
Abstract
RAB, ADP-ribosylation factors (ARFs) and ARF-like (ARL) proteins belong to the Ras superfamily of small GTP-binding proteins and are essential for various membrane-associated intracellular trafficking processes1,2. None of the ∼50 known members of this family are linked to human disease. Using a bioinformatic screen for ciliary genes in combination with mutational analyses, we identified ARL6 as the gene underlying Bardet-Biedl syndrome type 3, a multisystemic disorder characterized by obesity, blindness, polydactyly, renal abnormalities and cognitive impairment3,4. We uncovered four different homozygous substitutions in ARL6 in four unrelated families affected with Bardet-Biedl syndrome, two of which disrupt a threonine residue important for GTP binding5 and function5,6,7 of several related small GTP-binding proteins. Analysis of the Caenorhabditis elegans ARL6 homolog indicates that it is specifically expressed in ciliated cells, and that, in addition to the postulated cytoplasmic functions of ARL proteins, it undergoes intraflagellar transport. These findings implicate a small GTP-binding protein in ciliary transport and the pathogenesis of a pleiotropic disorder.
Similar content being viewed by others


Main
Cilia and flagella are ancient, evolutionarily conserved eukaryotic organelles that project from cells and have been adapted by organisms to carry out diverse biological functions8. The assembly, maintenance and function of cilia and flagella depend on intraflagellar transport (IFT), and defects in this microtubule-based transport process and the function of cilia are associated with several human diseases, including Bardet-Biedl syndrome (BBS)8,9,10. Genes underlying seven of the eight loci known to be associated with BBS have been identified4,11; only the gene mutated in BBS type 3 (called BBS3), previously mapped to 3p12 (refs. 12,13), remained unidentified. BBS is thought to result largely from ciliary dysfunction, because loss-of-function mutations in C. elegans bbs-7 and bbs-8 compromise cilia structure and function14 and RNA interference of Chlamydomonas BBS5 results in the loss of flagella11. Notably, all known C. elegans bbs genes are expressed exclusively in cells with cilia, owing to the presence of a DAF-19 RFX transcription factor binding site (X box) in their promoters10,11. We hypothesized that the C. elegans ortholog of human BBS3 would also contain this regulatory element, which would allow us to identify candidates from the >90 genes that map to the BBS3 critical interval12,13,15. We generated a consensus X-box sequence from a training set of 14 C. elegans genes containing X boxes that are known to be strictly expressed in ciliated cells and used them to scan the C. elegans genome. We identified 368 genes with an X-box sequence within 1.5 kb of the start codon, 168 of which had a bona fide human ortholog (E value ≤ 10−6); three of these fell in the BBS3 critical interval (Fig. 1a). The first gene, ESRRBL1, is probably the human ortholog of C. elegans che-13. che-13 is expressed exclusively in ciliated neurons and has an important role in IFT16, and so ESRRBL1 was an excellent candidate for BBS3. The second gene encodes the hypothetical protein DKFZp761H079, a member of the ARL family of small GTP-binding proteins1,2. Its sequence is closely related to that of ARL2, and we called it ARL2–like protein 1 (ARL2L1). The third gene encodes ARL6, another ARL family member17.

(a) The BBS3 critical region on chromosome 3 is flanked by the markers D3S1302 and D3S1595 (refs. 12,13), although a recombination in one of the pedigrees suggests that the qter marker is actually D3S1753 (ref. 12). The relative positions of the three candidate genes in the BBS3 critical region are shown along with information relating to their C. elegans homologs. (b,c) Expression of arl-6 and Y37E3.5 in C. elegans. Transcriptional GFP reporters arl-6p::gfp (b) and Y37E3.5p::gfp (c) were expressed in the ciliated neurons of the worm head (e.g., amphid and inner and outer labial neurons) and worm tail (phasmids). Shown in the panels are differential interference contrast (DIC), GFP fluorescence and merged images of the whole worm, as well as higher magnification images of the head and tail regions for worms expressing arl-6p::gfp. The ciliated head and phasmid neurons of the tail are indicated by square brackets and arrowheads, respectively. The fluorescent signals along the midbody of worms expressing arl-6p::gfp is due to nonspecific autofluorescence of gut granules.
To assess the likelihood that ARL6 or ARL2L1 is BBS3, we determined the expression patterns of their C. elegans orthologs (arl-6 and predicted gene Y37E3.5, respectively). We generated transgenic lines expressing promoter–green fluorescent protein (GFP) fusion constructs (arl-6p::gfp or Y37E3.5p::gfp) and analyzed GFP fluorescence. Like the che-13 promoter, the 5′ untranslated regions (UTRs) of arl-6 and Y37E3.5 directed expression to a small subset of sensory cells that are ciliated (Fig. 1b,c). For both transgenes, we observed GFP signals in the multiple ciliated amphid neurons in the head and both ciliated phasmid neurons (PHA and PHB) in the tail (Fig. 1b,c), as previously reported for other genes associated with BBS10,11. We also detected expression in other ciliated sensory neurons, including the inner and outer labial neurons (Fig. 1b,c) and male tail ray neurons (data not shown). In the case of the Y37E3.5p::gfp transgene, we also detected GFP fluorescence in the midbody PDE ciliated neuron and PQR ciliated tail neuron.
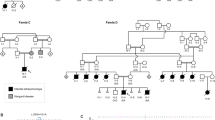
As the expression patterns of C. elegans che-13, arl-6 and Y37E3.5 are essentially identical to those of the five known C. elegans orthologs of genes associated with BBS10,11, we examined their human orthologs for mutations in families with BBS. We sequenced the complete reading frames and exon-intron boundaries of ESRRBL1 and ARL2L1 in DNA of an affected individual from a well-characterized family from Newfoundland with BBS3 (NF-B2; ref. 13) but found no pathogenic mutations. However, we did identify a homozygous missense mutation 859G → C (resulting in the amino acid substitution G169A) in ARL6 in this individual (Figs. 2 and 3a). The mutation segregated with the BBS phenotype in family NF-B2 (Fig. 2) and was absent from 100 control chromosomes. We next examined the genotypes of all available consanguineous families with respect to the eight BBS loci. The affected individual in a Saudi Arabian family (KK29) was homozygous with respect to markers across the BBS3 critical interval, and association of the disease with all other loci was excluded in this family. Sequencing and subsequent segregation in this family showed that the affected individuals, but none of the unaffected relatives, were homozygous with respect to the mutation 445C → T, resulting in the nonconservative amino acid change T31M (Figs. 2 and 3a). To investigate further whether ARL6 is BBS3 and to determine its contribution to BBS, we expanded our mutational analyses to a multiethnic cohort of 230 families with BBS and identified two more families with mutations that are probably pathogenic. In North American family AR390, of suspected consanguinity, the affected individual, but none of the unaffected relatives, was homozygous with respect to the mutation 862T → G, resulting in the nonconservative amino acid change L170W (Figs. 2 and 3a). In addition, we identified an Irish consanguineous family, PB140, who carried the homozygous mutation 445C → G affecting the Thr31 residue (resulting in the amino acid substitution T31R; Figs. 2 and 3a). None of these changes were detected in 184 ethnically matched control chromosomes.

Nucleotide positions for the above mutations are based on the ARL6 sequence from GenBank. WT, wild-type.

(a) Schematic of the gene ARL6 showing the locations of mutations found in affected individuals from four families with BBS3. Exons are shown as shaded boxes. Mutations refer to transcript that includes exon 2a but not exon 2b. (b) Sequence conservation of the nucleotide binding site (P loop) in small GTP-binding proteins. Thr31 of HsARL6 is marked by an asterisk. (c,d) Homology modeling of ARL6 based on the crystal structure of ARF6 (ref. 21) shows the putative structure of ARL6. Amino acids that hydrogen-bond with the nucleotide GTPγS (shaded orange) are shown in blue. Amino acid changes resulting from the mutations identified in the families with BBS3 are shown in red.
Mutations in ARL6 segregate with BBS in four independent families, indicating that ARL6 is BBS3. This idea is further supported by comparison of the amino acid sequences of ARL6 from divergent organisms (Supplementary Fig. 1 online). Gly169 is invariant, and the residues Thr31 and Leu170 are highly conserved: residue 31 is an amino acid with a hydroxyl side chain (threonine favored strongly over serine), and residue 170 is leucine (except for one valine) in 12 sequences (Supplementary Fig. 1 online). Of note, residue Thr31 lies in the highly conserved P loop of GTP-binding proteins (Fig. 3b). The analogous T31N mutation in ARF1 completely abrogates GTP binding in vitro5. Furthermore, overexpression of several small GTP-binding proteins mutated at the corresponding Thr31 residue produces cellular phenotypes consistent with loss of, or reduced, function. Therefore, such overexpression has been used extensively to study the functions of various ARF-ARL family members in vivo, including ARF1, ARF5, ARF6, ARL1, ARL3 and ARL7 (refs. 5–7,18–20). To determine the potential impact of the amino acid changes that we identified, we modeled ARL6 onto the structure of a homolog21, ARF6, which is 43% identical and 62% similar in amino acid sequence. All three altered residues cluster near, or are part of, the GTP binding site (Fig. 3c,d) and probably affect the GTP-binding activity and thus the function of ARL6. Taken together, these findings further indicate that the observed amino acid substitutions in ARL6 are pathogenic.
There is increasing evidence that the transmission of BBS is complex in some families, with a third mutation at a second locus acting as a modifier of either penetrance or expressivity4,22,23. We identified a family, NF-B10, in which two affected sisters are homozygous with respect to the mutation 1179T → G in BBS1 (resulting in the amino acid substitution M390R); one of them is also heterozygous with respect to the mutation 859G → C in BBS3 (Supplementary Fig. 1 online). Clinical assessment indicated that the sister with three mutations is more severely affected with respect to some clinical features of BBS (Table 1), suggesting that the additional mutation in BBS3 may act as a modifier.
RAB, ARF and ARL proteins are implicated in various aspects of biomembrane trafficking1,2, although the function of ARL6 has not been explored in much detail. Hemagglutinin-tagged ARL6 is predominantly cytosolic, and associates with membranes in vitro in the presence of the nonhydrolyzable GTP analog GTPγS17. ARL6 interacts with the β subunit of the heterotrimeric protein import channel SEC61 (ref. 17), but the physiological relevance of this finding is unclear. To shed light on the function of ARL6, we generated C. elegans transgenic lines expressing GFP-tagged ARL-6. Analysis of these lines showed that ARL-6–GFP is cytosolic, as it was found in the cell bodies, dendrites, transition zone regions (akin to basal bodies found at the base of cilia) and ciliated structures in sensory neurons (Fig. 4a). Its cell localization is therefore similar to that of GFP alone expressed in sensory neurons (Supplementary Video 1 online). To analyze whether ARL-6 may participate in dynamic, intracellular trafficking processes, we analyzed the transgenic lines by time-lapse microscopy. We observed that the GFP-tagged protein underwent IFT in the ciliaryaxoneme (Fig. 4b and Supplementary Videos 1,2,3,4,5,6 online). The movement of ARL-6–GFP occurred in both retrograde and anterograde directions along the cilium at 0.82 and 0.64 μm s−1, respectively. These rates are comparable to those of other dynein- and kinesin-associated IFT proteins8.

(a) Differential interference contrast (DIC), GFP fluorescence, and merged images of the head and tail regions of worms expressing a GFP-tagged ARL-6 fusion protein. ARL-6–GFP localized at the base of cilia in structures related to basal bodies (transition zones; Tz) and along the ciliary axonemes (Cil). Signals are also observed along the dendrites (Den) and axons (not shown) and within the main cell body (Cb). The cell bodies are shown only for the ciliated tail neurons. (b) Kymographs showing ARL-6–GFP particle movement. Scale bar is shown below each panel, and three time-frames (every 1,000 ms) show the movement of one particle (arrowheads) in the anterograde direction, away from the transition zones (intense staining on the left in each panel).
Our finding that BBS3 encodes a protein, ARL6, that is expressed specifically in ciliated cells and undergoes IFT in C. elegans has several important implications. ARL6 is the first BBS-associated protein to be identified whose evolutionarily conserved sequence immediately suggests a cellular function, namely that of a small GTP-binding protein involved in membrane-associated intracellular trafficking processes1. ARL6 is also the first known member of the RAB-SAR-ARF-ARL family of small GTP-binding proteins to be associated with an inherited human disease. The link between ARL6 and IFT extends the roles of this diverse protein family to include trafficking not only in the cytosol proper, but also in the ciliary axoneme. Given that BBS4 assists the targeting of a centrosomal protein, PCM1, in a dynein-dependent manner24, and that other C. elegans BBS-associated proteins undergo IFT14, we propose that the BBS-associated proteins generally share a common function related to intracellular trafficking processes, such as the transport of cellular components (e.g., vesicle-associated or other proteins) to the centrosome, to the basal body and in the ciliary apparatus. Such a shared cellular function for BBS-associated proteins is expected, as the phenotypes associated with mutations at different BBS loci are indistinguishable4. Other small GTP-binding proteins, such as ARL2L1 identified in this study, may have similar cilia-specific functions. Drosophila melanogaster ARL3 and ARL6 are expressed specifically in chemo- and mechanosensory ciliated cells25, and Leishmania ARL3 is required for flagellar integrity6. Similarly, mammalian ARL3 localizes to the photoreceptor connecting cilium in retinal rods and cones and, as such, could be implicated in retinitis pigmentosa26. Several RAB GTPases have also been detected in the flagella of the green alga Volvox carteri27. It will therefore be of particular interest to focus on the small GTP-binding protein family to determine how different members facilitate microtubule-based intracellular transport, and cilia- and flagella-related processes, especially in relation to human disorders that have a ciliary or neuronal component.
Methods
Identification of BBS3 candidate genes.
We created a profile Hidden Markov Model (HMM) using HMMER v1.8.4 (ref. 28) to identify C. elegans genes containing X-box regulatory sites in their upstream promoter regions. We constructed the profile HMM from a training set of 14 X box–containing genes whose expression is restricted to ciliated neurons: Y105E8A.5, F20D12.3, Y75B8A.12, T25F10.5, Y41G9A.1, T27B1.1, R31.3, F02D8.3, F38G1.1, F59C6.7, F33H1.1, K08D12.2, Y110A7A.20 and F40F9.1a. We used this profile HMM to scan the complete C. elegans genome (wormbase version WS110) and identified 368 genes with a consensus X-box sequence (raw score >14.0) present within 1.5 kb of the start codon. We downloaded from Ensembl a single FASTA format file containing the peptide sequences of all the C. elegans X box–containing genes that we identified using EnsMart. We downloaded a set of unique Homo sapiens clusters from build 119 of UniGene from National Center for Biotechnology Information. Using Stand-Alone BLAST from National Center for Biotechnology Information, we compared the C. elegans protein sequences with the H. sapiens UniGene clusters by TBLASTN with an E-value cutoff set at 1 × 10−6. For each C. elegans protein, we collected the top H. sapiens hit from the BLAST output and sorted these according to chromosome number and position. Three H. sapiens UniGene clusters fell within the BBS3 critical interval (Fig. 1a).
GFP fluorescence microscopy of C. elegans transcriptional and translational constructs.
To produce transcriptional gfp constructs for the candidate bbs-3 genes (arl-6 and Y37E3.5), we used fusion PCR to introduce the 5′ UTR sequence of each gene upstream of the full gfp coding sequence (including a nuclear localization signal sequence) and the unc-54 3′ UTR10. The arl-6p::gfp construct contained 1,197 bp of the 5′ UTR (and the first 14 bp of exon 1) and the Y37E3.5p::gfp construct contained 1,160 bp of the 5′ UTR (and the first 14 bp of exon 1). We created translational gfp constructs for bbs-3 (arl-6p::gfp) by fusing the entire exonic and intronic sequences of arl-6 (including 1,197 bp of its 5′ UTR) upstream of gfp (without a nuclear localization sequence) and the unc-54 3′ UTR. These GFP transgenes were expressed as extrachromosomal arrays in dpy-5(e907);Ex[dpy-5(+)] worms, generated as described previously10. For live imaging, we immobilized worms (using 15 mM levamisole), mounted them on agarose pads and visualized them on a Zeiss Axioskop 2+ compound fluorescent microscope. Images and videos were captured using Northern Eclipse version 6.0 software.
Subjects.
Individuals were diagnosed with BBS if they satisfied established criteria3. We assembled a large multiethnic cohort, comprising ∼300 families with BBS. We obtained blood samples with consent, in accordance with protocols approved by the appropriate human subjects ethics committees at each participating institution, and extracted DNA using standard methods.
Mutational analysis of BBS3 candidate genes.
We aligned the sequences of ARL6, ESRRBL1 and ARL2L1 with the corresponding human genome sequences and determined the exon-intron boundaries. We identified sequences flanking all coding exons and used them to design primers (sequences available on request) that amplify the exons and exon-intron boundaries of each gene. PCR amplification products were purified, sequenced and analyzed as described23.
Protein sequence analysis and homology modeling.
We obtained ARL6 protein sequences from GenBank and aligned them using the default settings with ClustalX. H. sapiens ARF6 was manually aligned to ARL6 based on a previous report2. Homology modeling of HsARL6 was done using Swiss-Model with HsARF6 as a template. The resulting ARL6 three-dimensional model was manipulated and rendered in PyMOL.
URLs.
Online Mendelian Inheritance in Man is available at http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?db=OMIM. Wormbase is available at http://www.wormbase.org/. Ensembl is available at http://www.ensemblorg/. Swiss-Model is available at http://www.expasy.org/swissmod/SWISS-MODEL.html. Protein Data Bank is available at http://www.rcsb.org/pdb/. The National Center for Biotechnology Information is available at http://www.ncbi.nlm.nih.gov/. Hidden Markov Model is available at http://hmmer.wustl.edu/. PyMOL is available at http://www.pymol.org.
Accession numbers.
GenBank: ESRRBL1, NM_018010; ARL2L1 (encoding hypothetical protein DKFZp761H079), NM_182896; ARL6, NM_032146. Protein Data Bank: HsARF6, 1HFV.
Note: Supplementary information is available on the Nature Genetics website.
References
Takai, Y., Sasaki, T. & Matozaki, T. Small GTP-binding proteins. Physiol. Rev. 81, 153–208 (2001).
Pasqualato, S., Renault, L. & Cherfils, J. Arf, Arl, Arp and Sar proteins: a family of GTP-binding proteins with a structural device for 'front-back' communication. EMBO Rep. 3, 1035–1041 (2002).
Beales, P.L., Elcioglu, N., Woolf, A.S., Parker, D. & Flinter, F.A. New criteria for improved diagnosis of Bardet-Biedl syndrome: results of a population survey. J. Med. Genet. 36, 437–446 (1999).
Katsanis, N. The oligogenic properties of Bardet-Biedl syndrome. Hum. Mol. Genet. 13, R65–R71 (2004).
Dascher, C. & Balch, W.E. Dominant inhibitory mutants of ARF1 block endoplasmic reticulum to Golgi transport and trigger disassembly of the Golgi apparatus. J. Biol. Chem. 269, 1437–1448 (1994).
Cuvillier, A. et al. LdARL-3A, a Leishmania promastigote-specific ADP-ribosylation factor-like protein, is essential for flagellum integrity. J. Cell Sci. 113, 2065–2074 (2000).
Engel, T. et al. ADP-ribosylation factor (ARF)-like 7 (ARL7) is induced by cholesterol loading and participates in apolipoprotein AI-dependent cholesterol export. FEBS Lett. 566, 241–246 (2004).
Rosenbaum, J.L. & Witman, G.B. Intraflagellar transport. Nat. Rev. Cell Biol. 3, 13–825 (2002).
Pazour, G.L. & Rosenbaum, J.L. Intraflagellar transport and cilia-dependent diseases. Trends Cell Biol. 12, 551–555 (2002).
Ansley, S.J. et al. Basal body dysfunction is a likely cause of pleiotropic Bardet-Biedl syndrome. Nature 425, 628–633 (2003).
Li, B.J. et al. Comparative genomic identification of conserved flagellar and basal body proteins that includes a novel gene for Bardet-Biedl syndrome. Cell 117, 541–552 (2004).
Sheffield, V.C. et al. Identification of a Bardet-Biedl syndrome locus on chromosome 3 and evaluation of an efficient approach to homozygosity mapping. Hum. Mol. Genet. 3, 1331–1335 (1994).
Young, T.L. et al. Canadian Bardet-Biedl syndrome family reduces the critical region of BBS3 (3p) and presents with a variable phenotype. Am. J. Med. Genet. 78, 461–467 (1998).
Blacque, O.E. et al. Loss of C. elegans BBS-7 and BBS-8 protein function results in cilia defects and compromised intraflagellar transport. Genes Dev. 18, 1630–1642 (2004).
Ghadami, M. et al. Bardet-Biedl syndrome type 3 in an Iranian family: clinical study and confirmation of disease localization. Am. J. Med. Genet. 94, 433–437 (2000).
Haycraft, C.J., Schafer, J.C., Zhang, Q., Taulman, P.D. & Yoder, B.K. Identification of CHE-13, a novel intraflagellar transport protein required for cilia formation. Exp. Cell Res. 284, 251–263 (2003).
Ingley, E. et al. A novel ADP-ribosylation like factor (ARL-6), interacts with the protein-conducting channel SEC61beta subunit. FEBS Lett. 459, 69–74 (1999).
Hickson, G.R. et al. Arfophilins are dual Arf/Rab 11 binding proteins that regulate recycling endosome distribution and are related to Drosophila nuclear fallout. Mol. Biol. Cell 14, 2908–2920 (2003).
Huang, C.F., Liu, Y.W., Tung, L., Lin, C.H. & Lee, F.J. Role for Arf3p in development of polarity, but not endocytosis, in Saccharomyces cerevisiae. Mol. Biol. Cell 14, 3834–3847 (2003).
Lu, L., Horstmann, H., Ng, C. & Hong, W.J. Regulation of Golgi structure and function by ARF-like protein 1 (Arl1). J. Cell Sci. 114, 4543–4555 (2001).
Pasqualato, S., Menetrey, J., Franco, M. & Cherfils, J. The structural GDP/GTP cycle of human Arf6. EMBO Rep. 2, 234–238 (2001).
Katsanis, N. et al. Triallelic inheritance in Bardet-Biedl Syndrome, a Mendelian recessive disorder. Science 293, 2256–2259 (2001).
Badano, J.L. et al. Heterozygous mutations in BBS1, BBS2 and BBS6 have a potential epistatic effect on Bardet-Biedl patients with two mutations at a second BBS locus. Hum. Mol. Genet. 12, 1651–1659 (2003).
Kim, J.C. et al. The Bardet-Biedl protein BBS4 targets cargo to the pericentriolar region and is required for microtubule anchoring and cell cycle progression. Nat. Genet. 36, 462–470 (2004).
Avidor-Reiss, T. et al. Decoding cilia function: defining specialized genes required for compartmentalized cilia biogenesis. Cell 117, 527–539 (2004).
Grayson, C. et al. Localization in the human retina of the X-linked retinitis pigmentosa protein RP2, its homologue cofactor C and the RP2 interacting protein Arl3. Hum. Mol. Genet. 11, 3065–3074 (2002).
Huber, H. et al. Small G proteins of two green algae are localized to exocytic compartments and to flagella. Plant Mol. Biol. 31, 279–293 (1996).
Eddy, S.R. Profile hidden Markov models. Bioinformatics 14, 755–763 (1998).
Acknowledgements
We thank the families who participated in this study. Financial support was provided by grants from the Canadian Institutes of Health Research (W.S.D.), the Heart and Stroke Foundation of B.C. & Yukon and National Cancer Institute of Canada (M.R.L.), the Janeway Children's Hospital Foundation and Memorial University Opportunities Fund (P.S.P.), Genome B.C. and Canada (D.L.B.), and in part by the National Institute of Child Health and Development, the National Institutes of Health and the March of Dimes (N.K.). M.R.L. is the recipient of Michael Smith Foundation for Health Research and Canadian Institutes of Health Research scholar awards. P.S.P. holds a Canadian Institutes of Health Research-RPP Distinguished Scientist Award. P.L.B. is a Wellcome Trust Senior Research Fellow. M.A.E. holds scholarships from Heart and Stroke Foundation Canada and Canadian Institutes of Health Research. H.M.-S. is supported by a Medical Research Council (UK) Cooperative studentship. O.E.B. is supported by fellowships from Canadian Institutes of Health Research and Michael Smith Foundation for Health Research.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Supplementary Fig. 1
Conservation of mutated residues in ARL6 proteins and pedigree analyses. (PDF 2 kb)
Supplementary Video 1
GFP-tagged ARL-6/BBS-3 undergoes anterograde and retrograde transport along ciliary axonemes. Shown in Videos 1–3 are one set of amphid cilia in the head (Video 1), one set of phasmid cilia in the tail (Video 2) and the ASER amphid cilium (Video 3) of worms expressing either a translational bbs-3:gfp transgene (Videos 1 and 2) or a transcriptional gcy-5p:gfp transgene (Video 3). GFP-tagged BBS-3 accumulates at the base of cilia (transition zones) and undergoes anterograde and retrograde transport along the ciliary axonemes (Videos 1 and 2). In contrast, the control worms (Video 3) show that although GFP alone localizes to the ciliary axoneme and accumulates at the transition zone of the ASER neuron, it does not undergo detectable transport (anterograde or retrograde) along the ciliary axoneme. Note that the ciliary axonemes extend upwards from the transition zones in Videos 1 and 3, and extend downwards from the transition zones in Video 2. Note also that Videos 1–3 are shown in real-time (i.e., 2 frames per second, where each frame was captured at 500 msec exposure). For further visual clarity of the anterograde and retrograde movement of GFP-tagged BBS-3 along the ciliary axonemes, we also show Videos 1–3 at a 3× real-time rate (Videos 4–6; note that Videos 4, 5 and 6 correspond to Videos 1, 2 and 3, respectively). (AVI 376 kb)
Rights and permissions
About this article
Cite this article
Fan, Y., Esmail, M., Ansley, S. et al. Mutations in a member of the Ras superfamily of small GTP-binding proteins causes Bardet-Biedl syndrome. Nat Genet 36, 989–993 (2004). https://doi.org/10.1038/ng1414
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ng1414
This article is cited by
-
Genetic kidney diseases: Caenorhabditis elegans as model system
Cell and Tissue Research (2017)
-
Small GTPase Arl6 controls RH30 rhabdomyosarcoma cell growth through ciliogenesis and Hedgehog signaling
Cell & Bioscience (2016)
-
A paneukaryotic genomic analysis of the small GTPase RABL2 underscores the significance of recurrent gene loss in eukaryote evolution
Biology Direct (2016)
-
Anatomical and ultrastructural study of PRAF2 expression in the mouse central nervous system
Brain Structure and Function (2016)
