to be derived from unligated fragments and were discarded. Ligation between the remaining read pairs is presumed to have occurred between the downstream DpnII sites. The number of read counts at each step for each of our Hi-C libraries is summarized in Supplementary Table 4.
Generation of interaction matrices. Count matrices were generated for chromosomes I, II and III. The genome was divided into equally sized bins (2 kb or 5 kb). Where chromosome length was not an exact multiple of bin length, the maximum number of non-overlapping bins was generated and the most telomeric regions of the chromosome were not analyzed. Few reads, if any, are accurately assigned to these regions owing to their non-unique sequences. Thus, no information is lost, while the equally sized bins ease subsequent statistical analyses. The count matrix is filled by ascribing individual DpnII ligation events to the bins containing their respective DpnII sites. When the ligation event occurs at a bin boundary, the event is ascribed to the bin containing the mapped read. To examine subsets of interactions, for example, inter-chromosome, intra-arm or inter-arm ligations, the count matrices for these regions were simply extracted from the larger count matrix.
Normalization of interaction matrices. The observed interaction frequency between two genomic regions is affected by many factors other than proximity. Normalization of the interaction matrices aims to remove these 'non-proximity' effects. Two techniques were used to normalize the matrices. The first technique, iterative correction43, does not attempt to model the source of these non-proximity factors. Instead, it operates on the principle that all genomic regions should be equally visible and partake in an equal number of interactions. The second technique, HiCNorm44, attempts to remove the impact of three non-proximity factors known to affect observed interactions—fragend frequency, GC content and mappability. The iteratively corrected normalized matrices were used for most of our analyses, except in Supplementary Figure 5a, where HiCNorm was used.
Prior to normalization, the intra-bin ligations were removed from the matrix by setting the diagonal of the count matrix to zero. Additionally, bins involved in fewer inter-bin interactions than a specified threshold were removed from the matrix to ensure optimal functioning of the normalization algorithm. A threshold of 500 interactions was used for the 2-kb bins (Supplementary Fig. 2a) and a threshold of 1,000 was used for the 5-kb bins. Matrices for the 2-kb bins were used for our analysis if not stated otherwise.
The iterative correction algorithm43 was used to normalize the altered count matrices such that all rows and columns were set to sum to 1. The resultant matrix can therefore be considered to be a probability matrix where the value at point (i,j) is the probability that region i is found to interact with region j, and vice versa. HiCNorm (version 08.05.2012)44 was provided with the mappability scores, GC content and active fragend size of all the 2-kb bins. Mappability scores and GC content were calculated from all regions within a bin that were within 200 bp of a DpnII cut site. Active fragend size is the cumulative length of these regions. Mappability scores were calculated by aligning tiled 50-bp reads (10-bp window shift), extracted from the regions of interest, and identifying the proportion of reads with a mapping quality of 10 or greater as defined by BWA-mem (version 0.7.12)42. A floor value for mappability scores was set as 0.1.
Interaction directionality and Euclidean clustering. To calculate the interaction directionality for a given bin, the interaction probabilities between the bin and its 50 upstream and 50 downstream bins were extracted from the intra-arm normalized matrix. The upstream and downstream probabilities were then paired by their absolute distance from the bin of interest. The interaction directionality value is the mean value of the log2 ratio of each pair of upstream and downstream probabilities. A value greater than 0 (a positive log2 directionality value) indicates that the bin preferentially interacts with upstream regions, whereas a value smaller than 0 (a negative log2 directionality value) indicates that downstream interactions are favored. Where one or more of the paired probability values were equal to zero, the pair was excluded from the calculation. Where fewer than two paired distance probabilities were available, owing to zero values or the end of the chromosome, no interaction directionality was calculated. Samples were clustered by calculating the Euclidean distance between vectors of log2 interaction directionality values and performing complete linkage agglomerative hierarchical clustering on the resulting distance matrix.
Histogram of contact probabilities. Contact probabilities in whole-genome normalized matrices were categorized into inter-chromosomal, intra-arm and inter-arm interactions according to the position of the bins. The contact probabilities categorized above were used to plot histograms of count distribution in normalized contact probabilities using ggplot2 in R. Contact probabilities in intra-arm normalized matrices were further divided into condensin-binding and non-binding groups according to the presence or absence of ChIP peaks, respectively (see below for detection of ChIP peak positions). Hi-C bins with ChIP peaks were identified using ChIPpeakAnno in Bioconductor45. Contact probabilities categorized as above were used to plot histograms.
Hi-C maps and Hi-C difference maps. Normalized matrices were plotted using ggplot2 in R. To prepare Hi-C difference maps, differential contact probability matrices were produced by subtraction of a normalized matrix in condition 1 (for example, interphase) from a normalized matrix in condition 2 (for example, mitosis). Then, matrix differences were plotted using ggplot2 in R. Normalized Hi-C maps with domain boundaries were prepared as below. Normalized Hi-C matrices from 5-kb bins were plotted, and the positions of the identified domain boundaries were then plotted on top using ggplot2 in R.
Calculation of fold change of inter-centromeric interactions. Bins encompassing centromere sequences and the surrounding 10 kb were identified as centromeric bins. Fold change in inter-centromeric interaction probabilities was calculated after adding up the Hi-C contact probabilities within the centromeric bins from whole-genome normalized matrices. Three biological replicates of Hi-C samples were used to generate box plots for each condition.
Contact probability as a function of distance. All the contact probabilities in intra-arm normalized matrices were classified according to the distance between the two underlying bins. The median contact probability was calculated for each distance and then plotted against distance for each condition. To calculate probability–distance quartiles, all the interaction probabilities of a bin of interest were extracted from an intra-arm normalized matrix or from the raw count matrix in the case of Supplementary Figure 5b. The bin probabilities were then ordered by their absolute distance from the bin of interest, and the cumulative probability of the bins was calculated. The 25th, 50th and 75th percentiles are the distances at which the cumulative probability first equaled or exceeded 0.25, 0.50 and 0.75, respectively. 25th-, 50th- (median) and 75th-percentile distances for the bins along chromosome II were plotted for each condition using ggplot2 in R.
4C-like plots. A bin in which a primer for 3C–qPCR analysis annealed on the right arm of chromosome II was selected as a viewpoint to generate 4C-like plots. Hi-C contact probabilities between the viewpoint and all other bins within the right arm of chromosome II were then plotted as dots in each condition. Lines of smoothed 4C-like contact probabilities, calculated using the loess-smoothing method within ggplot2 in R, were also plotted.
Determination of domain boundaries. Domain boundaries were determined using either log2 directionality or the TopDom software package (version 0.0.2)28 from the intra-arm normalized matrices for 2-kb bins. The log2 directionality values were calculated as below. A 40-kb sliding window was used to traverse the chromosome arm. For each window position, the maximum log2 value (log2 max) within the 20-kb upstream half of the window and the minimum log2 value (log2 min) within the 20-kb downstream half of the window were identified. A log2 shift value for the window was then calculated by subtracting log2 min from log2 max. A putative domain boundary was identified between the log2 min and log2 max values if log2 shift was greater or equal to 0.5. Putative boundaries were then filtered by grouping overlapping boundary domains and discarding all domains except the one with the highest log2 shift value. The domain boundary was then assigned to the midpoint between the log2 max and log2 min values for the remaining domain boundaries. To improve the reliability of domain boundary identification, only domain boundaries that were identified in the same location (±5 kb) in each of three biological replicates were classed as true boundary locations. To plot average log2 directionality at the boundaries, log2 directionality values within 40-kb upstream and downstream regions were normalized. The normalized log2 directionality values were plotted using the geom_smooth function provided by ggplot2 in R. Alternatively, TopDom was used as follows. A window size of ten bins was used to identify boundary positions. Common boundaries that were identified in three biological replicates were used for further analyses. Domain size was calculated as the distance between neighboring boundary positions.
Insulation scores. Insulation scores were generated from the intra-arm normalized matrices for 2-kb bins. Scores were calculated by taking the mean interaction probability across the two bins on either side of the bins of interest. To determine insulation score around the boundaries determined by log2 directionality, insulation scores within 50 kb upstream and downstream were normalized by log2 (insulation score/mean insulation score in the selected region). Average normalized insulation scores were calculated for all the normalized insulation scores at every 10-kb insulation distance. The average normalized insulation scores as a function of insulation distance were plotted using ggplot2 in R.
ChIP–seq data analysis.
Each read of the paired-end ChIP–seq library was trimmed to a length of 50 bases. Reads were aligned to the S. pombe genome (build ASM294v2.21) using the BWA-mem algorithm (version 0.7.12)42. ChIP peaks were identified from the alignments using version 2.1.0 of macs2 software with the input format set to BAMPE46. ChIP peaks are listed in Supplementary Table 1.
Code availability.
The computer code used to generate the results reported in this manuscript has been deposited with GitHub (see URLs).
Data availability.
The Hi-C and ChIP–seq data reported in this manuscript have been deposited with the Gene Expression Omnibus under accession GSE94478. A Life Science Reporting Summary is available.
Urls.
ParticleTracker plugin for tracking chromatin motility, https://bitbucket.org/djpbarry/particletracker; Gene Expression Omnibus, https://www.ncbi.nlm.nih.gov/geo/; computer code for analysis of Hi-C data at GitHub, https://github.com/adam-rabinowitz/Condensin-mediated-remodeling-of-the-mitotic-chromatin-landscape.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Accession codes
References
Hirano, T. Condensin-based chromosome organization from bacteria to vertebrates. Cell 164, 847–857 (2016).
Uhlmann, F. SMC complexes: from DNA to chromosomes. Nat. Rev. Mol. Cell Biol. 17, 399–412 (2016).
Belton, J.M. et al. Hi-C: a comprehensive technique to capture the conformation of genomes. Methods 58, 268–276 (2012).
Dekker, J., Marti-Renom, M.A. & Mirny, L.A. Exploring the three-dimensional organization of genomes: interpreting chromatin interaction data. Nat. Rev. Genet. 14, 390–403 (2013).
Saka, Y. et al. Fission yeast cut3 and cut14, members of a ubiquitous protein family, are required for chromosome condensation and segregation in mitosis. EMBO J. 13, 4938–4952 (1994).
Sutani, T. et al. Fission yeast condensin complex: essential roles of non-SMC subunits for condensation and Cdc2 phosphorylation of Cut3/SMC4. Genes Dev. 13, 2271–2283 (1999).
Hiraoka, Y., Toda, T. & Yanagida, M. The NDA3 gene of fission yeast encodes β-tubulin: a cold-sensitive nda3 mutation reversibly blocks spindle formation and chromosome movement in mitosis. Cell 39, 349–358 (1984).
Flemming, W. Zellsubstanz, Kern und Zelltheilung (F.C.W. Vogel, 1882).
Mizuguchi, T. et al. Cohesin-dependent globules and heterochromatin shape 3D genome architecture in S. pombe. Nature 516, 432–435 (2014).
Lieberman-Aiden, E. et al. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science 326, 289–293 (2009).
Naumova, N. et al. Organization of the mitotic chromosome. Science 342, 948–953 (2013).
Funabiki, H., Hagan, I., Uzawa, S. & Yanagida, M. Cell cycle–dependent specific positioning and clustering of centromeres and telomeres in fission yeast. J. Cell Biol. 121, 961–976 (1993).
Petrova, B. et al. Quantitative analysis of chromosome condensation in fission yeast. Mol. Cell. Biol. 33, 984–998 (2013).
Tanizawa, H. et al. Mapping of long-range associations throughout the fission yeast genome reveals global genome organization linked to transcriptional regulation. Nucleic Acids Res. 38, 8164–8177 (2010).
Nagasaka, K., Hossain, M.J., Roberti, M.J., Ellenberg, J. & Hirota, T. Sister chromatid resolution is an intrinsic part of chromosome organization in prophase. Nat. Cell Biol. 18, 692–699 (2016).
Kanke, M. et al. Auxin-inducible protein depletion system in fission yeast. BMC Cell Biol. 12, 8 (2011).
Petersen, J. & Hagan, I.M.S. S. pombe aurora kinase/survivin is required for chromosome condensation and the spindle checkpoint attachment response. Curr. Biol. 13, 590–597 (2003).
Hauf, S. et al. Aurora controls sister kinetochore mono-orientation and homolog bi-orientation in meiosis-I. EMBO J. 26, 4475–4486 (2007).
Tada, K., Susumu, H., Sakuno, T. & Watanabe, Y. Condensin association with histone H2A shapes mitotic chromosomes. Nature 474, 477–483 (2011).
Nakazawa, N., Mehrotra, R., Ebe, M. & Yanagida, M. Condensin phosphorylated by the Aurora-B-like kinase Ark1 is continuously required until telophase in a mode distinct from Top2. J. Cell Sci. 124, 1795–1807 (2011).
Oliveira, R.A., Coelho, P.A. & Sunkel, C.E. The condensin I subunit Barren/CAP-H is essential for the structural integrity of centromeric heterochromatin during mitosis. Mol. Cell. Biol. 25, 8971–8984 (2005).
Ribeiro, S.A. et al. Condensin regulates the stiffness of vertebrate centromeres. Mol. Biol. Cell 20, 2371–2380 (2009).
Gerlich, D., Hirota, T., Koch, B., Peters, J.M. & Ellenberg, J. Condensin I stabilizes chromosomes mechanically through a dynamic interaction in live cells. Curr. Biol. 16, 333–344 (2006).
Dixon, J.R. et al. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature 485, 376–380 (2012).
Crane, E. et al. Condensin-driven remodelling of X chromosome topology during dosage compensation. Nature 523, 240–244 (2015).
Sofueva, S. et al. Cohesin-mediated interactions organize chromosomal domain architecture. EMBO J. 32, 3119–3129 (2013).
Kim, K.-D., Tanizawa, H., Iwasaki, O. & Noma, K. Transcription factors mediate condensin recruitment and global chromosomal organization in fission yeast. Nat. Genet. 48, 1242–1252 (2016).
Shin, H. et al. TopDom: an efficient and deterministic method for identifying topological domains in genomes. Nucleic Acids Res. 44, e70 (2016).
Cheng, T.M. et al. A simple biophysical model emulates budding yeast chromosome condensation. eLife 4, e05565 (2015).
Fudenberg, G. et al. Formation of chromosomal domains by loop extrusion. Cell Reports 15, 2038–2049 (2016).
Ganier, O. et al. Synergic reprogramming of mammalian cells by combined exposure to mitotic Xenopus egg extracts and transcription factors. Proc. Natl. Acad. Sci. USA 108, 17331–17336 (2011).
Li, Y.C., Cheng, T.H. & Gartenberg, M.R. Establishment of transcriptional silencing in the absence of DNA replication. Science 291, 650–653 (2001).
Martins-Taylor, K., Dula, M.L. & Holmes, S.G. Heterochromatin spreading at yeast telomeres occurs in M phase. Genetics 168, 65–75 (2004).
Shintomi, K., Takahashi, T.S. & Hirano, T. Reconstitution of mitotic chromatids with a minimum set of purified factors. Nat. Cell Biol. 17, 1014–1023 (2015).
Bähler, J. et al. Heterologous modules for efficient and versatile PCR-based gene targeting in Schizosaccharomyces pombe. Yeast 14, 943–951 (1998).
Moreno, S., Klar, A. & Nurse, P. Molecular genetic analysis of fission yeast Schizosaccharomyces pombe. Methods Enzymol. 194, 795–823 (1991).
Nishimura, K., Fukagawa, T., Takisawa, H., Kakimoto, T. & Kanemaki, M. An auxin-based degron system for the rapid depletion of proteins in nonplant cells. Nat. Methods 6, 917–922 (2009).
Basi, G., Schmid, E. & Maundrell, K. TATA box mutations in the Schizosaccharomyces pombe nmt1 promoter affect transcription efficiency but not the transcription start point or thiamine repressibility. Gene 123, 131–136 (1993).
Kakui, Y. et al. Module-based construction of plasmids for chromosomal integration of the fission yeast Schizosaccharomyces pombe. Open Biol. 5, 150054 (2015).
Schindelin, J. et al. Fiji: an open-source platform for biological-image analysis. Nat. Methods 9, 676–682 (2012).
Abella, J.V. et al. Isoform diversity in the Arp2/3 complex determines actin filament dynamics. Nat. Cell Biol. 18, 76–86 (2016).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25, 1754–1760 (2009).
Imakaev, M. et al. Iterative correction of Hi-C data reveals hallmarks of chromosome organization. Nat. Methods 9, 999–1003 (2012).
Hu, M. et al. HiCNorm: removing biases in Hi-C data via Poisson regression. Bioinformatics 28, 3131–3133 (2012).
Zhu, L.J. et al. ChIPpeakAnno: a Bioconductor package to annotate ChIP–seq and ChIP–chip data. BMC Bioinformatics 11, 237 (2010).
Zhang, Y. et al. Model-based analysis of ChIP-Seq (MACS). Genome Biol. 9, R137 (2008).
Acknowledgements
We would like to thank J. Abella and M. Way for their help with high-speed microscopy, A. Stewart for bioinformatic support, C. Haering (EMBL, Heidelberg) for Slp1 shutoff and chromosomal loci–tagged strains and H. Masukata (Osaka University) for the Skp1–Tir1 strain and AID plasmids, and P. Bates, E. Wershof, B. Khatri, Y. Murayama, T. Toda and our laboratory members for discussions and critical reading of the manuscript. This work was supported by the European Research Council and the Francis Crick Institute, which receives its core funding from Cancer Research UK (FC001198), the UK Medical Research Council (FC001198) and the Wellcome Trust (FC001198). Y.K. was supported by the Japanese Society for the Promotion of Science (JSPS Overseas Research Fellowships).
Author information
Authors and Affiliations
Contributions
Y.K. and F.U. conceived the study, Y.K. performed the experiments, Y.K., A.R. and D.J.B. analyzed the data, and Y.K. and F.U. wrote the manuscript with input from A.R.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Integrated supplementary information
Supplementary Figure 1 Confirmation of mitotic arrest and condensin depletion.
(a) Chromosome morphology in mitotically arrested wild-type cells (WT; left) and following condensin depletion (cut14SO; right). DNA was stained with DAPI and visualized together with microtubules (GFP-Atb2) to confirm mitotic arrest. Dotted squares indicate the areas of the magnified images on the right. Scale bar, 1 μm. (b) Depletion of the condensin subunits Cut14 or Cnd3 in the presence of thiamine to repress gene transcription and auxin (NAA) to promote protein degradation via an auxin-inducible degron (aid)16, was confirmed by western blotting. Tat1 (α-tubulin) served as a loading control. (c) Schematic of the positions on chromosome I, where LacO repeats (green) and TetO repeats (red) were inserted and visualized by LacI-GFP and TetR-tdTomato repressor fusion proteins, respectively13. (d) Examples of images to visualize LacO and TetO localization in mitotically arrested wild-type cells and following condensin depletion. Nuclear DNA was counterstained with DAPI. Dotted squares indicate areas corresponding to the individual color images on the right. Scale bar, 1 μm. (e) Box plots of the LacO–TetO distance distributions (n ≥ 155 cells for each condition). The box shows the 25th, 50th and 75th percentiles of distance between two dots. Whiskers indicate 1.58 times the interquantile distance divided by the square roots of total cell number. Distances more than 1.5 times the 75th percentile or less than 1.5 times the 25th percentile are shown as outliers. (f) Chromosome morphology in interphase, mitosis and mitosis following chemical inhibition of Ark1/Aurora B kinase (+1NM-PP1). A Cnd3-GFP fusion visualizes condensin, DNA was stained with DAPI. Magnified individual color images of the highlighted squares are shown at the bottom. Scale bar, 1 μm.
Supplementary Figure 2 Quality control of the Hi-C data sets.
(a) Distribution of read counts within each 2-kb bin. The blue dotted line demarcates bins containing less than 500 reads that were discounted from the analysis. The red dashed line indicates the median read count. Distributions of one replicate of wild-type cells in interphase (WT Interphase Replicate 1) and in mitosis (WT Mitosis Replicate 1) are shown. (b) log2 directionality plots, with the identified boundary positions indicated (red lines), along the chromosome I left arm of the same samples shown in a. (c) Clustering of Euclidean distances between the interaction directionality plots of all the samples included in this study. (d) Smoothed mean normalized log2 directionalities at the identified boundaries. Gray areas show the 95% confidential interval. (e) Average normalized log2 insulation scores around boundaries determined by log2 directionality are shown as a function of insulation distance in interphase (left) and mitosis (right).
Supplementary Figure 3 Schematic Hi-C map, illustrating chromosomal interactions.
(a) The different types of chromosomal interactions studied in our Hi-C analyses are illustrated. Interactions within a chromosome arm (Intra-arm) are shown in red; those between the two arms of one chromosome (Inter-arm) are shown in dark blue. Interactions between different chromosomes (Inter-chr) are represented in light blue. Inter-centromeric and inter-telomeric interactions (Inter-cen and Inter-tel) are highlighted in yellow and green, respectively. (b) Schematic Hi-C map, highlighting the areas corresponding to each of the interactions shown in a, identified by the use of the same color. Note that rDNA repeats are found at the ends of chromosome III. Because of their repetitive nature, we could not analyze chromosome interactions close to the ends of chromosome III.
Supplementary Figure 4 The effect of cnd3SO and mitotic Ark1 inhibition on contact probability changes between interphase and mitosis.
(a,b) Hi-C difference maps from experiments comparing interphase to mitosis following depletion of the condensin subunit Cnd3 (cnd3SO) or mitotic Ark1 inhibition (+1NM-PP1). (c) Distribution of normalized contact probabilities between chromosomes (Inter-chr), within chromosome arms (Intra-arm) or between the two arms of the same chromosome (Inter-arm). (d) Median contact probabilities in interphase and mitosis as a function of genomic distance along the chromosome II right arm are shown, corresponding to the above comparisons.
Supplementary Figure 5 Moving intra-arm median interacting distances along chromosome II.
As in Figure 3g, the moving intra-arm median interacting distances along chromosome II (solid lines) are shown together with shaded areas representing the 25th and 75th percentiles in each indicated condition. In contrast to the iteratively normalized matrices used in Figure 3g, interacting distances were calculated from HiCNorm-normalized matrices (a) or from raw count matrices (b).
Supplementary Figure 6 Chromatin domain boundaries following Cnd3 depletion or mitotic Ark1 inhibition.
(a,b) Hi-C contact probability maps along a section of chromosome I are shown under the indicated conditions. Domain boundaries are indicated (black triangles). (c) Effect of Cnd3 depletion or mitotic Ark1 inhibition on domain size distribution.
Supplementary Figure 7 Numbers and overlap of boundary positions in interphase and mitosis.
(a) Numbers and overlap of boundary positions in interphase and mitosis. Boundary positions were determined using either log2 directionality or the TopDom algorithm. (b) Correlation between boundaries determined by log2 directionality and by the TopDom algorithm in each of the indicated conditions.
Supplementary Figure 8 Boundaries overlap with condensin peaks.
(a–c) Correlation between condensin peaks in mitosis and boundary positions in mitosis (a), in interphase (b) and in mitosis following depletion of condensin (c).
Supplementary Figure 9 Chromatin motility in interphase and mitosis.
(a) Schematic of the locations where LacO (green) or TetO (red) repeats were inserted in chromosomes I and II, respectively. (b) A typical image of a cell harboring LacO and TetO repeats bound by LacI-GFP and TetR-tdTomato, respectively. (c) Mean square displacement (MSD) of the TetO locus in wild-type interphase and mitosis. Mean ± s.e.m. is shown (n = 22 interphase, n = 26 mitosis).
Supplementary information
Supplementary Text and Figures
Supplementary Figures 1–9 and Supplementary Tables 2 and 3. (PDF 1974 kb)
Supplementary Table 1
Condensin ChIP peaks along fission yeast chromosomes I–III. (XLSX 80 kb)
Supplementary Table 4
Hi-C library sequencing metrics. (XLSX 47 kb)
Rights and permissions
About this article

Cite this article
Kakui, Y., Rabinowitz, A., Barry, D. et al. Condensin-mediated remodeling of the mitotic chromatin landscape in fission yeast. Nat Genet 49, 1553–1557 (2017). https://doi.org/10.1038/ng.3938
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ng.3938
This article is cited by
-
Orchestrating chromosome conformation capture analysis with Bioconductor
Nature Communications (2024)
-
A phase transition for chromosome transmission when cells divide
Nature (2022)
-
Guiding functions of the C-terminal domain of topoisomerase IIα advance mitotic chromosome assembly
Nature Communications (2021)
-
Potential roles of condensin in genome organization and beyond in fission yeast
Journal of Microbiology (2021)
-
Fission yeast condensin contributes to interphase chromatin organization and prevents transcription-coupled DNA damage
Genome Biology (2020)
