
Abstract
Immune cells and glia interact with neurons to alter pain sensitivity and to mediate the transition from acute to chronic pain. In response to injury, resident immune cells are activated and blood-borne immune cells are recruited to the site of injury. Immune cells not only contribute to immune protection but also initiate the sensitization of peripheral nociceptors. Through the synthesis and release of inflammatory mediators and interactions with neurotransmitters and their receptors, the immune cells, glia and neurons form an integrated network that coordinates immune responses and modulates the excitability of pain pathways. The immune system also reduces sensitization by producing immune-derived analgesic and anti-inflammatory or proresolution agents. A greater understanding of the role of the immune system in pain processing and modulation reveals potential targets for analgesic drug development and new therapeutic opportunities for managing chronic pain.
Similar content being viewed by others

Main
Injury to tissue and nerves initiates an inflammatory response that is intended to contain pathogens, clear damaged tissues and promote repair. As one of the five cardinal signs of inflammation, pain (dolor) is initially protective and beneficial to recuperation. However, under certain conditions, the pain lingers and becomes chronic even after the injury has healed. Chronic pain affects millions of people and is difficult to treat. The mechanisms by which chronic pain emerges after acute injury remain unclear.
Although pain is processed in the nervous system, the immune system, astrocytes and microglia also contribute to chronic pain hypersensitivity1,2,3,4,5,6. An emerging concept is that the immune cells, glia and neurons form an integrated network in which activation of an immune response modulates the excitability of pain pathways. In a manner analogous to neurons, immune cells and glia show dynamic, activity-dependent plasticity and contribute to neuronal hyperexcitability in pain transmission pathways. Once activated by injury, immune and immune-related cells such as keratinocytes and vascular endothelial cells also synthesize and secrete anti-inflammatory cytokines, proresolution lipid mediators and opioid peptides to suppress pain7,8.
Inflammation and peripheral nociceptor sensitization
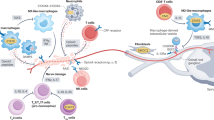
Upon injury, inflammation is triggered by innate immune activation of pattern-recognition receptors including Toll-like receptors (TLRs) that recognize and bind invading pathogens or endogenous molecules released from damaged cells, such as heat shock proteins and high mobility group box 1 protein9,10 (Fig. 1). TLRs are expressed in immune cells, including monocytes or macrophages and dendritic cells, and in immune-related cells such as keratinocytes. Binding to TLRs is followed by activation of nuclear factor-κB (NF-κB) signaling and the release of inflammatory cytokines. Resident immune cells, mast cells and macrophages are also activated within minutes of injury and release proinflammatory cytokines, chemokines, effectors of the complement cascade (C3a and C5a) and vasodilators, including vasoactive amines and bradykinin. Blood-borne neutrophils, monocytes and T lymphocytes adhere to the vessel walls, extravasate and accumulate at the site of injury. These immune cells contribute to peripheral nociceptive sensitization by releasing soluble factors and interacting directly with nociceptors.

Injury initiates the release of mediators that activate TLRs on keratinocytes (top) and mast cells (MC) close to the nerve terminal. Vasodilators are also released, promoting adhesion and transmigration of immune cells including T cells (T), neutrophils (N) and monocytes (MN), and recruitment of macrophages (Mφ). These cells, once activated, release a battery of inflammatory mediators that act on receptors expressed on adjacent nociceptor nerve terminals, leading to peripheral nociceptor sensitization. Targets include cytokine receptors (CytR), G protein–coupled receptors (GPCR), ligand-gated channels (LGC) and tyrosine kinase receptor type 1 (TrkA). Three examples of interactions between immune cells and nerve terminals are depicted. (1) Mast cell degranulation requires direct contact between mast cells and nerve terminals, mediated by N-cadherin (N-cad). The metalloproteinase MMP-24 prevents mast cell degranulation by digesting N-cad. (2) Release of TNF-α and IL-15 by peripheral nerves and Schwann cells activates MMP-9 and facilitates recruitment of macrophages. (3) Nociceptive nerve terminals can secrete substance P (SP) and CGRP through antidromic activation of neighboring nerve terminal branches (see text). Substance P and CGRP promote vasodilation and extravasation of immune cells. Neutral endopeptidase (NEP) restrains neuroinflammation by degrading substance P and CGRP.
Interactions between immune cells and nociceptors. Mast cells are granulated resident immune cells that are divided into mucosal and connective tissue subtypes and are found close to capillaries. They participate in innate host defense and allergic reactions and are degranulated within minutes of an inflammatory reaction, resulting in the release of histamine, bradykinin and other mediators that contribute to vasodilation11.
Recent work by Folgueras et al.12 suggests that degranulation of mast cells requires direct interaction between mast cells and peripheral nerve terminals, which is mediated by the calcium-dependent cell adhesion molecule N-cadherin (Fig. 1). N-cadherin is expressed in both mast cells and primary sensory neurons and is cleaved by metalloproteinase MT5-MMP (MMP-24), which is expressed by neurons13. Expression of N-cadherin is increased in MT5-MMP–deficient mice, and this gives rise to increased interactions between mast cells and nerve terminals, enhanced mast cell degranulation and increased thermal pain sensitivity (thermal hyperalgesia). Interestingly, MT5-MMP mutant mice do not develop inflammatory thermal hyperalgesia. This may be due in part to preemptive degranulation of mast cells in the absence of suppression of mast cell–nerve terminal interactions by MT5-MMP12.
Mast cells are found close to primary nociceptive neurons and contribute to nociceptor sensitization in a number of contexts. Injection of the secretagogue compound 48/80 promotes degranulation of mast cells in the dura and leads to excitation of meningeal nociceptors14. Mast cell degranulation also contributes to the rapid onset of nerve growth factor–induced thermal hyperalgesia15. Pelvic pain associated with neurogenic cystitis was eliminated in mice lacking mast cells16. Although these findings indicate that resident mast cells sensitize peripheral nociceptors, it is unclear which chemical mediator(s) derived from mast cells are essential for this effect. Histamine has an important role in mediating mast cell-induced nociceptor activation16,17, but surprisingly, tumor necrosis factor-α (TNF-α) seems not to be required for mast cell–dependent pelvic pain16.
Macrophages are derived from circulating monocytes and are maintained by the local proliferation and maturation of blood monocytes after diapedesis. Migrating blood monocytes are recruited to the site of injury and mature within hours to increase the proportion of macrophages in the inflamed area within days to weeks. Resident macrophages become phagocytic almost immediately after injury.
The number of macrophages is increased at the site of nerve injury (as indicated by increased staining for ED1 (the rat homolog of human CD68))18,19 and correlates with the development of mechanical allodynia, pain induced by a normally non-noxious stimulus, after nerve injury18. The recruitment of macrophages after nerve injury is mediated by several inflammatory cytokines. TNF-α, which is released from Schwann cells immediately after nerve injury, induces MMP-9. In turn, MMP-9 promotes migration of macrophages to the injured site via breakdown of the blood-brain barrier20,21 (Fig. 1). Interleukin 15 (IL-15), which acts on B cells and promotes T cell proliferation, is upregulated in nerves a few hours after injury. Intraneural injection of IL-15 into the sciatic nerve induces the infiltration of macrophages and T cells into the nerve, an effect that is blocked by antibodies to IL-15 and by the ganglioside 9-O-Ac GD1b (neurostatin), an IL-15 modulator that binds IL-15 with high affinity22. Like TNF-α, IL-15 can activate MMP-9 (ref. 23) (Fig. 1). Consistent with their pro-nociceptive roles, TNF-α induces peripheral nociceptor sensitization24, and intraplantar injection of IL-15 induces mechanical hyperalgesia25.
Following recruitment and activation, macrophages contribute to nociceptor sensitization by releasing several soluble mediators. Expression of the chemokine macrophage inflammatory protein-1α (MIP-1α) and its receptors CCR1 and CCR5 is increased in macrophages and Schwann cells after partial ligation of the sciatic nerve and contributes to the development of neuropathic pain26.
Depletion of circulating monocytes and macrophages by liposome-encapsulated clodronate partially reduces thermal and mechanical hyperalgesia19,27 without altering mechanical allodynia28 in models of neuropathic pain. Although this suggests that macrophages may have only a minor role in neuropathic pain, caution is warranted in interpreting these results, as clodronate does not effectively deplete resident macrophages29. Resident macrophages have an important role in polymorphonuclear leukocyte infiltration and acute inflammation, as demonstrated by conditional macrophage ablation30. This strategy could be used to clarify the role of resident macrophages in chronic pain.
Neutrophils are the most abundant polymorphonuclear leukocytes. Neutrophil migration is associated with inflammatory pain31,32. Within the first hour of the onset of inflammation, neutrophils migrate through the vascular endothelium and accumulate at the site of injury. Nerve terminals influence neutrophil recruitment through neurogenic inflammation, which is also called sterile inflammation because no pathogens are involved. During neurogenic inflammation, primary afferent neurons generate impulses that spread through neighboring nerve terminals, resulting in the release of the vasoactive neuropeptides substance P and calcitonin gene-related peptide (CGRP) at peripheral branches (Fig. 1). IL-1 can also bind nerve terminals33 and induce substance P release and migration of polymorphonuclear leukocytes7,33. Notably, mast cell degranulation is also facilitated by substance P and CGRP34. Synergistic neuroimmune interactions, in which multiple soluble mediators can amplify a response and increase the recruitment of cells, facilitate sensitization and the emergence of a chronic pain state. By knocking out neutral endopeptidase, a key enzyme that controls neurogenic inflammation, both neurogenic inflammation and neuropathic pain behavior can be enhanced in mice35 (Fig. 1).
Lymphocytes contribute to the sensitization of peripheral nociceptors, but the data regarding their contribution are less conclusive than for other immune cells. T cells infiltrate the sciatic nerve and dorsal root ganglion (DRG) after nerve injury18,36. Hyperalgesia and allodynia induced by nerve injury are markedly attenuated or abrogated in rodents lacking T cells37,38,39 and the immunosuppressant rapamycin attenuates neuropathic pain in rats, partly owing to an effect on T cells40. Among the subsets of T cells, type 1 and 2 helper T cells (TH1 and TH2 cells) have been shown to have different roles in neuropathic pain. TH1 cells facilitate neuropathic pain behavior by releasing proinflammatory cytokines (IL-2 and interferon-γ (IFNγ)), whereas TH2 cells inhibit it by releasing anti-inflammatory cytokines (IL-4, IL-10 and IL-13)37. It is noteworthy that the concentration of IL-17 in the spinal cord of rats is increased after nerve injury38. Although natural killer (NK) cells are recruited to the injured sciatic nerve in rats, they do not seem to be involved in neuropathic pain because there is no difference in the number of NK cells between allodynic and nonallodynic rats18. B cells also show no change in people with chronic pain41,42 and do not seem to contribute to the development of neuropathic pain in mice38,39.
The complement system is an important part of the innate defense43. Effectors of the complement cascade attack microbes, activate mast cells and basophils, and promote chemotaxis of leukocytes. These proteins are normally present in the blood but can leak out to inflamed tissue.
The complement system also has a role in inflammatory hyperalgesia and neuropathic pain31,44,45,46. C5a, an anaphylatoxin, is an important effector of the complement cascade and upon binding to C5aR1 receptors on neutrophils it becomes a potent neutrophil attractant. Injection of C5a and C3a into the hindpaw of rats or mice induces behavioral hyperalgesia31,46,47, whereas PMX53, a C5a receptor antagonist, suppresses it31,48. Zymosan-induced recruitment of neutrophils is inhibited by PMX53 and C5a-induced hyperalgesia is reduced in neutrophil-depleted rats31.
Complement components also have a direct effect on nociceptors. Application of C5a or C3a to peripheral nerves ex vivo sensitizes C fiber nociceptors46. This effect might be mediated by a direct effect of binding C5a receptors, as C5a receptor mRNA is expressed in primary sensory neurons46. Although these observations suggest that complement proteins have multiple parallel effects on immune cells and nociceptors, one plausible scenario is that activation of C nociceptors by the complement fragment leads to neurogenic inflammation, which facilitates neutrophil migration and hyperalgesia (Fig. 1). C5a is also involved in neuropathic pain, as it activates spinal microglia in neuropathic pain49 and blockade of the complement cascade in the spinal cord reverses neuropathic pain behavior50. Although C5a has a role in pain hypersensitivity, the formation of the membrane attack complex, another end product of the complement cascade for cell lysis, does not seem to contribute to neuropathic pain44,49.
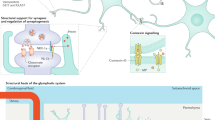
Interactions in sensory ganglia. Peripheral nerve fibers and their cell bodies in the DRG and trigeminal ganglion relay injury-related primary afferent input to the spinal and medullary dorsal horn. The cell bodies of DRG and trigeminal ganglion neurons are surrounded by small satellite glial cells (SGCs). Like astrocytes in the CNS, SGCs are connected by gap junctions and support DRG neurons by supplying nutrients and buffering extracellular ion and neurotransmitter levels. An estimated 15,000 major histocompatibility complex II–positive cells, probably macrophages, are found in each of the lumbar DRGs36 and provide immune protection. Blood-derived macrophages and T cells invade the DRG after nerve injury36,51. Macrophages then gradually move through satellite cells and migrate closer to the neuronal soma (Fig. 2). These macrophages eventually form perineuronal rings under the satellite cells around medium-to-large neurons after constriction of the sciatic nerve36,51. Close opposition of satellite cells and neurons favors interactions through paracrine signaling, an important mechanism in the DRG that underlies peripheral sensitization6.

(a) Nerve injury reduces Kir4.1 expression in SGCs, resulting in reduced K+ buffering and increased neuronal excitability. (b) A reciprocal paracrine signaling loop involving NO, COX, PGE2, CGRP and IL-1β. Macrophages infiltrate into the space between SGCs and neurons and secrete inflammatory mediators. (c) Chemokine-mediated regulation of neuronal TRP channels through paracrine (Schwann cell–derived CCL3 and neuronal CCR1) and autocrine (neuron-derived CCL2 and neuronal CCR2) signaling. (d) P2X7R in SGC tonically inhibits P2X3R in neurons by activating neuronal P2Y1.
New evidence has emerged on how these interactions promote the transition to a chronic pain state. Satellite cells in the DRG show increased gap junction coupling after injection of complete Freund's adjuvant (CFA) into the hindpaw, an effect that parallels a reduction in the pain threshold52,53. In the trigeminal ganglion, injection of the retrograde tracer True Blue into the temporomandibular joint capsule leads to accumulation of the dye in SGCs after injection of capsaicin into the temporomandibular joint54. Importantly, there is no gap junction coupling between DRG neurons or between neurons and SGCs in the absence of noxious stimulation53. Thus, increased communication between SGCs and between neurons and SGCs after peripheral noxious stimulation increases neuronal excitability and enhances primary afferent input. Increased communication can also spread into neighboring neurons and SGCs. Thalokoti et al.55 showed that activation of the sensory neurons that innervate the mandibular territory led to pain-related cellular changes not only in neurons and SGCs of the mandibular division but also in the maxillary and ophthalmic divisions of the trigeminal ganglion. This cross-excitation within the sensory ganglion provides a mechanism for extraterritorial pain that occurs outside the injured dermatome54.
SGCs can also affect neuronal excitability through reduced potassium buffering (Fig. 2a). Extracellular potassium homeostasis influences neuronal excitability. When extracellular potassium is increased, the activation threshold is lowered and neuronal excitability increases. SGCs, but not neurons, in trigeminal ganglia express the inward rectifying K+ channel Kir4.1, which has a key role in buffering K+ concentration in the ganglion56. Ten days after injury of the infraorbital nerve—the time at which neuropathic pain behavior develops—Kir4.1 expression is reduced by 40% in the trigeminal ganglion57. Silencing of Kir4.1 in the trigeminal ganglion by small interfering RNA (siRNA) is also sufficient to induce mechanical hypersensitivity in the corresponding peripheral field57.
A reciprocal paracrine signaling loop between neurons and SGCs in the trigeminal ganglion also contributes to sensitization of nociceptors58 (Fig. 2b). Release of CGRP by neurons induces the production of IL-1β in SGCs. IL-1β, but not IFN-γ or TNF-α, increases the production of prostaglandin E2 (PGE2) by activating the cyclooxygenase-2 (COX2) pathway in SGCs. Nitric oxide (NO) produced in trigeminal ganglion neurons also induces PGE2 production, probably by activating COX1. PGE2 in turn stimulates the production of CGRP in trigeminal ganglion neurons, completing the positive feedback loop (Fig. 2). Although this feedback loop can increase sensitization of nociceptors6, further study is needed to confirm whether this increases pain hypersensitivity in vivo.
The accumulation of pro- and anti-inflammatory cytokines and chemokines in the DRG after injury also contributes to the sensitization of sensory neurons. TNF-α, IL-1β, IL-10 and several chemokines are upregulated in the DRG shortly after injury45,59. TNF-α is also transported retrogradely to the DRG60. The chemokine monocyte chemoattractant protein-1 (MCP-1, or CCL2) and its receptor CCR2 are also upregulated in DRG neurons in models of neuropathic pain26,61,62. These cytokines and chemokines act on their respective receptors on DRG neurons and, by coupling to transient receptor potential (TRP) and sodium channels, generate ectopic discharges and enhance primary afferent input to the spinal dorsal horn (reviewed in ref. 6) (Fig. 2c).
Inhibitory effect of immune cells on pain. Upon injury, the immune system also releases factors that promote tissue recovery, suppress inflammation and reduce pain. Leukocytes and keratinocytes release opioids, mainly β-endorphin, after injury7,63,64. Recent studies indicate that endomorphins are expressed in T cells, macrophages and fibroblasts from the synovial tissue of patients with osteroarthritis and rheumatoid arthritis65. The inflammation-evoked release of chemokines such as CXCL1 and CXCL2 not only facilitates leukocyte recruitment but also induces the release of opioid peptides from migrating leukocytes7. Activation of the endothelin-B receptor can also trigger the release of β-endorphin from keratinocytes63.
The peripheral purinergic system negatively regulates immune responses and pain. Purinergic receptors consist of G protein–coupled P2Y and P1 receptors and the ligand-gated P2X family. The P1 receptor, which is activated by adenosine, facilitates analgesia at the spinal level66. P2Y receptors are activated by ATP, ADP and UTP, whereas P2X receptors are activated by ATP alone. When it binds P2Y receptors, ATPγS, a slowly hydrolyzing ATP analog, inhibits secretion of TNF-α and CCL2 and increases release of IL-10 after TLR activation in human monocytes67. In the DRG, P2X7R in satellite cells tonically inhibits P2X3R expression in neurons by activation of neuronal P2Y1Rs via SGC-derived ATP, which prevents the development of inflammatory pain in rats68 (Fig. 2d).
Several classes of lipid mediators including lipoxins, resolvins and neuroprotectins69 are produced by neutrophils, vascular endothelial cells and other immune cells when they are activated. These lipid mediators actively promote resolution of inflammation. Lipoxins suppress inflammatory pain70. Resolvins are derived endogenously from omega-3 essential polyunsaturated fatty acids. Two series of resolvins, the D and E series, have been identified. Resolvin D1 (RvD1) inhibits IL-1β production in microglia, and RvD2 attenuates neutrophil migration to the site of inflammation by inhibiting leukocyte-endothelial interactions in vivo69,71. The analgesic properties of resolvins are not limited to their anti-inflammatory actions. Both RvE1 and RvD1 reduce hyperalgesia in the formalin, carrageenan and CFA models of inflammatory pain8. RvE1 also inhibits mechanical allodynia and potentiation of NMDA receptor currents induced by TNF-α8.
CNS responses to peripheral injury
Central glial responses to peripheral injury. Glia show increased activity in multiple pain processing pathways in response to peripheral injury72,73,74,75. Activation signals are relayed to the brain by peripheral immune activation and through afferent nerve input76,77,78, circulating cytokines79 and immune cell trafficking38,39. In several animal models of inflammatory pain, injection of formalin80,81, zymosan81,82 or carrageenan83,84 into the hindpaw induces activation of spinal glia (as assessed by increased expression of CD11b, ionized calcium-binding adapter molecule (Iba1), glial fibrillary acid protein (GFAP) or S100 calcium-binding protein B) and behavioral hyperalgesia. However, conflicting results have been obtained following injection of CFA; some groups have reported activation of spinal glia72,85, whereas others have not82,86,87. Consistent activation of glia is observed in studies on deep tissue injury of the muscle76,88, joint89, nerve trunk90 or viscera91,92, with consistent time-dependent and somatotopically relevant glial hyperactivity related to inflammatory injury and pain. It seems that glial activity is more sensitive to deep tissue injury. Skin incision, a model of postoperative pain induced by cutaneous tissue injury, produces much weaker effects on glial marker expression than spinal nerve injury93. These findings are clinically relevant because most debilitating inflammatory pain conditions involve deep tissues or organs. Glial modulators that are designed to inhibit glial activation attenuate persistent hyperalgesia94, and importantly, basal pain threshold is usually not affected by glial inhibitors83,84, suggesting that glia selectively promote sensitization after injury.
Role of afferent nerve input. Increased primary afferent input not only activates postsynaptic neurons in the spinal dorsal horn and spinal trigeminal nucleus but also can alter central glial activity. Blocking peripheral nerve conduction abolishes masseter muscle inflammation-induced upregulation of GFAP in the spinal trigeminal nucleus, suggesting that activation of central glia in response to peripheral inflammation depends on nerve input4,76. Similarly, glial activation in models of neuropathic pain is associated with primary afferent input77,78. Electrical stimulation of the rat sciatic nerve or dorsal root at a noxious intensity stimulates the release of CX3CL1 (fractalkine), increases microglial activation (as assessed by Iba1 immunoreactivity) in the spinal dorsal horn and increases pain sensitivity95,96. However, not all forms of nociceptive input upregulate glial function. Acute tissue injury produced by mustard oil irritant does not increase glial activation (as assessed by OX-42 and GFAP immunoreactivity) in the spinal cord97, which suggests that glial responses are selective to different forms of primary afferent input. With regard to inflammatory injury, sustained input from peripheral tissues, particularly those from deep muscle, joints and viscera, may be more likely to activate glia.
Peripheral immune signaling to the brain. Among the prototypical proinflammatory cytokines, IL-6 has been shown to act as a messenger in conveying peripheral immune signals to the CNS. As early as 3 h after carrageenan-induced inflammation in the rat, the blood levels of IL-6, but not of IL-1β or TNF-α, are increased. The increase in circulating IL-6 is associated with induction of COX-2 activity and PGE2 release in vascular endothelial cells of the brain (which express the IL-6 receptor)98,99. These responses are markedly attenuated by treatment with antibodies to IL-6, and neutralization of IL-6 attenuates inflammatory hyperalgesia79.
Infiltration of immune cells into the CNS facilitates the induction of chronic pain. Migration of immune cells to the CNS is selective. A subpopulation of neutrophils that express the calcium-binding proteins S100A8 and S100A9 migrate to the spinal cord after hindpaw inflammation and accumulate in the intraluminal and perivascular spaces100. CD4+ T cells infiltrate into the spinal cord, whereas NK cells and B lymphocytes are not found in the spinal cord after L5 spinal nerve transection39 (but see ref. 38). Macrophages were not detected in the spinal cord after hindpaw inflammation and L5 spinal nerve transection39,100. However, after partial sciatic nerve ligation, peripheral macrophages or monocytes invade the spinal cord and differentiate into cells with a microglial phenotype101, suggesting that blood-borne immune cells make a direct contribution to glial responses in the CNS.
It is unclear whether the permeability of the blood-brain and blood-spinal cord barriers is altered after tissue or nerve injury and whether this facilitates the migration of immune cells and inflammatory mediators into the CNS102,103,104. Infiltration of immune cells into the CNS is initiated by chemotactic signals. C5a is upregulated in spinal microglia after nerve injury49, and small inducible cytokine A2 (SCYA2), CCL2 and endothelial leukocyte adhesion molecule-1 are substantially upregulated in the choroid plexus in response to inflammation in the hindpaw105. Neutralization of CCL2 in the spinal cord abolishes the infiltration of monocytes or macrophages after nerve injury101.
Activation of neurons and glia in chronic pain
Neuron-to-glia signaling. Neurotransmitters, neuromodulators and inflammatory mediators are released from primary afferent terminals into the spinal cord61,106. CCL2 is packaged into large, dense-core vesicles in DRG neurons61, suggesting that it can be released in a manner similar to a neurotransmitter6. Upon arrival of a nerve impulse, neural and immune mediators such as glutamate, ATP, substance P, CGRP, brain-derived neurotrophic factor (BDNF), IL-6 and CCL2 are released. These act on receptors on the postsynaptic nerve terminal and on microglia and astrocytes, modulating glial activity (Fig. 3). This has been the topic of numerous recent reviews1,2,3,4,5,6,107.

(a) Microglia-neuron interactions. Upon activation, afferent nerve terminals release neurotransmitters, substance P, CGRP, glutamate (Glu), ATP and BDNF, as well as inflammatory mediators including IL-6 and CCL2 and the growth and differentiation factor neuregulin-1 (NRG-1), into the spinal cord. Three examples are shown. (1) Neuronal NRG-1 acts on microglial erbB2, leading to IL-1β release. (2) Microglial cathepsin S (catS) cleaves neuronal CX3CL1, which binds CX3CR1 and stimulates phosphorylation of p38 MAPK in microglia. This pathway may be inhibited by protein-coupled receptor kinase 2 (GRK2). (3) ATP binds P2X4 and induces BDNF release from microglia, which upon binding TrkB receptor induces a shift in the chloride anion gradient and GABAA receptor-mediated depolarization in dorsal horn neurons. (b) Astrocyte-neuron interactions. (1) Astrocytes release glutamate and D-serine, which bind extrasynaptic and synaptic NMDA receptors on neurons, respectively. (2) Injury-induced downregulation of astrocytic GLT-1 alters glutamate homeostasis in the synaptic cleft. (3) TNF-α activates the JNK1 pathway, which leads to release of CCL2 and alterations in NMDAR and AMPAR activity. (c) Cross-talk between nerve terminals, astrocytes and glia. (1) TLR priming and purinergic signaling increase IL-1β release by glia, which modulates NMDA receptor activity on postsynaptic neurons. TIMPs in astrocytes inhibit MMP-mediated cleavage of pro–IL-1β. (2) Microglial IL-18 binds IL18R on astrocytes and induces NF-κB activity and upregulation of inflammatory cytokines. Dashed lines represent multiple intermediate signaling events.
Neurons can regulate the activity of microglia through multiple cellular pathways. A recent study has described a microglia-specific signaling pathway that is mediated through neuregulin-1 (NRG-1), a growth and differentiation factor that is released from primary afferent terminals and binds to the receptor tyrosine kinase erbB2 on microglia108. This leads to activation of spinal microglia, release of proinflammatory cytokines (including IL-1β), chemotaxis and the development of pain hypersensitivity108 (Fig. 3a). Interestingly, microglial TLR4 might act as an atypical, nonstereoselective opioid receptor107. Morphine seems to bind the same domain of TLR4 as lipopolysaccharide and induces the release of proinflammatory cytokines from glia. This raises the possibility that in addition to their analgesic properties, endogenous opioids might directly stimulate microglial activity. Activation of the Janus kinase–signal transducer and activator of transcription-3 (JAK-STAT3) pathway in microglia by IL-6 has also been shown to be important for allodynia after nerve injury109. However, the effect of IL-6 on microglia is probably mediated by neurons because the IL-6 receptor is abundantly expressed on neurons98,99,110.
There is evidence that microglia are subtly involved in suppressing pain. G protein–coupled receptor kinase 2 (GRK2) is a ubiquitously expressed negative regulator of G protein–coupled receptors111. Mice in which GRK2 expression is reduced by 50% (Grk2+/−) show enhanced and prolonged hyperalgesia after carrageenan-induced hindpaw inflammation83, which suggests that GRK2 suppresses inflammatory hyperalgesia. Selective knockdown of GRK2 in microglia and macrophages (by crossing Grk2fl/+ mice with mice expressing Cre under the control of the LysM (microglia-, macrophage- and granulocyte-specific promoter) increases the duration of hyperalgesia112. These results suggest that GRK2 in microglia or macrophages controls the duration of inflammatory hyperalgesia (Fig. 3a).
The activation of astrocytes is modulated by neuronal activity after peripheral injury76. Inhibition of neuronal activity reduces GFAP expression in the spinal cord after nerve injury113. Garrison et al.114 showed that nerve injury–induced upregulation of GFAP depends on NMDA receptor activity, mediated directly by glutamatergic synaptic input115,116. In an ex vivo medullary slice preparation, application of substance P or CGRP induced a substantial increase in GFAP in the spinal trigeminal complex76, although it is unclear whether this is a direct effect of binding to astrocytes. Furthermore, in the absence of κ-opioid receptors or their endogenous ligand dynorphin, nerve injury–induced upregulation of GFAP in the spinal dorsal horn is abrogated, which suggests that opioid signaling can also modulate astrocyte activation117. Upon activation, several signaling events in astrocytes mediated by NF-κB118,119,120, c-Jun N-terminal kinase-1 (JNK1)121 and tissue inhibitors of metalloproteinases (TIMPs)106,122 contribute to the development of hyperalgesia (Fig. 3c).
Glia-cytokine-neuron interactions. Both microglia and astrocytes release substances that influence neuronal activity. Activated microglia release several mediators that act on neurons and sensitive nociceptors4,123,124,125. One example of a reciprocal interaction between microglia and neurons involves the chemokine CX3CL1 (Fig. 3a). CX3CL1 is expressed in primary sensory neurons and dorsal horn neurons95,123,124. CX3CL1 is normally anchored to the cell membrane by a mucin stalk that can be cleaved by protease activity. Upon primary afferent stimulation the lysosomal cysteine protease cathepsin S is released from microglia and cleaves CX3CL1, which is located on the surface of dorsal horn neurons. CX3CL1 in turn activates its receptor CX3CR1 on microglia, which leads to phosphorylation of p38 MAPK in microglia95,126. Another example involves purinergic signaling (Fig. 3a). ATP, which can be derived from multiple sources including nerve terminals, induces BDNF release from microglia by activating P2X4R127. BDNF from microglia binds to the TrkB receptor on neurons and induces a shift in the chloride anion gradient in dorsal horn nociceptive neurons. This increases the excitability of lamina I nociceptive neurons through GABAA receptor–mediated depolarization125 (Fig. 3a).
Given their close proximity and intimate association with neurons, astrocytes are in a unique position to interact with neurons in regulating synaptic activity. The release of glutamate at nerve terminals activates metabotropic glutamate receptors on astrocytes, which increases Ca2+ mobilization in astrocytes. This leads to the release from astrocytes of a battery of mediators, including glutamate, D-serine and ATP, which in turn modulate neuronal activity128. NMDA receptors have an important role in synaptic plasticity and persistent pain. D-serine, a co-agonist of NMDA receptors, is released from astrocytes along with glutamate. D-serine acts on synaptic NMDA receptors while astrocytic glutamate binds to extrasynaptic NMDA receptors (Fig. 3b). The astrocytic glutamate transporter (GLT-1) buffers glutamate released into synapses to prevent excessive activation of postsynaptic glutamate receptors. However, GLT-1 is downregulated after injury129, and the glutamate-glutamine shuttle between astrocytes and neurons is altered130,131. These changes in synaptic glutamate homeostasis lead to increased dorsal horn excitability and contribute to the development of persistent pain4,132,133 (Fig. 3b).
Among the many immune- or glia-derived mediators that are related to pain hypersensitivity, IL-1β is a key cytokine that modulates microglia, astrocytes and neurons76,134,135. ATP induces the release of IL-1β from microglia in spinal cord slices in a manner that requires P2X7 receptors135. ATP-induced release of IL-1β requires priming of TLRs by lipopolysaccharide, suggesting that this occurs only upon injury. IL-1β release is also mediated by CX3CL1 signaling and activation of p38 MAPK in microglia136,137. IL-1β is also selectively upregulated in astrocytes in the spinal cord, spinal trigeminal nucleus and rostral ventromedial medulla in models of cancer pain, inflammation and nerve injury74,76,106,138,139, which suggests that astrocytes can act as an alternative source of this inflammatory cytokine. After spinal nerve ligation injury, pro–IL-1β is cleaved by MMP-9 in microglia and MMP-2 in astrocytes, but not by the cysteine protease caspase-1, a key enzyme that is responsible for the production of mature IL-1β106. IL-1β is also an important messenger between glia and neurons. The IL-1 receptor colocalizes with NMDA receptors on neurons74,76. Activation of IL-1 receptors facilitates NMDA receptor phosphorylation, induces changes in synaptic strength and results in behavioral hyperalgesia74,76,133 (Fig. 3c). However, the role of IL-1β in persistent pain also involves NMDA receptor–independent mechanisms139.
TNF-α is upregulated in pain pathways after injury and secreted by immune and glial cells72,74. TNFα induces the phosphorylation of JNK1 and activates NF-κB in astrocytes, leading to CCL2 release118,121. CCL2 then acts on CCR2 receptors on neurons and interacts positively with neuronal NMDA and AMPA receptors121 (Fig. 3b). In the rostral ventromedial medulla, which is responsible for descending pain modulation, TNFα is induced after nerve injury and facilitates NMDA receptor phosphorylation74. TNFα also stimulates phosphorylation of the GluA1 subunit of the AMPA receptor and its trafficking to the membrane in dorsal horn neurons140. These findings strengthen the view that glia-derived proinflammatory cytokines interact with excitatory amino acid receptors.
IL-18, an inflammatory cytokine of the IL-1 family, acts as a messenger between microglia and astrocytes141 (Fig. 3c). After spinal nerve injury, IL-18 is upregulated in microglia, and its receptor IL-18R is upregulated selectively on astrocytes in the spinal cord. IL-18 signaling leads to activation of NF-κB in astrocytes and the development of neuropathic pain behavior in rats. IL-18 may also contribute to descending pain facilitation in the brainstem. Activation of spinal 5-HT3 receptors increases pain hypersensitivity by a mechanism that involves IL-18, microglia and astrocytes (M. Gu, R.D., K.R. and F. Wei, unpublished observations).
Therapeutic opportunities
Insight into the roles of various cell types and soluble mediators in pain has spurred the development of potential analgesic targets. However, findings from preclinical studies have yet to be translated into the clinical setting. Here we discuss some recent developments in targeting cytokines, chemokines, resolvins and glial modulators as analgesics.
Anti-inflammatory cytokines and cytokine inhibitors. Kineret (anakinra, Amgen), a recombinant human IL-1 receptor antagonist (IL-1ra), and two TNF inhibitors, Enbrel (etanercept, Pfizer and Amgen) and Remicade (infliximab, Centoco), inhibit the pain associated with rheumatoid arthritis and other inflammatory conditions142. Arcalyst (rilonacept, Regeneron) binds IL-1α and IL-1β with high affinity and has shown promise in treating inflammatory diseases143,144. The drug is a fusion protein that comprises the extracellular domain and accessory protein of the human IL-1 receptor. As an IL-1 blocker, rilonacept has advantages over monomeric soluble IL-1 receptors, which bind IL-1 with low affinity and in some cases can act as an agonist. Rilonacept suppresses hyperalgesia and inflammation in a model of arthritis that is induced by injecting monosodium urate crystals into the mouse ankle joint144. In a pilot study that involved 10 patients with chronic active gouty arthritis, rilonacept significantly reduced pain after subcutaneous injection and was well tolerated143.
IL-10, an anti-inflammatory cytokine that is secreted by monocytes and TH2 cells, reverses neuropathic pain behavior in animal studies145. To improve delivery and increase the duration of effect, a new formulation was designed comprised of plasmid DNA encoding IL-10 (pDNA–IL-10) encapsulated with microparticles of PLGA (poly(lactic-co-glycolic-acid)), a synthetic degradable polymer145. This formulation permits slow release of IL-10 and increases IL-10 production compared to unencapsulated pDNA–IL-10. In a model that involves chronic constriction of the sciatic nerve, intrathecal injection of these microparticles abrogated mechanical allodynia for more than 70 d after a single administration145. PLGA microparticles can also induce the recruitment of macrophages and stimulate phagocytic activity, so further studies are required to elucidate their precise mechanism of action.
Proresolution resolvins. The therapeutic utility of resolvins as analgesics has recently gained interest8. One advantage of these lipid mediators is that they act by both suppressing inflammation and by inhibiting the mechanisms of synaptic plasticity that are involved in the transition to chronic pain. Another desirable property of resolvins is that they do not affect the baseline pain threshold but suppress injury-induced pain in preclinical studies8. In contrast with broad anti-inflammatory agents, proresolution lipid mediators do not alter protective inflammatory responses and are less likely to increase the risk of infection.
Complement receptor antagonists. PMX53, a cyclic hexapeptide compound, is a complement C5a receptor antagonist and inhibits inflammatory hyperalgesia in preclinical studies31 without altering the baseline pain threshold. However, in a double-blind, placebo-controlled clinical study of individuals with rheumatoid arthritis, PMX53 did not reduce synovial inflammation146. Whether PMX53 attenuated pain in these patients was not determined.
Glial modulators. A wealth of preclinical studies support a role for glia in the development of chronic pain1,2,3,4,5,6,147. The most commonly used glial modulators include minocycline (a semisynthetic tetracycline), fluorocitrate (a cell metabolic toxin), propentofylline (a xanthine derivative and phosphodiesterase inhibitor), methionine sulfoximine (an astroglial glutamine synthetase inhibitor) and Mac-1-saporin (a CD11b receptor and saporin conjugate; Advanced Targeting Systems). Among these agents, minocycline is used clinically as an antibiotic and propentofylline has been used in clinical trials for Alzheimer's disease. The potential analgesic efficacy of these glial modulators in patients has not been established. AV411 (ibudilast, Avigen/MediciNova), a glial inhibitor and inhibitor of phosphodiesterase activity, potentiates opioid analgesia in rats148 and in a phase 2 clinical trial (NCT00576277) has shown promise in treating neuropathic pain. The plasma concentration of AV411 plasma correlated with decreased pain and the drug was well tolerated (http://www.globenewswire.com/newsroom/news.html?d=130518).
Concluding remarks
We now fully appreciate the importance of interactions between the immune and nervous systems in pain. The body's innate immune cells respond to injury with an inflammatory response that activates pain pathways. Soluble mediators released by immune and glial cells act on nociceptors, increasing synaptic strength and altering pain sensitivity. After activation of peripheral immune cells and nociceptors, the initial acute pain response, if left unabated, can develop into chronic pathological pain.
There remain crucial gaps in our knowledge. The role of innate immune activation in pain is quite clear, but relatively little is known about the role of the adaptive immune system in chronic inflammatory conditions and their contribution to chronic pain. Furthermore, most studies have investigated only the protective phase of pain associated with initial injury. Current animal models are limited by an acute inflammatory response and short-lived hyperalgesia, which attenuate over time. Better models are needed to explore the contributions of immune cells in a chronic setting and their role in maintaining a chronic pain state. Further studies of the involvement of immune cells and glia in pain should make it possible to identify novel targets and more selective inhibitors.
References
DeLeo, J.A., Tanga, F.Y. & Tawfik, V.L. Neuroimmune activation and neuroinflammation in chronic pain and opioid tolerance/hyperalgesia. Neuroscientist 10, 40–52 (2004).
Watkins, L.R. & Maier, S.F. Immune regulation of central nervous system functions: from sickness responses to pathological pain. J. Intern. Med. 257, 139–155 (2005).
Scholz, J. & Woolf, C.J. The neuropathic pain triad: neurons, immune cells and glia. Nat. Neurosci. 10, 1361–1368 (2007).
Ren, K. & Dubner, R. Neuron-glia crosstalk gets serious: role in pain hypersensitivity. Curr. Opin. Anaesthesiol. 21, 570–579 (2008).
Thacker, M.A., Clark, A.K., Marchand, F. & McMahon, S.B. Pathophysiology of peripheral neuropathic pain: immune cells and molecules. Anesth. Analg. 105, 838–847 (2007).
Miller, R.J., Jung, H., Bhangoo, S.K. & White, F.A. Cytokine and chemokine regulation of sensory neuron function. Handb. Exp. Pharmacol 194, 417–449 (2009).
Rittner, H.L., Brack, A. & Stein, C. Pain and the immune system. Br. J. Anaesth. 101, 40–44 (2008).
Xu, Z.-Z. et al. Resolvins RvE1 and RvD1 attenuate inflammatory pain via central and peripheral actions. Nat. Med. 16, 592–597 (2010).
Guo, L.H. & Schluesener, H.J. The innate immunity of the central nervous system in chronic pain: the role of Toll-like receptors. Cell. Mol. Life Sci. 64, 1128–1136 (2007).
Takeuchi, O. & Akira, S. Pattern recognition receptors and inflammation. Cell 140, 805–820 (2010).
Lawrence, T., Willoughby, D.A. & Gilroy, D.W. Anti-inflammatory lipid mediators and insights into the resolution of inflammation. Nat. Rev. Immunol. 2, 787–795 (2002).
Folgueras, A.R. et al. Metalloproteinase MT5-MMP is an essential modulator of neuro-immune interactions in thermal pain stimulation. Proc. Natl. Acad. Sci. USA 106, 16451–16456 (2009).
Suzuki, A., Suzuki, R., Furuno, T., Teshima, R. & Nakanishi, M. N-cadherin plays a role in the synapse-like structures between mast cells and neurites. Biol. Pharm. Bull. 27, 1891–1894 (2004).
Levy, D., Burstein, R., Kainz, V., Jakubowski, M. & Strassman, A.M. Mast cell degranulation activates a pain pathway underlying migraine headache. Pain 130, 166–176 (2007).
Lewin, G.R., Rueff, A. & Mendell, L.M. Peripheral and central mechanisms of NGF-induced hyperalgesia. Eur. J. Neurosci. 6, 1903–1912 (1994).
Rudick, C.N., Bryce, P.J., Guichelaar, L.A., Berry, R.E. & Klumpp, D.J. Mast cell-derived histamine mediates cystitis pain. PLoS ONE 3, e2096 (2008).
Barbara, G. et al. Mast cell-dependent excitation of visceral-nociceptive sensory neurons in irritable bowel syndrome. Gastroenterology 132, 26–37 (2007).
Cui, J.G., Holmin, S., Mathiesen, T., Meyerson, B.A. & Linderoth, B. Possible role of inflammatory mediators in tactile hypersensitivity in rat models of mononeuropathy. Pain 88, 239–248 (2000).
Liu, T., van Rooijen, N. & Tracey, D.J. Depletion of macrophages reduces axonal degeneration and hyperalgesia following nerve injury. Pain 86, 25–32 (2000).
Shubayev, V.I. et al. TNFα-induced MMP-9 promotes macrophage recruitment into injured peripheral nerve. Mol. Cell. Neurosci. 31, 407–415 (2006).
Shubayev, V.I. & Myers, R.R. Upregulation and interaction of TNFα and gelatinases A and B in painful peripheral nerve injury. Brain Res. 855, 83–89 (2000).
Gómez-Nicola, D., Valle-Argos, B., Suardíaz, M., Taylor, J.S. & Nieto-Sampedro, M. Role of IL-15 in spinal cord and sciatic nerve after chronic constriction injury: regulation of macrophage and T-cell infiltration. J. Neurochem. 107, 1741–1752 (2008).
Constantinescu, C.S., Grygar, C., Kappos, L. & Leppert, D. Interleukin 15 stimulates production of matrix metalloproteinase-9 and tissue inhibitor of metalloproteinase-1 by human peripheral blood mononuclear cells. Cytokine 13, 244–247 (2001).
Sorkin, L.S., Xiao, W.H., Wagner, R. & Myers, R.R. Tumour necrosis factor-alpha induces ectopic activity in nociceptive primary afferent fibres. Neuroscience 81, 255–262 (1997).
Verri, W.A. Jr. et al. IL-15 mediates immune inflammatory hypernociception by triggering a sequential release of IFN-γ, endothelin, and prostaglandin. Proc. Natl. Acad. Sci. USA 103, 9721–9725 (2006).
Kiguchi, N., Maeda, T., Kobayashi, Y., Fukazawa, Y. & Kishioka, S. Macrophage inflammatory protein-1α mediates the development of neuropathic pain following peripheral nerve injury through interleukin-1beta up-regulation. Pain 149, 305–315 (2010).
Barclay, J. et al. Role of the cysteine protease cathepsin S in neuropathic hyperalgesia. Pain 130, 225–234 (2007).
Rutkowski, M.D., Pahl, J.L., Sweitzer, S., van Rooijen, N. & DeLeo, J.A. Limited role of macrophages in generation of nerve injury-induced mechanical allodynia. Physiol. Behav. 71, 225–235 (2000).
Brück, W., Huitinga, I. & Dijkstra, C.D. Liposome-mediated monocyte depletion during wallerian degeneration defines the role of hematogenous phagocytes in myelin removal. J. Neurosci. Res. 46, 477–484 (1996).
Cailhier, J.F. et al. Resident pleural macrophages are key orchestrators of neutrophil recruitment in pleural inflammation. Am. J. Respir. Crit. Care Med. 173, 540–547 (2006).
Ting, E. et al. Role of complement C5a in mechanical inflammatory hypernociception: potential use of C5a receptor antagonists to control inflammatory pain. Br. J. Pharmacol. 153, 1043–1053 (2008).
Guerrero, A.T. et al. Involvement of LTB4 in zymosan-induced joint nociception in mice: participation of neutrophils and PGE2. J. Leukoc. Biol. 83, 122–130 (2008).
Perretti, M., Ahluwalia, A., Flower, R.J. & Manzini, S. Endogenous tachykinins play a role in IL-1-induced neutrophil accumulation: involvement of NK-1 receptors. Immunology 80, 73–77 (1993).
Ottosson, A. & Edvinsson, L. Release of histamine from dural mast cells by substance P and calcitonin gene-related peptide. Cephalalgia 17, 166–174 (1997).
Krämer, H.H., He, L., Lu, B., Birklein, F. & Sommer, C. Increased pain and neurogenic inflammation in mice deficient of neutral endopeptidase. Neurobiol. Dis. 35, 177–183 (2009).
Hu, P. & McLachlan, E.M. Macrophage and lymphocyte invasion of dorsal root ganglia after peripheral nerve lesions in the rat. Neuroscience 112, 23–38 (2002).
Moalem, G., Xu, K. & Yu, L. T lymphocytes play a role in neuropathic pain following peripheral nerve injury in rats. Neuroscience 129, 767–777 (2004).
Costigan, M. et al. Fitzgerald M.T-cell infiltration and signaling in the adult dorsal spinal cord is a major contributor to neuropathic pain-like hypersensitivity. J. Neurosci. 29, 14415–14422 (2009).
Cao, L. & DeLeo, J.A. CNS-infiltrating CD4+ T lymphocytes contribute to murine spinal nerve transection-induced neuropathic pain. Eur. J. Immunol. 38, 448–458 (2008).
Orhan, C.E., Onal, A. & Ulker, S. Antihyperalgesic and antiallodynic effect of sirolimus in neuropathic pain and the role of cytokines in this effect. Neurosci. Lett. 481, 17–20 (2010).
Brennan, P.C., Graham, M.A., Triano, J.J., Hondras, M.A. & Anderson, R.J. Lymphocyte profiles in patients with chronic low back pain enrolled in a clinical trial. J. Manipulative Physiol. Ther. 17, 219–227 (1994).
Gilman-Sachs, A., Robbins, L. & Baum, L. Flow cytometric analysis of lymphocyte subsets in peripheral blood of chronic headache patients. Headache 29, 290–294 (1989).
Carroll, M.C. The complement system in regulation of adaptive immunity. Nat. Immunol. 5, 981–986 (2004).
Li, M., Peake, P.W., Charlesworth, J.A., Tracey, D.J. & Moalem-Taylor, G. Complement activation contributes to leukocyte recruitment and neuropathic pain following peripheral nerve injury in rats. Eur. J. Neurosci. 26, 3486–3500 (2007).
Levin, M.E. et al. Complement activation in the peripheral nervous system following the spinal nerve ligation model of neuropathic pain. Pain 137, 182–201 (2008).
Jang, J.H. et al. Nociceptive sensitization by complement C5a and C3a in mouse. Pain 148, 343–352 (2010).
Levine, J.D., Gooding, J., Donatoni, P., Borden, L. & Goetzl, E.J. The role of the polymorphonuclear leukocyte in hyperalgesia. J. Neurosci. 5, 3025–3029 (1985).
Clark, J.D. et al. Blockade of the complement C5a receptor reduces incisional allodynia, edema, and cytokine expression. Anesthesiology 104, 1274–1282 (2006).
Griffin, R.S. et al. Complement induction in spinal cord microglia results in anaphylatoxin C5a-mediated pain hypersensitivity. J. Neurosci. 27, 8699–8708 (2007).
Twining, C.M. et al. Activation of the spinal cord complement cascade might contribute to mechanical allodynia induced by three animal models of spinal sensitization. J. Pain 6, 174–183 (2005).
Hu, P., Bembrick, A.L., Keay, K.A. & McLachlan, E.M. Immune cell involvement in dorsal root ganglia and spinal cord after chronic constriction or transection of the rat sciatic nerve. Brain Behav. Immun. 21, 599–616 (2007).
Dublin, P. & Hanani, M. Satellite glial cells in sensory ganglia: their possible contribution to inflammatory pain. Brain Behav. Immun. 21, 592–598 (2007).
Ledda, M., Blum, E., De Palo, S. & Hanani, M. Augmentation in gap junction-mediated cell coupling in dorsal root ganglia following sciatic nerve neuritis in the mouse. Neuroscience 164, 1538–1545 (2009).
Durham, P.L. & Garrett, F.G. Emerging importance of neuron-satellite glia interactions within trigeminal ganglia in craniofacial pain. Open Pain J. 3, 3–13 (2010).
Thalakoti, S. et al. Neuron-glia signaling in trigeminal ganglion: implications for migraine pathology. Headache 47, 1008–1023 (2007).
Tang, X., Schmidt, T.M., Perez-Leighton, C.E. & Kofuji, P. Inwardly rectifying potassium channel Kir4.1 is responsible for the native inward potassium conductance of satellite glial cells in sensory ganglia. Neuroscience 166, 397–407 (2010).
Vit, J.P., Ohara, P.T., Bhargava, A., Kelley, K. & Jasmin, L. Silencing the Kir4.1 potassium channel subunit in satellite glial cells of the rat trigeminal ganglion results in pain-like behavior in the absence of nerve injury. J. Neurosci. 28, 4161–4171 (2008).
Capuano, A. et al. Proinflammatory-activated trigeminal satellite cells promote neuronal sensitization: relevance for migraine pathology. Mol. Pain 5, 43 (2009).
Uçeyler, N., Tscharke, A. & Sommer, C. Early cytokine expression in mouse sciatic nerve after chronic constriction nerve injury depends on calpain. Brain Behav. Immun. 21, 553–560 (2007).
Deruddre, S. et al. Effects of a bupivacaine nerve block on the axonal transport of tumor necrosis factor-α (TNF-α) in a rat model of carrageenan-induced inflammation. Brain Behav. Immun. 24, 652–659 (2010).
Jung, H., Toth, P.T., White, F.A. & Miller, R.J. Monocyte chemoattractant protein-1 functions as a neuromodulator in dorsal root ganglia neurons. J. Neurochem. 104, 254–263 (2008).
Sun, J.H., Yang, B., Donnelly, D.F., Ma, C. & LaMotte, R.H. MCP-1 enhances excitability of nociceptive neurons in chronically compressed dorsal root ganglia. J. Neurophysiol. 96, 2189–2199 (2006).
Khodorova, A. et al. Endothelin-B receptor activation triggers an endogenous analgesic cascade at sites of peripheral injury. Nat. Med. 9, 1055–1061 (2003).
Hua, S. & Cabot, P.J. Mechanisms of peripheral immune-cell-mediated analgesia in inflammation: clinical and therapeutic implications. Trends Pharmacol. Sci. 31, 427–433 (2010).
Jessop, D.S. et al. Endomorphins in rheumatoid arthritis, osteoarthritis, and experimental arthritis. Ann. NY Acad. Sci. 1193, 117–122 (2010).
Zylka, M.J. et al. Prostatic acid phosphatase is an ectonucleotidase and suppresses pain by generating adenosine. Neuron 60, 111–122 (2008).
Kaufmann, A. et al. “Host tissue damage” signal ATP promotes non-directional migration and negatively regulates toll-like receptor signaling in human monocytes. J. Biol. Chem. 280, 32459–32467 (2005).
Chen, Y. et al. Activation of P2X7 receptors in glial satellite cells reduces pain through downregulation of P2X3 receptors in nociceptive neurons. Proc. Natl. Acad. Sci. USA 105, 16773–16778 (2008).
Serhan, C.N., Chiang, N. & Van Dyke, T.E. Resolving inflammation: dual anti-inflammatory and pro-resolution lipid mediators. Nat. Rev. Immunol. 8, 349–361 (2008).
Svensson, C.I., Zattoni, M. & Serhan, C.N. Lipoxins and aspirin-triggered lipoxin inhibit inflammatory pain processing. J. Exp. Med. 204, 245–252 (2007).
Spite, M. et al. Resolvin D2 is a potent regulator of leukocytes and controls microbial sepsis. Nature 461, 1287–1291 (2009).
Raghavendra, V., Tanga, F.Y. & DeLeo, J.A. Complete Freunds adjuvant-induced peripheral inflammation evokes glial activation and proinflammatory cytokine expression in the CNS. Eur. J. Neurosci. 20, 467–473 (2004).
Zhao, P., Waxman, S.G. & Hains, B.C. Modulation of thalamic nociceptive processing after spinal cord injury through remote activation of thalamic microglia by cysteine cysteine chemokine ligand 21. J. Neurosci. 27, 8893–8902 (2007).
Wei, F., Guo, W., Zou, S., Ren, K. & Dubner, R. Supraspinal glial-neuronal interactions contribute to descending pain facilitation. J. Neurosci. 28, 10482–10495 (2008).
Roberts, J., Ossipov, M.H. & Porreca, F. Glial activation in the rostroventromedial medulla promotes descending facilitation to mediate inflammatory hypersensitivity. Eur. J. Neurosci. 30, 229–241 (2009).
Guo, W. et al. Glial-cytokine-neuronal interactions underlying the mechanisms of persistent pain. J. Neurosci. 27, 6006–6018 (2007).
Wen, Y.R. et al. Nerve conduction blockade in the sciatic nerve prevents but does not reverse the activation of p38 mitogen-activated protein kinase in spinal microglia in the rat spared nerve injury model. Anesthesiology 107, 312–321 (2007).
Xie, W., Strong, J.A. & Zhang, J.M. Early blockade of injured primary sensory afferents reduces glial cell activation in two rat neuropathic pain models. Neuroscience 160, 847–857 (2009).
Oka, Y. et al. Interleukin-6 is a candidate molecule that transmits inflammatory information to the CNS. Neuroscience 145, 530–538 (2007).
Fu, K.Y., Light, A.R. & Maixner, W. Relationship between nociceptor activity, peripheral edema, spinal microglial activation and long-term hyperalgesia induced by formalin. Neuroscience 101, 1127–1135 (2000).
Sweitzer, S.M., Colburn, R.W., Rutkowski, M. & DeLeo, J.A. Acute peripheral inflammation induces moderate glial activation and spinal IL-1beta expression that correlates with pain behavior in the rat. Brain Res. 829, 209–221 (1999).
Clark, A.K., Gentry, C., Bradbury, E.J., McMahon, S.B. & Malcangio, M. Role of spinal microglia in rat models of peripheral nerve injury and inflammation. Eur. J. Pain 11, 223–230 (2007).
Eijkelkamp, N. et al. GRK2: a novel cell-specific regulator of severity and duration of inflammatory pain. J. Neurosci. 30, 2138–2149 (2010).
Hua, X.Y. et al. Intrathecal minocycline attenuates peripheral inflammation-induced hyperalgesia by inhibiting p38 MAPK in spinal microglia. Eur. J. Neurosci. 22, 2431–2440 (2005).
Chen, Y., Willcockson, H.H. & Valtschanoff, J.G. Influence of the vanilloid receptor TRPV1 on the activation of spinal cord glia in mouse models of pain. Exp. Neurol. 220, 383–390 (2009).
Honore, P. et al. Murine models of inflammatory, neuropathic and cancer pain each generates a unique set of neurochemical changes in the spinal cord and sensory neurons. Neuroscience 98, 585–598 (2000).
Zhang, J. et al. Induction of CB2 receptor expression in the rat spinal cord of neuropathic but not inflammatory chronic pain models. Eur. J. Neurosci. 17, 2750–2754 (2003).
Chacur, M., Lambertz, D., Hoheisel, U. & Mense, S. Role of spinal microglia in myositis-induced central sensitisation: an immunohistochemical and behavioural study in rats. Eur. J. Pain 13, 915–923 (2009).
Sun, S. et al. New evidence for the involvement of spinal fractalkine receptor in pain facilitation and spinal glial activation in rat model of monoarthritis. Pain 129, 64–75 (2007).
Ledeboer, A. et al. Minocycline attenuates mechanical allodynia and proinflammatory cytokine expression in rat models of pain facilitation. Pain 115, 71–83 (2005).
Riazi, K. et al. Microglial activation and TNFalpha production mediate altered CNS excitability following peripheral inflammation. Proc. Natl. Acad. Sci. USA 105, 17151–17156 (2008).
Feng, Q.X. et al. Astrocytic activation in thoracic spinal cord contributes to persistent pain in rat model of chronic pancreatitis. Neuroscience 167, 501–509 (2010).
Romero-Sandoval, A., Chai, N., Nutile-McMenemy, N. & DeLeo, J.A. A comparison of spinal Iba1 and GFAP expression in rodent models of acute and chronic pain. Brain Res. 1219, 116–126 (2008).
Meller, S.T., Dykstra, C., Grzybycki, D., Murphy, S. & Gebhart, G.F. The possible role of glia in nociceptive processing and hyperalgesia in the spinal cord of the rat. Neuropharmacology 33, 1471–1478 (1994).
Clark, A.K., Yip, P.K. & Malcangio, M. The liberation of fractalkine in the dorsal horn requires microglial cathepsin S. J. Neurosci. 29, 6945–6954 (2009).
Hathway, G.J., Vega-Avelaira, D., Moss, A., Ingram, R. & Fitzgerald, M. Brief, low frequency stimulation of rat peripheral C-fibres evokes prolonged microglial-induced central sensitization in adults but not in neonates. Pain 144, 110–118 (2009).
Molander, C., Hongpaisan, J., Svensson, M. & Aldskogius, H. Glial cell reactions in the spinal cord after sensory nerve stimulation are associated with axonal injury. Brain Res. 747, 122–129 (1997).
Schöbitz, B., de Kloet, E.R., Sutanto, W. & Holsboer, F. Cellular localization of interleukin 6 mRNA and interleukin 6 receptor mRNA in rat brain. Eur. J. Neurosci. 5, 1426–1435 (1993).
Vallières, L. & Rivest, S. Regulation of the genes encoding interleukin-6, its receptor, and gp130 in the rat brain in response to the immune activator lipopolysaccharide and the proinflammatory cytokine interleukin-1beta. J. Neurochem. 69, 1668–1683 (1997).
Mitchell, K. et al. Localization of S100A8 and S100A9 expressing neutrophils to spinal cord during peripheral tissue inflammation. Pain 134, 216–231 (2008).
Zhang, J. et al. Expression of CCR2 in both resident and bone marrow-derived microglia plays a critical role in neuropathic pain. J. Neurosci. 27, 12396–12406 (2007).
Willis, C.L. & Davis, T.P. Chronic inflammatory pain and the neurovascular unit: a central role for glia in maintaining BBB integrity? Curr. Pharm. Des. 14, 1625–1643 (2008).
Hemley, S.J., Biotech, B., Tu, J. & Stoodley, M.A. Role of the blood-spinal cord barrier in posttraumatic syringomyelia. J. Neurosurg. Spine 11, 696–704 (2009).
Lu, P. et al. CNS penetration of small molecules following local inflammation, widespread systemic inflammation or direct injury to the nervous system. Life Sci. 85, 450–456 (2009).
Mitchell, K., Yang, H.Y., Berk, J.D., Tran, J.H. & Iadarola, M.J. Monocyte chemoattractant protein-1 in the choroid plexus: a potential link between vascular pro-inflammatory mediators and the CNS during peripheral tissue inflammation. Neuroscience 158, 885–895 (2009).
Kawasaki, Y. et al. Distinct roles of matrix metalloproteases in the early- and late-phase development of neuropathic pain. Nat. Med. 14, 331–336 (2008).
Milligan, E.D. & Watkins, L.R. Pathological and protective roles of glia in chronic pain. Nat. Rev. Neurosci. 10, 23–36 (2009).
Calvo, M. et al. Neuregulin-ErbB signaling promotes microglial proliferation and chemotaxis contributing to microgliosis and pain after peripheral nerve injury. J. Neurosci. 30, 5437–5450 (2010).
Dominguez, E., Mauborgne, A., Mallet, J., Desclaux, M. & Pohl, M. SOCS3-mediated blockade of JAK/STAT3 signaling pathway reveals its major contribution to spinal cord neuroinflammation and mechanical allodynia after peripheral nerve injury. J. Neurosci. 30, 5754–5766 (2010).
Kawasaki, Y., Zhang, L., Cheng, J.K. & Ji, R.R. Cytokine mechanisms of central sensitization: distinct and overlapping role of interleukin-1β, interleukin-6, and tumor necrosis factor-α in regulating synaptic and neuronal activity in the superficial spinal cord. J. Neurosci. 28, 5189–5194 (2008).
Premont, R.T. & Gainetdinov, R.R. Physiological roles of G protein–coupled receptor kinases and arrestins. Annu. Rev. Physiol. 69, 511–534 (2007).
Willemen, H.L. et al. Microglial/macrophage GRK2 determines duration of peripheral IL-1β–induced hyperalgesia: Contribution of spinal cord CX3CR1, p38 and IL-1 signaling. Pain 150, 550–560 (2010).
Wang, W. et al. Crosstalk between spinal astrocytes and neurons in nerve injury-induced neuropathic pain. PLoS ONE 4, e6973 (2009).
Garrison, C.J., Dougherty, P.M. & Carlton, S.M. GFAP expression in lumbar spinal cord of naive and neuropathic rats treated with MK-801. Exp. Neurol. 129, 237–243 (1994).
Lalo, U., Pankratov, Y., Kirchhoff, F., North, R.A. & Verkhratsky, A. NMDA receptors mediate neuron-to-glia signaling in mouse cortical astrocytes. J. Neurosci. 26, 2673–2683 (2006).
Ikeda, H., Tsuda, M., Inoue, K. & Murase, K. Long-term potentiation of neuronal excitation by neuron-glia interactions in the rat spinal dorsal horn. Eur. J. Neurosci. 25, 1297–1306 (2007).
Xu, M., Bruchas, M.R., Ippolito, D.L., Gendron, L. & Chavkin, C. Sciatic nerve ligation-induced proliferation of spinal cord astrocytes is mediated by kappa opioid activation of p38 mitogen-activated protein kinase. J. Neurosci. 27, 2570–2581 (2007).
Tchivileva, I.E. et al. Characterization of NF-κB–mediated inhibition of catechol-O-methyltransferase. Mol. Pain 5, 13 (2009).
Diatchenko, L. et al. Catechol-O-methyltransferase gene polymorphisms are associated with multiple pain-evoking stimuli. Pain 125, 216–224 (2006).
Fu, E.S. et al. Transgenic inhibition of glial NF-κB reduces pain behavior and inflammation after peripheral nerve injury. Pain 148, 509–518 (2010).
Gao, Y.J., Xu, Z.Z., Liu, Y.C., Wen, Y.R., Decosterd, I. & Ji, R.R. The c-Jun N-terminal kinase 1 (JNK1) in spinal astrocytes is required for the maintenance of bilateral mechanical allodynia under a persistent inflammatory pain condition. Pain 148, 309–319 (2010).
Gardner, J. et al. Potential mechanisms for astrocyte-TIMP-1 downregulation in chronic inflammatory diseases. J. Neurosci. Res. 83, 1281–1292 (2006).
Verge, G.M. et al. Fractalkine (CX3CL1) and fractalkine receptor (CX3CR1) distribution in spinal cord and dorsal root ganglia under basal and neuropathic pain conditions. Eur. J. Neurosci. 20, 1150–1160 (2004).
Zhuang, Z.Y. et al. Role of the CX3CR1/p38 MAPK pathway in spinal microglia for the development of neuropathic pain following nerve injury-induced cleavage of fractalkine. Brain Behav. Immun. 21, 642–651 (2007).
Coull, J.A. et al. BDNF from microglia causes the shift in neuronal anion gradient underlying neuropathic pain. Nature 438, 1017–1021 (2005).
Clark, A.K. et al. Inhibition of spinal microglial cathepsin S for the reversal of neuropathic pain. Proc. Natl. Acad. Sci. USA 104, 10655–10660 (2007).
Trang, T., Beggs, S., Wan, X. & Salter, M.W. P2X4-receptor–mediated synthesis and release of brain-derived neurotrophic factor in microglia is dependent on calcium and p38-mitogen-activated protein kinase activation. J. Neurosci. 29, 3518–3528 (2009).
Hamilton, N.B. & Attwell, D. Do astrocytes really exocytose neurotransmitters? Nat. Rev. Neurosci. 11, 227–238 (2010).
Sung, B., Lim, G. & Mao, J. Altered expression and uptake activity of spinal glutamate transporters after nerve injury contribute to the pathogenesis of neuropathic pain in rats. J. Neurosci. 23, 2899–2910 (2003).
Chiang, C.Y. et al. Astroglial glutamate-glutamine shuttle is involved in central sensitization of nociceptive neurons in rat medullary dorsal horn. J. Neurosci. 27, 9068–9076 (2007).
Okada-Ogawa, A. et al. Astroglia in medullary dorsal horn (trigeminal spinal subnucleus caudalis) are involved in trigeminal neuropathic pain mechanisms. J. Neurosci. 29, 11161–11171 (2009).
Nie, H. & Weng, H.R. Glutamate transporters prevent excessive activation of NMDA receptors and extrasynaptic glutamate spillover in the spinal dorsal horn. J. Neurophysiol. 101, 2041–2051 (2009).
Ren, K. Emerging role of astroglia in pain hypersensitivity. Jpn. Dent. Sci. Rev. 46, 86–92 (2010).
Samad, T.A. et al. Interleukin-1β-mediated induction of Cox-2 in the CNS contributes to inflammatory pain hypersensitivity. Nature 410, 471–475 (2001).
Clark, A.K. et al. P2X7-dependent release of interleukin-1β and nociception in the spinal cord following lipopolysaccharide. J. Neurosci. 30, 573–582 (2010).
Johnston, I.N. et al. A role for proinflammatory cytokines and fractalkine in analgesia, tolerance, and subsequent pain facilitation induced by chronic intrathecal morphine. J. Neurosci. 24, 7353–7365 (2004).
Clark, A.K. et al. Rapid co-release of interleukin 1β and caspase 1 in spinal cord inflammation. J. Neurochem. 99, 868–880 (2006).
Zhang, R.X. et al. Spinal glial activation in a new rat model of bone cancer pain produced by prostate cancer cell inoculation of the tibia. Pain 118, 125–136 (2005).
Weyerbacher, A.R., Xu, Q., Tamasdan, C., Shin, S.J. & Inturrisi, C.E. N-Methyl-D-aspartate receptor (NMDAR) independent maintenance of inflammatory pain. Pain 148, 237–246 (2010).
Choi, J.I., Svensson, C.I., Koehrn, F.J., Bhuskute, A. & Sorkin, L.S. Peripheral inflammation induces tumor necrosis factor dependent AMPA receptor trafficking and Akt phosphorylation in spinal cord in addition to pain behavior. Pain 149, 243–253 (2010).
Miyoshi, K., Obata, K., Kondo, T., Okamura, H. & Noguchi, K. Interleukin-18–mediated microglia/astrocyte interaction in the spinal cord enhances neuropathic pain processing after nerve injury. J. Neurosci. 28, 12775–12787 (2008).
Listing, J. et al. Infections in patients with rheumatoid arthritis treated with biologic agents. Arthritis Rheum. 52, 3403–3412 (2005).
Terkeltaub, R. et al. The interleukin 1 inhibitor rilonacept in treatment of chronic gouty arthritis: results of a placebo-controlled, monosequence crossover, non-randomised, single-blind pilot study. Ann. Rheum. Dis. 68, 1613–1617 (2009).
Torres, R. et al. Hyperalgesia, synovitis and multiple biomarkers of inflammation are suppressed by interleukin 1 inhibition in a novel animal model of gouty arthritis. Ann. Rheum. Dis. 68, 1602–1608 (2009).
Soderquist, R.G. et al. Release of plasmid DNA encoding IL-10 from PLGA microparticles facilitates long-term reversal of neuropathic pain following a single intrathecal administration. Pharm. Res. 27, 841–854 (2010).
Vergunst, C.E. et al. Blocking the receptor for C5a in patients with rheumatoid arthritis does not reduce synovial inflammation. Rheumatology (Oxford) 46, 1773–1778 (2007).
Romero-Sandoval, E.A., Horvath, R.J. & DeLeo, J.A. Neuroimmune interactions and pain: focus on glial-modulating targets. Curr. Opin. Investig. Drugs 9, 726–734 (2008).
Hutchinson, M.R. et al. Reduction of opioid withdrawal and potentiation of acute opioid analgesia by systemic AV411 (ibudilast). Brain Behav. Immun. 23, 240–250 (2009).
Acknowledgements
The authors' work is supported by US National Institutes of Health grants R01-DE11964, R01-NS060735 and R01-NS059028.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Rights and permissions
About this article
Cite this article
Ren, K., Dubner, R. Interactions between the immune and nervous systems in pain. Nat Med 16, 1267–1276 (2010). https://doi.org/10.1038/nm.2234
Published:
Issue Date:
DOI: https://doi.org/10.1038/nm.2234
This article is cited by
-
Exploring the association between dietary Inflammatory Index and chronic pain in US adults using NHANES 1999–2004
Scientific Reports (2024)
-
Relationship between muscle sympathetic nerve activity and rapid increases in circulating leukocytes during experimental muscle pain
Clinical Autonomic Research (2024)
-
Gateway reflexes describe novel neuro-immune communications that establish immune cell gateways at specific vessels
Bioelectronic Medicine (2023)
-
Regulatory role of PI3K/Akt/WNK1 signal pathway in mouse model of bone cancer pain
Scientific Reports (2023)
-
β-Sitosterol Alleviates Neuropathic Pain by Affect Microglia Polarization through Inhibiting TLR4/NF-κB Signaling Pathway
Journal of Neuroimmune Pharmacology (2023)
