
Abstract
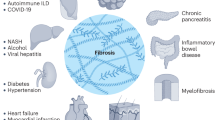
Fibrosis is a pathological feature of most chronic inflammatory diseases. Fibrosis, or scarring, is defined by the accumulation of excess extracellular matrix components. If highly progressive, the fibrotic process eventually leads to organ malfunction and death. Fibrosis affects nearly every tissue in the body. Here we discuss how key components of the innate and adaptive immune response contribute to the pathogenesis of fibrosis. We also describe how cell-intrinsic changes in important structural cells can perpetuate the fibrotic response by regulating the differentiation, recruitment, proliferation and activation of extracellular matrix–producing myofibroblasts. Finally, we highlight some of the key mechanisms and pathways of fibrosis that are being targeted as potential therapies for a variety of important human diseases.
Similar content being viewed by others

Main
Fibrosis is the final, common pathological outcome of many chronic inflammatory diseases. Although collagen deposition is an indispensable and, typically, reversible part of wound healing, normal tissue repair can evolve into a progressively irreversible fibrotic response if the tissue injury is severe or repetitive or if the wound-healing response itself becomes dysregulated. Fibrosis is defined by the excessive accumulation of fibrous connective tissue (components of the extracellular matrix (ECM) such as collagen and fibronectin) in and around inflamed or damaged tissue, which can lead to permanent scarring, organ malfunction and, ultimately, death, as seen in end-stage liver disease, kidney disease, idiopathic pulmonary fibrosis (IPF) and heart failure1,2. Fibrosis is also a major pathological feature of many chronic autoimmune diseases, including scleroderma, rheumatoid arthritis, Crohn's disease, ulcerative colitis, myelofibrosis and systemic lupus erythematosus. Fibrosis also influences tumor invasion and metastasis, chronic graft rejection and the pathogenesis of many progressive myopathies. Although fibrogenesis is becoming increasingly recognized as a major cause of morbidity and mortality in most chronic inflammatory diseases, there are few—if any—treatment strategies available that specifically target the pathogenesis of fibrosis.
Many distinct triggers can contribute to the development of progressive fibrotic disease. Examples include, to name just a few, inherited genetic disorders; persistent infections; recurrent exposure to toxins, irritants or smoke; chronic autoimmune inflammation; minor human leukocyte antigen mismatches in transplants; myocardial infarction; high serum cholesterol; obesity; and poorly controlled diabetes and hypertension3. Regardless of the initiating events, a feature common to all fibrotic diseases is the activation of ECM-producing myofibroblasts, which are the key mediators of fibrotic tissue remodeling4,5. For example, the left-ventricular hypertrophy that accompanies chronic hypertension is caused by an abnormal accumulation of collagen and other ECM components in the extracellular space. This reactive and progressive interstitial fibrosis contributes to myocardial stiffness and, ultimately, ventricular dysfunction, and it is believed to result from the persistent activation of cardiac myofibroblasts. Liver cirrhosis caused by chronic hepatitis C virus (HCV) infection, alcohol abuse or nonalcoholic steatohepatitis (NASH) is similarly induced by activated myofibroblasts1. In the liver, excessive collagen deposition distorts normal tissue architecture, leading to hepatocellular dysfunction and increased hepatic resistance to blood flow, which cause hepatic insufficiency and portal hypertension. Because ECM-secreting myofibroblasts are central to the pathogenesis of all fibrotic diseases, fibrosis research has focused on elucidating the molecular and immunological mechanisms that initiate, maintain and terminate the differentiation of quiescent fibroblasts into actively proliferating, ECM-producing myofibroblasts.
It is now clear that many elements of the innate and adaptive immune response participate in the differentiation and activation of fibroblasts (Fig. 1). In this Review, we describe some of the important mechanisms that contribute to the progression of fibrosis. We also present experimental findings from several different model systems, with the goal of identifying common pathways of fibrosis that could be targeted to facilitate the development of broadly effective antifibrotic drugs. Finally, we discuss some of the major obstacles in antifibrotic-drug development and highlight a few of the important pathways of fibrogenesis that have progressed to clinical testing.

Epithelial and/or endothelial damage caused by various insults triggers complex interconnected wound-healing programs to quickly restore homeostasis. The coagulation pathway, which functions to stem blood loss, is triggered first, followed by acute inflammation and activation of innate immune mediators such as resident macrophages, neutrophils and dendritic cells. Epithelial and innate immune cell–derived cytokines subsequently influence the activation of the adaptive immune response. The tissue damage can also directly activate the adaptive immune response. Inflammatory and immune mediators (cytokines, chemokines and free radicals) attempt to eliminate the inciting factor while activating the resident quiescent fibroblasts into myofibroblasts that orchestrate angiogenesis and production of ECM components. Failure to adequately contain or eliminate the inciting factors can exacerbate the inflammatory response and lead to a chronic wound-healing response, with continued tissue damage, repair and regeneration, ultimately resulting in fibrosis. TSLP, thymic stromal lymphopoietin; Ab, antibody; PMN, polymorphonuclear leukocyte; EOS, eosinophil; Baso, basophil; Mast, mast cell.
Innate immunity and fibrosis
Acute inflammatory reactions play an important part in triggering fibrosis in many different organ systems. For example, in bleomycin-induced pulmonary fibrosis and carbon tetrachloride–induced liver fibrosis, brief exposure to these drugs causes epithelial-cell apoptosis and hepatocyte necrosis, respectively, activating an inflammatory wound-healing response that can lead to a temporary excess in deposition of ECM components in the affected tissues6,7. Low-grade but persistent inflammation is also thought to contribute to the progression of fibrosis in cardiovascular disease and hypertension. Indeed, in many fibrotic disorders, a persistent inflammatory trigger is crucial to the activation of the wound-healing program that leads to fibrosis. Consequently, removal of the inflammatory trigger is the most straightforward way to halt the progression of tissue remodeling and allow the normal tissue architecture to be restored after injury, as has been observed in individuals with chronic hepatitis B infection who are treated with entecavir, a highly effective oral antiviral drug. This may be easy to accomplish when the tissue-damaging mechanism is known, but in many fibrotic disorders, the tissue-damaging irritant is either unknown or cannot be easily eliminated. In this case, researchers have focused on pinpointing the innate and adaptive mechanisms that control inflammation, with the goal of identifying key mediators that could be targeted to attenuate fibrosis.
Innate wound-healing mechanisms initiate the fibrotic response. The coagulation response is the first wound-healing mechanism activated after injury. When the endothelium is damaged, circulating platelets are activated upon encountering exposed collagen and von Willebrand factor in the subendothelial layer8. Coagulation factor II (prothrombin) is also proteolytically cleaved to form thrombin. Thrombin, in turn, functions as a serine protease that converts soluble fibrinogen into insoluble strands of fibrin, which helps platelets clump together to form the fibrin clot that ensures quick hemostasis. Activated platelets also release growth factors such as platelet-derived growth factor (PDGF), a potent chemoattractant for inflammatory cells, and transforming growth factor-β1 (TGF-β1), which stimulates ECM synthesis by local fibroblasts9. Consequently, any prolonged disturbance in the coagulation cascade can lead to fibrosis10. For example, a recent study demonstrated that activated coagulation factor X (FXa) contributes to fibrosis after acute lung injury by inducing myofibroblast differentiation11. The authors showed that bleomycin-induced pulmonary fibrosis could be inhibited in mice when FXa was neutralized. The coagulation response is also believed to be a major driver in the pathogenesis of liver fibrosis. In addition to its role in the coagulation cascade, thrombin can directly promote fibrosis by inducing production of chemokine (C-C motif) ligand 2 (CCL2) and by signaling through protease-activated receptors expressed on liver fibroblasts (hepatic stellate cells)12. In support of this hypothesis, antagonists of protease-activated receptor 1 and anticoagulant drugs have each been shown to protect against experimental liver fibrosis in mice13,14. However, although the coagulation cascade can be an important initiator of fibrosis, deficiencies in the clotting pathway have also been shown to contribute to fibrosis. Procoagulant deficiencies are common in patients with cirrhosis and portal hypertension. Here, obliterative lesions in the portal and hepatic veins develop in response to microthrombi, which cause tissue ischemia, endothelial-cell death and fibrosis through parenchymal extinction15. Together, these findings illustrate how disturbances in the coagulation cascade can substantially contribute to the development of progressive fibrotic disease.
Inflammatory myeloid cells have a role in fibrosis. In addition to activating the coagulation cascade, platelets and damaged epithelial and endothelial cells release a variety of chemotactic factors that recruit inflammatory monocytes and neutrophils to the site of tissue damage (Fig. 2). These circulating myeloid cells respond to a gradient of CCL2 and are recruited to damaged tissues, where they differentiate into macrophages that phagocytose the fibrin clot and cellular debris. ECM fragments, including hyaluronan, have also been shown to be important drivers of fibrosis by stimulating chemokine and proinflammatory cytokine production by inflammatory monocytes and macrophages16. Neutrophils are also recruited quickly after injury and participate in removal of tissue debris and the killing of invading bacteria. Although the recruitment of inflammatory monocytes and neutrophils at the site of tissue injury is important for the wound-healing process, these cells also secrete a variety of toxic mediators, including reactive oxygen and nitrogen species that are harmful to the surrounding tissues. Consequently, if the inflammatory macrophages and neutrophils are not quickly eliminated, they can further exacerbate the tissue-damaging inflammatory response that leads to scarring. Indeed, proper wound healing can occur only after these inflammatory cells are controlled, as demonstrated by a recent study in which early macrophage depletion was shown to substantially reduce the development of liver fibrosis in mice17. Other studies have identified a similar profibrotic role for neutrophils in bleomycin- and hypersensitivity pneumonitis–induced pulmonary fibrosis in mice, and possibly in IPF18. It has also been suggested that liver dendritic cells have a role in the development of liver fibrosis, by promoting inflammation and activating hepatic stellate cells19. Thus, limiting the proinflammatory activity of innate myeloid-lineage cells might prove highly beneficial in a variety of chronic inflammatory and fibrotic diseases.

The macrophage is the prototypical innate immune cell involved in chronic inflammation and fibrosis. Macrophages are generated from blood monocytes that differentiate into macrophages as they enter tissues or, in some cases, from the local proliferation of tissue-resident macrophages. Depending on their etiology, macrophages are activated by a variety of triggers. IFN-γ and/or Toll-like receptor ligands such as lipopolysaccharide (LPS) and low molecular weight hyaluronic acid (LMWHA) lead to classical activation, which is characterized by the production of reactive oxygen and nitrogen species; IL-4, IL-13 and granulocyte-macrophage colony–stimulating factor (GM-CSF) mediate alternative activation, leading to the production of polyamines and L-proline by L-arginine catabolism. Certain triggers (such as extracellular bacteria and tissue damage) also elicit persistent or recurrent neutrophil infiltration mediated by IL-17 and other neutrophil-recruiting and/or neutrophil-activating signals that can substantially augment the microbicidal and tissue-damaging activities by free radicals. Likewise, helminth antigens and allergens mediate infiltration of eosinophils that assist in parasite killing that can result in substantial collateral damage to host tissues if not tightly confined inside fibrotic granulomata. EPO, erythropoietin; MBP, myelin basic protein; EDN, eosinophil-derived neurotoxin; AAM, alternatively activated macrophage; MMP2, metalloproteinase-2; MMP9, metalloproteinase-9; CAM, classically activated macrophage; iNOS, inducible nitric oxide synthase; ROS, reactive oxygen species; RNS, reactive nitrogen species.
Innate inflammatory mediators regulate fibrosis. Various growth factors and cytokines secreted by innate inflammatory cells (including macrophages, neutrophils, mast cells and eosinophils) have emerged as potential targets for antifibrotic therapy. Tumor necrosis factor-α (TNF-α) and interleukin-1β (IL-1β), in particular, have been identified as important targets in a variety of fibrotic diseases20. Mice that overexpress TNF-α or IL-1β in the lung develop highly progressive pulmonary fibrosis21,22. Studies have also shown an essential role for TNF-α in the development of silica- and bleomycin-induced pulmonary fibrosis in mice23,24. In support of these experimental findings, patients with idiopathic or systemic sclerosis–associated pulmonary fibrosis have high levels of TNF-α25. TNF-α has also been shown to play a crucial part in radiation-induced fibrosis, Crohn's disease–induced intestinal fibrosis, CCL4- and cholestasis-induced liver fibrosis and NASH26,27,28. Consequently, clinical trials have recently been initiated to evaluate whether TNF pathway inhibitors such as etanercept or infliximab could be beneficial for the treatment of pulmonary fibrosis and other fibrotic diseases29. Similarly to the TNF-α studies, other studies have documented profibrotic activity for IL-1β and NALP3/ASC inflammasome signaling in macrophages30. Pulmonary fibrosis induced by bleomycin and silica, liver fibrosis in hypercholesterolemic mice, renal interstitial fibrosis resulting from unilateral ureteric obstruction and cardiovascular fibrosis after myocardial infarction are all reduced in IL-1β–deficient mice31,32,33. Like TNF-α, IL-1β is a potent proinflammatory mediator that exacerbates parenchymal-cell injury. It also induces epithelial-mesenchymal transition (EMT) and myofibroblast activation through a TGF-β1–mediated mechanism34, confirming that it functions as a potent upstream driver of fibrosis. IL-1β and TNF-α also increase expression of IL-6, which shows autocrine growth-factor activity in fibroblasts. IL-6 is an important mediator of fibrosis in diffuse systemic sclerosis, liver fibrosis after CCL4 exposure and fibrosis in chronic cardiac allograft rejection35,36. Thus, many innate proinflammatory cytokines have crucial roles in the pathogenesis of fibrosis.
TGF-β1 has both anti-inflammatory and profibrotic activity. Macrophages that appear early in the wound-healing response are also major producers of TGF-β, which is, indisputably, one of the key drivers of fibrosis. TGF-β production correlates with the progression of liver, lung, kidney, skin and cardiac fibrosis, and inhibition of the TGF-β1 signaling pathway has been shown to reduce the development of fibrosis in many experimental models. In addition to its role as a profibrotic cytokine that can directly induce the differentiation of fibroblasts into collagen-secreting myofibroblasts, TGF-β1 is now widely described as a multifunctional cytokine with broad modulatory activities that affect numerous important biological pathways. These include pathways involved in the regulation of embryogenesis, immunity, carcinogenesis, cell proliferation and migration, wound healing, inflammation and fibrosis, among others37. Studies have suggested that the cellular source of TGF-β1 dictates its activity, with TGF-β1 derived from macrophages generally showing wound-healing and profibrotic activity and TGF-β1 secreted from CD4+ T regulatory cells (Treg cells) functioning as an anti-inflammatory and antifibrotic mediator38. Mice that are deficient in TGF-β1 develop numerous autoimmune disorders and are more susceptible to cancer. Therefore, it remains unclear whether antagonism of the TGF-β1 signaling pathway will prove beneficial in humans, as patients with progressive fibrosis are likely to require prolonged antifibrotic therapy.
Alternatively activated M2 macrophages suppress fibrosis. A unique functional subset of macrophages, called M2 or alternatively activated macrophages, has also been implicated in the pathogenesis of fibrosis39. Gordon and his colleagues first noted that macrophages activated with IL-4 (or IL-13) develop an alternative activation state that is distinct from that of classically activated macrophages (M1) induced by interferon-γ (IFN-γ)40. In vitro and in vivo studies in mice have shown that this phenotype is characterized by elevated expression of the mannose receptor (CD206), chitinase-3–like protein-3 (Chi3l3, also known as Ym1), resistin-like molecule-α (Relm-α, also known as FIZZ-1), major histocompatibility complex class II antigens and the enzyme arginase-1 (Arg1). Expression of Arg1 by M2 cells is of particular interest because this enzyme controls L-proline production, which is required for collagen synthesis by activated myofibroblasts41. M2 cells have also been implicated in the development of T helper type 2 (TH2) effector responses, production of fibrogenic cytokines, suppression of M1 responses and recruitment of fibrocytes42,43. Because they are commonly observed during the peak of the profibrotic immune response, M2 macrophages have been hypothesized to be important inducers of wound healing and fibrosis. However, mechanistic studies conducted with LysMCreIL-4Rα−/flox mice, in which Cre-mediated recombination results in deletion of the IL-4Rα chain in the myeloid cell lineage, including in M2 cells, have demonstrated that M2 macrophages are not required for the development of TH2 responses or liver fibrosis following infection with Schistosoma mansoni44. In fact, related studies done in mice with a conditional Arg1 deficiency suggested that Arg1-expressing M2 cells are required for the suppression and resolution of fibrosis45. These later experiments suggested that M2 cells compete with TH2 cells and fibroblasts for L-arginine, which is required for the production of polyamines and L-proline, which regulate cell growth and collagen synthesis, respectively. The inhibitory activity of M2 cells is also consistent with studies showing them to be important inducers of Treg cells46, which have also been implicated in the suppression of fibrosis. Thus, rather than promoting fibrosis, M2 cells seem to exploit multiple mechanisms to inhibit ECM synthesis by myofibroblasts. Given this modified view of the role of M2 macrophages, it will be important to investigate whether M2 cells have a similar inhibitory role in other models of fibrosis.
These findings suggest that M2 cells could be exploited to ameliorate fibrotic disease. Indeed, drugs that modulate the activation status of macrophages could emerge as a general therapeutic strategy to treat fibrosis. For example, peroxisome proliferator–activated receptor-α (PPAR-α) and PPAR-γ are induced in inflammatory macrophages, and endogenous and synthetic ligands that engage these receptors have been shown to inhibit the development of fibrosis by inducing the differentiation of alternatively activated macrophages that decrease proinflammatory cytokine production by M1 cells47,48. PPAR ligands also directly inhibit fibroblast activation induced by TGF-β1 (ref. 49). In support of these observations, PPAR-α and PPAR-γ activators have proven efficacious in a variety of experimental models of fibrosis, including hypertension-induced cardiac fibrosis, bile duct ligation–induced liver fibrosis, unilateral ureteral obstruction–induced renal interstitial fibrosis and bleomycin-induced pulmonary fibrosis50,51,52,53. The M2 macrophages that develop spontaneously during helminth infection have also been shown to reduce inflammation and slow the progression of liver fibrosis44,45. Thus, the conditional IL-4Rα– and Arg1-deficient mice described above will be important tools to further investigate the therapeutic potential of M2 cells in fibrotic disease.
Other innate myeloid-lineage cells have roles in fibrosis. Although it is widely recognized that monocytes, macrophages and neutrophils have important roles in the progression and resolution of fibrosis39, other myeloid-lineage cells (such as mast cells, eosinophils and basophils) have also been implicated in the pathogenesis of fibrosis in multiple organ systems and are viewed as potential therapeutic targets. For example, studies in mice have identified mast cells as inducers of systemic sclerosis, renal fibrosis after kidney transplantation and left-ventricular fibrosis in hypertensive hearts. Mechanistic studies in rats have suggested that mast cells promote fibrosis by recruiting inflammatory leukocytes and by producing profibrotic mediators54. Eosinophils seem to function in a similar fashion and are considered to be important sources of TGF-β1 and IL-13 (refs. 55,56). Although eosinophils have been most commonly associated with the development of pulmonary fibrosis57, increased numbers of eosinophils have also been linked with the activation of myofibroblasts in skin and liver fibrosis as well as idiopathic retroperitoneal fibrosis55,58. Bronchoalveolar-lavage eosinophilia has also been identified as a predictive biomarker of progressive lung disease in IPF and pulmonary fibrosis associated with collagen vascular disorder59. Finally, although basophils have a less clear role in the development of fibrosis than the other myeloid-cell populations, they have been implicated in the pathogenesis of myelofibrosis and are frequently found in greater numbers in patients with interstitial lung disease60. Basophils are also thought to be an important source of type 2 cytokines, suggesting that they might serve as important drivers of IL-4– and/or IL-13–dependent fibrosis.
Adaptive immunity and fibrosis
TH17-type immunity is proinflammatory and profibrotic. CD4+ T cells are divided into unique subsets on the basis of the cytokines they secrete and their distinct functional abilities (Fig. 3). The CD4+ TH17 cell subset that expresses the proinflammatory cytokine IL-17A is emerging as an important driver of fibrosis. IL-17A expression has been implicated in the pathogenesis of pulmonary fibrosis61, chronic allograft rejection62, fibrosis in orthotopic lung transplantation63, myocardial fibrosis64 and hepatitis-induced hepatic fibrosis65. In many cases, IL-17A expression is associated with persistent neutrophilia66, and it has been suggested that exaggerated neutrophil recruitment contributes to the development of tissue damage and fibrosis by inducing apoptosis in vascular endothelial cells67. Neutrophil recruitment is also an important predictor of early mortality in IPF patients68. Mechanistic studies investigating the IL-17 pathway of fibrosis in mice have identified the proinflammatory cytokines IL-1β and IL-23 as important upstream initiators of profibrotic TH17 responses61,69. A link between IL-17A and TGF-β1 has also been identified61. In addition to its role in promoting neutrophilic inflammation, IL-17A has been shown to directly induce expression of matrix metalloproteinase-1 in primary human cardiac fibroblasts70, suggesting that IL-17A promotes fibrosis by both exacerbating the upstream inflammatory response and regulating the downstream activation of fibroblasts. Together, these data identify the IL-1β–IL-17A–TGF-β1 cytokine axis as an important pathway in inflammation-driven fibrosis.

Naive CD4+ T cells differentiate into various distinct functional lineages driven by cues produced by injured epithelial cells and activated antigen presenting cells (dendritic cells and macrophages). Intracellular infections trigger IL-12–driven TH1 responses that produce IFN-γ, which activates microbicidal and cytotoxic activities that aid in pathogen clearance. Extracellular bacteria and certain fungi can lead to inflammasome activation and IL-6 production and can, in the presence of TGF-β1, drive TH17 differentiation. IL-17 from TH17 cells helps recruit neutrophils to clear the infection and exacerbates inflammation. Infection with extracellular, tissue-dwelling helminth parasites drives TH2 differentiation, with IL-4, IL-25, IL-33 and thymic stromal lymphopoietin (TSLP) from innate and epithelial sources guiding the differentiation of CD4+ TH2 cells. IL-13, when not competed for by the higher-affinity decoy receptor IL-13Rα2, binds its signaling receptor IL-13Rα1, leading to alternative activation of macrophages as well as epithelial apoptosis and myofibroblast activation. Treg cells are crucial in limiting the magnitude of TH cell responses and thereby ensure proper regulation of the wound-healing response. There is also a great deal of cross-regulation among TH cell subsets. For example, IL-13 suppresses TH17 differentiation, whereas IFN-γ can suppress IL-13–induced fibrosis by inducing classical macrophage activation and suppressing IL-13 and TGF-β1–induced collagen synthesis in myofibroblasts. Rx, pathways being targeted for therapy, either preclinically or in clinical trials; iNOS, inducible nitric oxide synthase; ROS, reactive oxygen species; PMN, polymorphonuclear leukocyte; MUC5AC, mucin-5AC.
TH2-type immunity is a potent driver of progressive fibrosis. Numerous studies have suggested that the type 2 cytokine response is a key driver of progressive fibrosis71. TH2 responses are defined by the production of IL-4, IL-5 and IL-13 (ref. 72), and although all three have been linked to the development of fibrosis55,73,74, IL-13 has emerged as a dominant mediator of fibrotic tissue remodeling in several experimental and natural models of fibrosis74. IL-13 production has been implicated in the development of fibrosis in chronic asthma75, IPF76, models of experimental lung fibrosis77, systemic sclerosis78, atopic dermatitis–induced skin fibrosis79, radiation-induced fibrosis80, fibrosis associated with ulcerative colitis81 and liver fibrosis resulting from persistent infections and NASH82,83. Mechanistically, IL-13 has been hypothesized to induce fibrosis by stimulating the production and activation of TGF-β84. However, other studies have suggested that IL-13 can promote fibrosis independently of TGF-β85,86 by directly activating the synthetic and proliferative properties of fibroblasts, epithelial cells and smooth-muscle cells87,88. Consequently, unlike IL-17A—which seems to promote fibrosis indirectly by inducing tissue damage and inflammation—IL-13 and TGF-β show direct fibrotic activity. TH2 cells that produce IL-13 and Treg cells that express TGF-β are also known to inhibit TH17 responses89, suggesting dual roles for IL-13 and TGF-β in the wound-healing response, as both cytokines suppress inflammation while promoting fibrosis. The profibrotic activity of IL-13 is controlled by the abundance of the IL-13Rα1 signaling receptor and IL-13Rα2 decoy receptor expressed on important target cells such as myofibroblasts90,91. When decoy receptor expression is low or absent, IL-13–dependent fibrosis is exacerbated92. However, mice deficient in IL-13Rα2 are more resistant to IL-1β– and IL-17–driven inflammation, probably because of the enhanced IL-13 activity89, suggesting that IL-13Rα2 functions as a key regulator of both TH17-mediated inflammation and TH2-driven fibrosis93.
TH1-type immunity shows antifibrotic activity. Numerous studies have suggested that TH1 effector T cells, defined by their production of IFN-γ, have antifibrotic activity94,95,96. IFN-γ–producing natural killer and natural killer T cells have also been shown to have similar roles97,98. Mechanistically, IFN-γ is believed to inhibit fibrosis, at least in part, by antagonizing the profibrotic activity of TGF-β1. IFN-γ inhibits the TGF-β–induced phosphorylation of the signal transducer Smad3 and subsequent activation of TGF-β–responsive genes99. IFN-γ also acts through a pathway dependent on Janus-associated kinase (Jak1) and the transcription factor Stat1 and induces expression of Smad7, which can prevent the interaction of Smad3 with the TGF-β receptor, thus further attenuating TGF-β–induced signaling. IFN-γ also directly inhibits fibroblast proliferation, TGF-β1–induced expression of the genes encoding procollagen I and procollagen III, and collagen synthesis in activated myofibroblasts100. IFN-γ also prevents the TH2 cytokine–induced differentiation of CD14+ peripheral blood monocytes into fibroblast-like cells called fibrocytes, which are believed to participate in the development of fibrosis in many organ systems101. Finally, by virtue of its ability to stimulate IFN-γ production in TH1 and natural killer cells, IL-12 has shown similar antifibrotic activity in vivo in mice71,102. But despite an abundance of in vitro and in vivo evidence supporting an antifibrotic role for TH1-type immunity, clinical studies investigating the therapeutic potential of IFN-γ in the treatment of IPF, systemic sclerosis and other fibrotic disorders have so far been mostly disappointing103.
Treg cells either suppress or promote fibrosis. Treg cells are induced in a variety of fibrotic diseases, but their role in tissue fibrogenesis is less clear than that of some of the other subsets of TH cells. Foxp3-expressing Treg cells are important producers of immunosuppressive cytokines that control Treg cell function, such as IL-10 and TGF-β1. Thus, one might predict that if Treg cells were present in sufficient numbers, they would inhibit progressive fibrotic disease by negatively regulating the inflammatory response that drives ECM deposition. Indeed, several groups exploring the mechanisms of fibrosis have documented a suppressive role for Treg cells in tissue fibrogenesis. Treg cells have been linked to the amelioration of fibrosis in IPF104, dystrophic mouse muscle105, cardiovascular disease106, chronic graft-versus-host disease–induced lupus nephritis107 and chronic HCV- and HIV-induced liver fibrosis108. One study has suggested that Treg cells protect mice from fibrosis by secreting IL-10 (ref. 38). However, Treg cells are also an important source of TGF-β1, with at least a few studies demonstrating that TGF-β–producing Treg cells induce rather than inhibit fibrosis109,110. Therefore, it remains uncertain whether Treg cells can be effectively exploited to ameliorate progressive fibrotic disease. It is unclear why Treg cells would have antifibrotic activity in some situations and profibrotic activity in others, but the development and recruitment of Treg cells relative to other effector T cell populations is likely to have a decisive role. It also seems plausible that Treg cells could suppress TH17- and TH 2-driven fibrosis but exacerbate TGF-β1–dependent fibrosis.
Intrinsic, autocrine and epigenetic mechanisms regulate fibrosis
Mechanical modifications to the ECM and cell-intrinsic changes in fibroblasts and epithelial cells have also been shown to contribute to the progression of fibrosis by maintaining the activation of key fibrogenic pathways.
Mechanisms that perpetuate myofibroblast activation. The Wnt–β-catenin signaling pathway, which regulates cell growth and is intimately involved in tumorigenesis, is constitutively activated in alveolar epithelial type II (ATII) cells in mouse models of pulmonary fibrosis and in patients diagnosed with IPF and chronic obstructive pulmonary disease111. Activation of Wnt–β-catenin signaling has also been reported in kidney, liver, skin and cardiac fibrosis112,113. Mechanistically, Wnt-1–inducible signaling protein 1 (WISP-1) has been shown in mice to increase the proliferation of ATII cells, promote EMT in the lung and kidney and mediate TGF-β1–driven renal fibrosis114 (Fig. 4). WISP-1 also increases the synthesis of ECM components in mouse and human lung fibroblasts and regulates activation of hepatic stellate cells in the liver115. Blocking studies demonstrated that bleomycin-induced pulmonary fibrosis is highly dependent on the Wnt-1 pathway116. Thus, Wnt signaling has emerged as a potential therapeutic target for IPF and several other chronic fibrotic disorders.

In mouse models of renal and hepatic fibrosis, up to 40% of α-SMA–positive, collagen-secreting myofibroblasts have been shown to arise from the differentiation of local epithelial progenitors via EMT. Repression of the transcription factors Snail1, Snail2, Zeb1 and Zeb2 are important for the maintenance of epithelial morphology. Several factors that are upregulated in the context of inflammation, including nuclear factor-κB (NF-κB), TGF-β1, bone morphogenetic proteins (BMPs), Wnt and Notch signaling proteins, can activate the Snail-Zeb pathway, leading to mesenchymal differentiation in these cells. Rx, pathways being targeted for therapy, either preclinically or in clinical trials; HA, hyaluronic acid; FSP-1, fibroblast-specific protein-1.
As tissues become more fibrotic, the increased tissue stiffness and decreased elasticity result in mechanical stress, which has been shown to exacerbate tissue injury and perpetuate the activation of local fibroblasts expressing α-smooth muscle actin (α-SMA)117. Two recent in vitro studies in mouse and porcine cells have suggested that mechanical stress contributes to aberrant wound healing and fibrosis by inducing EMT in alveolar type II epithelial cells via a mechanism driven by TGF-β1, Wnt–β-catenin and hyaluronan118,119. Fibroblasts that are activated as a result of increased tissue or substrate stiffness also seem to maintain their activated phenotype when returned to healthy 'soft' tissues120, suggesting that mechanical sensing by fibroblasts can permanently alter their behavior in favor of a fibrotic phenotype. Indeed, it has been suggested that the differentiation of fibroblasts into ECM-producing myofibroblasts is controlled by the combined actions of IL-1β, TGF-β1 and mechanical tension121. Increased compression, shear forces and hydrostatic pressures associated with portal hypertension and vascular remodeling can also perpetuate myofibroblast activation.
Epigenetic modifications in fibroblasts also contribute to the pathogenesis of fibrosis by stably altering the activation status of myofibroblasts. A recent genome-wide methylation scan of fibroblasts revealed several DNA methylation modifications that were unique to collagen-secreting myofibroblasts obtained from fibrotic mouse kidneys122. One of the modifications contributed to the epigenetic silencing of Rasal1 (encoding a suppressor of the Ras oncoprotein), which increased Ras activity and led to spontaneous, growth factor–independent proliferation of fibroblasts. These findings were important because they suggested that intrinsic changes in fibroblasts could lead to the uncontrolled expansion of myofibroblasts seen in some cancers. They also provided a new molecular explanation for the sustained and heritable activation of fibroblasts that is often seen in IPF and other highly progressive fibrotic diseases. On the basis of these observations, it may be predicted that the targeted repression of known antifibrotic genes by hypermethylation could contribute to the development of fibrosis, as was recently documented in IPF123. DNA methyltransferase inhibitors were found to reverse epigenetic modifications and protect mice from pulmonary fibrosis124. Therefore, because epigenetic modifications seem to be reversible, they could emerge as important targets for antifibrotic therapy.
Endoplasmic reticulum stress and the unfolded protein response. Intrinsic mechanisms that contribute to cell death have also been hypothesized to regulate the progression of fibrosis. Endoplasmic reticulum (ER) stress leading to apoptosis of key structural cells, such as epithelial cells, endothelial cells and hepatocytes, is believed to be an important driver of fibrosis. For example, excess accumulation of unfolded or misfolded proteins in the ER activates cellular stress pathways. The stress response, in turn, activates a complex counterresponse, which is typically protective for the cell. However, if the counter-response is delayed, inadequate or associated with substantial mitochondrial dysfunction, it can lead to apoptosis and sustained activation of the wound-healing response. Hepatocyte apoptosis driven by ER stress is believed to play a major part in the pathogenesis of liver fibrosis in NASH125. ER stress leading to apoptosis in alveolar epithelial cells is also thought to have a key role in sporadic IPF126,127. Therefore, a better understanding of the mechanisms that induce ER stress or lead to its resolution could reveal new strategies to prevent or treat progressive fibrotic disease.
Telomere shortening in mesenchymal cells promotes injury leading to fibrosis. Telomere shortening has been hypothesized to be an important contributor to the development of adult-onset and familial forms of pulmonary fibrosis128,129. Indeed, circulating peripheral leukocytes with shortened telomeres are found in a large percentage of sporadic, familial and idiopathic cases of pulmonary fibrosis. This shortening has been attributed to mutations in the gene encoding telomerase reverse transcriptase (TERT), an enzyme crucial to the maintenance of telomere length in adult stem cells130,131,132. Alveolar type 2 cells and lung-resident mesenchymal stem cells express telomerase, and mutations in this enzyme have been shown to contribute to their premature death, resulting in decreased lung regeneration after injury. Telomerase-null mice also show compromised lung alveolar integrity and decreased myofibroblast differentiation as a result of telomere shortening133,134. Profibrotic mediators, such as TGF-β1 and reactive oxygen intermediates, also participate in telomere shortening135,136, suggesting that a vicious cycle of profibrotic-mediator production, impaired telomerase activity and increased senescence of stem cells and/or AT2 cells contributes to the pathogenesis of fibrosis. Nevertheless, studies with TERT-deficient mice have raised questions about whether TERT has only a protective role in fibrosis, as telomerase activity was found to be necessary for development of bleomycin-induced pulmonary fibrosis in mice137.
MicroRNAs regulate fibroblast growth and activation. MicroRNAs (miRNAs) include a broad class of small evolutionarily conserved noncoding RNAs that have important roles in a variety of pathophysiological processes by blocking translation or promoting degradation of complementary target mRNAs. Although unique subsets of miRNAs have been identified in various fibrotic diseases, a much smaller subset of miRNAs have emerged as key regulators of the fibrotic process. For example, miR-21 is expressed in the lungs of individuals with IPF, and mice treated with miR-21 antisense probes were protected from bleomycin-induced pulmonary fibrosis138 and cardiac fibrosis induced by pressure overload139. Mechanistically, miR-21 is thought to promote fibrosis by regulating TGF-β1 and MAP kinase signaling in activated myofibroblasts138,139. miR-21 suppresses several tumor suppressor genes, suggesting that MIR21 may also function as an oncogene. miR-21 also operates as an antiapoptotic factor in tumor cells140. Aberrant expression of miR-21 or other key miRNAs in fibroblasts could, therefore, contribute to their survival and/or differentiation into pathogenic ECM-producing myofibroblasts. Nevertheless, a recent study with miR-21–null mice has raised questions about the overall importance of miR-21 in pathological cardiac remodeling141. In rat and mouse studies, miR-192, miR-216a and miR-217 have, like miR-21, been identified as key triggers of fibrosis driven by TGF-β and Smad3 (refs. 142,143). miR-29 has also been identified in human and mouse studies as a potential therapeutic target in systemic sclerosis, liver fibrosis and cardiac fibrosis144,145,146. miR-29 seems to promote fibrosis in human cells by directly regulating type I collagen expression147. Other miRNAs are constitutively expressed in healthy tissues but are downregulated as fibrosis develops, suggesting that they might have a protective antifibrotic role such as that shown recently for the miRNA let-7d in IPF148. Decreases in miR-133 and miR-30 have also been linked to increased activity of connective tissue growth factor (CTGF), which resulted in increased myocardial matrix remodeling149. Downregulation of miRNA-150 and miRNA-194 in hepatic stellate cells has been hypothesized to contribute to stellate cell activation and increased ECM production in the liver150. Finally, expression of miR-200a has been shown to prevent renal fibrogenesis by repressing TGF-β2 expression151. Thus, identifying specific miRNAs that inhibit important fibrogenic pathways or promote tissue healing or regeneration could prove highly beneficial in the treatment of fibrosis148. Indeed, the targeting of specific miRNAs with small-molecule inhibitors or miRNA mimics could be a general strategy to treat fibrotic disease.
Key fibrogenic pathways for therapeutic targeting
Mechanistic studies of tissue fibrogenesis have revealed a variety of potential therapeutic approaches to combating progressive fibrotic disease. Some of the unique strategies and targets that have progressed to the clinical-trial phase are discussed briefly in the following sections.
Myofibroblasts and the TGF-β pathway. Because activated myofibroblasts are the key pathogenic cells in all fibrotic diseases, a number of experimental antifibrotic strategies are attempting to target activation, proliferation and/or recruitment of fibroblasts (Box 1 and Table 1). Pirfenidone, marketed under the names Esbriet and Pirespa, is the first targeted antifibrotic drug to be approved for the treatment of IPF in Europe and Japan152. Although its exact mechanism of action remains unclear, pirfenidone is believed to attenuate fibroblast proliferation and the production by activated myofibroblasts of fibrosis-associated mediators and ECM components. Pirfenidone has also shown efficacy in preclinical models of liver fibrosis, renal fibrosis, hypertrophic cardiomyopathy and radiation-induced fibrosis, suggesting that it may have broad antifibrotic activity. Therapeutic antibodies to TGF-β1 (ref. 153), a key cytokine involved in the activation of myofibroblasts; CTGF, a matrix-associated, heparin-binding protein that mirrors the profibrotic activity of TGF-β on fibroblasts; and integrin αvβ6, which is responsible for the activation of constitutively expressed latent TGF-β154, are also being investigated for their antifibrotic activity. A humanized monoclonal antibody to αvβ6 developed by Stromedix and Biogen Idec is being investigated as a treatment for interstitial fibrosis and tubular atrophy in kidney-transplant recipients and as a therapy for IPF155. Genzyme (owned by Sanofi-Aventis) is also exploring a humanized pan–TGF-β inhibitor (fresolimumab) as a treatment for patients with early-stage diffuse systemic sclerosis, focal segmental glomerulosclerosis, IPF and myelofibrosis156,157, and antibodies and antisense drugs targeting CTGF are being investigated in IPF and scar-revision surgery158. Antagonists of the lysophosphatidic acid-1 receptor, a growth factor that induces CTGF and TGF-β1 expression, are being considered as treatments for kidney fibrosis, IPF and systemic sclerosis159,160. Bone morphogenetic protein-7 has also been identified as a potential therapeutic agent for chronic renal injury because it can counteract TGF-β1–induced EMT161. An antagonist of the endothelin receptor, which promotes myofibroblast contraction and migration, is being explored in cardiovascular disease and IPF162. A humanized monoclonal antibody targeting lysyl oxidase–like-2, an enzyme that catalyzes the cross-linking of collagen, is being explored by Gilead Sciences as a treatment for cardiac fibrosis, IPF and liver fibrosis163. Other matrix assembly proteins, such as prolyl hydroxylases, are being investigated preclinically for antifibrotic activity164. Bortezomib, a proteasomal inhibitor approved by the US Food and Drug Administration, inhibits TGF-β1 signaling in vitro and has been shown to protect mice from bleomycin-induced skin and lung fibrosis165. It also induces apoptosis of hepatic stellate cells166. Consequently, bortezomib may prove efficacious for diseases in which TGF-β1, ER stress and activated myofibroblasts have been identified as key pathogenic mediators. Studies are also under way to examine whether a serine/threonine protein kinase inhibitor reduces the number of circulating fibrocytes in individuals with IPF167. Because ECM-producing myofibroblasts are the final common pathogenic cell in all fibrotic diseases, any therapy that successfully ablates their activity could have broad antifibrotic activity.
Proinflammatory pathways. Targeting key inflammatory pathways might also prove beneficial in the treatment of fibrosis. Because TNF-α has emerged as a key driver of fibrosis in many experimental studies, clinical trials have been initiated to examine whether inhibitors of the TNF-α pathway could be used to treat IPF and other scarring disorders29. Although the small-molecule tyrosine kinase inhibitor imatinib mesylate (Gleevec), which inhibits signaling of PDGF receptor and vascular endothelial growth factor receptor, did not affect survival or lung function in patients with IPF168, ongoing trials are investigating its potential as a treatment for nephrogenic systemic fibrosis169. Boehringer Ingelheim also has ongoing clinical studies with another investigational tyrosine kinase inhibitor, BIBF 1120, which showed promise in slowing lung-function decline in patients with IPF170. Inhibitors of Jaks, a family of intracellular, nonreceptor tyrosine kinases that transduce cytokine-mediated signals via the Jak-Stat pathway, are also being investigated for their therapeutic activity in liver fibrosis and myelofibrosis171. Finally, given that recent studies have directly linked IL-17A with TGF-β1 (ref. 61), therapeutic antibodies that disrupt IL-17 signaling might also prove beneficial for the treatment of fibrosis. Targeting upstream proinflammatory pathways would probably be required in situations in which sustained inflammation is a major driving force in the development of fibrosis. However, it is important to point out that although IFN-γ is a well-known antifibrotic cytokine, IFN-γ1b showed little efficacy in the treatment of IPF or chronic HCV-induced liver fibrosis172,173. Numerous anti-inflammatory drugs, including corticosteroids, immunomodulatory agents and cytotoxic drugs, have also been tested and found to have little to no therapeutic benefit in IPF or other fibrotic diseases, so it remains uncertain whether the newer class of 'anti-inflammatory' drugs will ultimately prove beneficial for fibrosis.
The profibrotic type 2 immune response. The TH2-associated cytokine IL-13 has emerged as a key driver of infection and allergen-driven fibrosis74,82,84. IL-13 and its receptors have also been detected at high levels in the lungs and blood of patients with IPF76,174. On the basis of these results, Novartis has initiated clinical studies to determine whether humanized IL-13–specific monoclonal antibodies could be used to treat IPF. Sanofi is also exploring the role of type 2 cytokines in IPF using an antibody that targets both IL-4 and IL-13. Support for the potential utility of an IL-13 blocker comes from a recent study in which lebrikizumab, a humanized monoclonal antibody to IL-13 developed by Genentech, was shown to be effective in a subset of adults with poorly controlled asthma175. Patients showing the greatest improvement in lung function after lebrikizumab therapy had high pretreatment levels of serum periostin, an IL-13–induced protein176. Thus, it will be interesting to determine whether this circulating biomarker could be used to identify individuals with IL-13–dependent fibrosis who might also benefit from anti–IL-13 therapy. Because a growing number of chronic fibrotic diseases are characterized by the excess production of IL-13 and/or increased expression of IL-13–inducible genes, many individuals with fibrosis might benefit from the neutralization of IL-13.
Myeloid cells and Treg cells. Numerous studies have suggested that macrophages have stage-specific roles in fibrosis2. Macrophages show profibrotic activity in the early phases of the wound-healing response by producing inflammatory mediators that can exacerbate tissue injury, such as IL-1β, TNF-α and reactive oxygen and nitrogen species. They also produce profibrotic mediators such as TGF-β1. But in the later stages of the wound-healing response, a subset of macrophages converts into a suppressive phenotype that expresses a variety of anti-inflammatory mediators, such as IL-10, Arg1, programmed death ligand-2 and Relm-α, which promote wound healing and direct the resolution of the inflammatory response46. Given the central role that macrophages have in fibrosis, a number of experimental antifibrotic strategies are being designed to regulate the activation and/or recruitment of distinct myeloid-cell populations. For example, Johnson & Johnson is conducting a phase 1 study in individuals with IPF of a human monoclonal antibody to CCL2 (also known as MCP-1), which is involved in the recruitment of inflammatory monocytes177. Researchers at Promedior are investigating the protective effects of recombinant human serum amyloid P (SAP; also known as pentraxin 2) in IPF and post-surgical scarring in patients treated for glaucoma178. Their preclinical studies have suggested that SAP reduces inflammation and fibrosis by inducing the production of IL-10 in 'proresolution' macrophages179. Ono Pharmaceutical is examining whether a competitive inhibitor of human neutrophil elastase would be beneficial in the treatment of pulmonary fibrosis and idiopathic interstitial pneumonia. Inhibitors of histone deacetylases, enzymes that regulate gene transcription, have been hypothesized to prevent cardiac fibrosis by inducing Treg cells and reducing numbers of fibrocytes180. Finally, ImmuneWorks is examining whether Treg cells could be induced to treat IPF. It has been suggested that IPF is an autoimmune disease driven by an immune response against collagen type V181. ImmuneWorks investigators are examining whether an orally administered antigen will induce oral tolerance by expanding a population of collagen type V–specific Treg cells104. Thus, therapies that modulate the activation and/or recruitment of Treg cells or distinct macrophage populations could emerge as viable treatments for fibrotic disease. These strategies are particularly attractive because they are based on alteration of the character of the immune response rather than simple disruption of it, which could have a more durable antifibrotic effect.

Conclusions and future directions
There are many distinct immunological and molecular mechanisms that can contribute to the progression of fibrotic disease. Dysregulated innate and adaptive immune responses are major contributors to fibrosis. However, cell-intrinsic modifications in fibroblasts and other structural cells can also contribute to fibrosis and should be considered in the design and testing of new antifibrotic therapies. Because fibrosis is often characterized by the activation of several profibrotic pathways, a multipronged approach will probably be needed to slow the progression of fibrosis. Therapeutics that target key epigenetic modifications or miRNAs could emerge as viable strategies because they regulate large families of genes rather than a single target. However, a more integrated antifibrotic strategy that simultaneously targets important inflammatory mediators, profibrotic cytokines and epigenetic and cell and/or tissue intrinsic changes will probably emerge as the most successful way to treat this highly complex and difficult-to-manage pathology. Thus, we need to begin devising and testing strategies that incorporate combination therapies. One of the major obstacles slowing the development of antifibrotic drugs is the lack of disease-specific biomarkers that can be used to identify patients who might benefit from a specific therapy. Therefore, it will be important to incorporate genetic and biological phenotyping in the clinical staging of patients diagnosed with fibrosis, so that more homogenous patient populations can be selected for specific trials. It will also be important to design clinical trials with well-defined clinical endpoints and to develop techniques that can quickly, noninvasively and accurately evaluate the efficacy of new antifibrotic therapies. Examples of such techniques are the 6-minute walk test, measurements of forced vital capacity and patient-recorded outcomes (such as dyspnea and quality-of-life changes) used in some IPF trials. New and improved preclinical models that more closely replicate fibrotic diseases in humans are also needed, as was recently demonstrated in a mouse model of liver cirrhosis92. Studies are increasingly suggesting that, although fibrosis is often linked with a strong inflammatory response initially, there are specific mediators and pathways contributing to the pathogenesis of fibrosis that are distinct from the mechanisms driving inflammation. Thus, to design effective therapeutics for fibrotic disease, we need to begin viewing fibrosis as a pathological process distinct from inflammation. Further, because inflammatory mediators are intimately involved in wound repair and regulate both the initiation and resolution of fibrosis, we need to figure out how to harness the beneficial aspects of inflammation so that fibrosis can be slowed or reversed and normal tissue regenerated, which is the ultimate goal of all fibrosis research.
References
Bataller, R. & Brenner, D.A. Liver fibrosis. J. Clin. Invest. 115, 209–218 (2005).
Wynn, T.A. Integrating mechanisms of pulmonary fibrosis. J. Exp. Med. 208, 1339–1350 (2011).
Wynn, T.A. Cellular and molecular mechanisms of fibrosis. J. Pathol. 214, 199–210 (2008).
Gabbiani, G. The myofibroblast in wound healing and fibrocontractive diseases. J. Pathol. 200, 500–503 (2003).
Kalluri, R. & Zeisberg, M. Fibroblasts in cancer. Nat. Rev. Cancer 6, 392–401 (2006).
Fujii, T. et al. Mouse model of carbon tetrachloride–induced liver fibrosis: histopathological changes and expression of CD133 and epidermal growth factor. BMC Gastroenterol. 10, 79 (2010).
Chen, J. & Stubbe, J. Bleomycins: towards better therapeutics. Nat. Rev. Cancer 5, 102–112 (2005).
Esmon, C.T. The interactions between inflammation and coagulation. Br. J. Haematol. 131, 417–430 (2005).
Barrientos, S., Stojadinovic, O., Golinko, M.S., Brem, H. & Tomic-Canic, M. Growth factors and cytokines in wound healing. Wound Repair Regen. 16, 585–601 (2008).
Chambers, R.C. Procoagulant signalling mechanisms in lung inflammation and fibrosis: novel opportunities for pharmacological intervention? Br. J. Pharmacol. 153, S367–S378 (2008).
Scotton, C.J. et al. Increased local expression of coagulation factor X contributes to the fibrotic response in human and murine lung injury. J. Clin. Invest. 119, 2550–2563 (2009).
Coughlin, S.R. Thrombin signalling and protease-activated receptors. Nature 407, 258–264 (2000).
Fiorucci, S. et al. PAR1 antagonism protects against experimental liver fibrosis. Role of proteinase receptors in stellate cell activation. Hepatology 39, 365–375 (2004).
Anstee, Q.M. et al. Coagulation status modulates murine hepatic fibrogenesis: implications for the development of novel therapies. J. Thromb. Haemost. 6, 1336–1343 (2008).
Wanless, I.R. et al. Hepatic and portal vein thrombosis in cirrhosis: possible role in development of parenchymal extinction and portal hypertension. Hepatology 21, 1238–1247 (1995).
Li, Y. et al. Severe lung fibrosis requires an invasive fibroblast phenotype regulated by hyaluronan and CD44. J. Exp. Med. 208, 1459–1471 (2011).
Duffield, J.S. et al. Selective depletion of macrophages reveals distinct, opposing roles during liver injury and repair. J. Clin. Invest. 115, 56–65 (2005).
Pardo, A. et al. Increase of lung neutrophils in hypersensitivity pneumonitis is associated with lung fibrosis. Am. J. Respir. Crit. Care Med. 161, 1698–1704 (2000).
Connolly, M.K. et al. In liver fibrosis, dendritic cells govern hepatic inflammation in mice via TNF-α. J. Clin. Invest. 119, 3213–3225 (2009).
Zhang, Y., Lee, T.C., Guillemin, B., Yu, M.C. & Rom, W.N. Enhanced IL-1β and tumor necrosis factor-α release and messenger RNA expression in macrophages from idiopathic pulmonary fibrosis or after asbestos exposure. J. Immunol. 150, 4188–4196 (1993).
Miyazaki, Y. et al. Expression of a tumor necrosis factor-α transgene in murine lung causes lymphocytic and fibrosing alveolitis. A mouse model of progressive pulmonary fibrosis. J. Clin. Invest. 96, 250–259 (1995).
Kolb, M., Margetts, P.J., Anthony, D.C., Pitossi, F. & Gauldie, J. Transient expression of IL-1β induces acute lung injury and chronic repair leading to pulmonary fibrosis. J. Clin. Invest. 107, 1529–1536 (2001).
Piguet, P.F., Collart, M.A., Grau, G.E., Sappino, A.P. & Vassalli, P. Requirement of tumour necrosis factor for development of silica-induced pulmonary fibrosis. Nature 344, 245–247 (1990).
Piguet, P.F., Collart, M.A., Grau, G.E., Kapanci, Y. & Vassalli, P. Tumor necrosis factor/cachectin plays a key role in bleomycin-induced pneumopathy and fibrosis. J. Exp. Med. 170, 655–663 (1989).
Piguet, P.F., Ribaux, C., Karpuz, V., Grau, G.E. & Kapanci, Y. Expression and localization of tumor necrosis factor-α and its mRNA in idiopathic pulmonary fibrosis. Am. J. Pathol. 143, 651–655 (1993).
Bahcecioglu, I.H. et al. Hepatoprotective effect of infliximab, an anti–TNF-α agent, on carbon tetrachloride–induced hepatic fibrosis. Inflammation 31, 215–221 (2008).
Nawroth, I. et al. Intraperitoneal administration of chitosan/DsiRNA nanoparticles targeting TNF-α prevents radiation-induced fibrosis. Radiother. Oncol. 97, 143–148 (2010).
Tomita, K. et al. Tumour necrosis factor-α signalling through activation of Kupffer cells plays an essential role in liver fibrosis of non-alcoholic steatohepatitis in mice. Gut 55, 415–424 (2006).
Raghu, G. et al. Treatment of idiopathic pulmonary fibrosis with etanercept: an exploratory, placebo-controlled trial. Am. J. Respir. Crit. Care Med. 178, 948–955 (2008).
Gasse, P. et al. IL-1R1/MyD88 signaling and the inflammasome are essential in pulmonary inflammation and fibrosis in mice. J. Clin. Invest. 117, 3786–3799 (2007).
Bujak, M. & Frangogiannis, N.G. The role of IL-1 in the pathogenesis of heart disease. Arch. Immunol. Ther. Exp. (Warsz.) 57, 165–176 (2009).
Jones, L.K. et al. IL-1RI deficiency ameliorates early experimental renal interstitial fibrosis. Nephrol. Dial. Transplant. 24, 3024–3032 (2009).
Kamari, Y. et al. Lack of interleukin-1α or interleukin-1β inhibits transformation of steatosis to steatohepatitis and liver fibrosis in hypercholesterolemic mice. J. Hepatol. 55, 1086–1094 (2011).
Fan, J.M. et al. Interleukin-1 induces tubular epithelial-myofibroblast transdifferentiation through a transforming growth factor-β1–dependent mechanism in vitro. Am. J. Kidney Dis. 37, 820–831 (2001).
Diaz, J.A. et al. Critical role for IL-6 in hypertrophy and fibrosis in chronic cardiac allograft rejection. Am. J. Transplant. 9, 1773–1783 (2009).
Natsume, M. et al. Attenuated liver fibrosis and depressed serum albumin levels in carbon tetrachloride–treated IL-6–deficient mice. J. Leukoc. Biol. 66, 601–608 (1999).
Verrecchia, F. & Mauviel, A. Transforming growth factor-β and fibrosis. World J. Gastroenterol. 13, 3056–3062 (2007).
Kitani, A. et al. Transforming growth factor (TGF)-β1–producing regulatory T cells induce Smad-mediated interleukin-10 secretion that facilitates coordinated immunoregulatory activity and amelioration of TGF-β1–mediated fibrosis. J. Exp. Med. 198, 1179–1188 (2003).
Wynn, T.A. & Barron, L. Macrophages: master regulators of inflammation and fibrosis. Semin. Liver Dis. 30, 245–257 (2010).
Gordon, S. & Taylor, P.R. Monocyte and macrophage heterogeneity. Nat. Rev. Immunol. 5, 953–964 (2005).
Hesse, M. et al. Differential regulation of nitric oxide synthase-2 and arginase-1 by type 1/type 2 cytokines in vivo: granulomatous pathology is shaped by the pattern of l-arginine metabolism. J. Immunol. 167, 6533–6544 (2001).
Song, E. et al. Influence of alternatively and classically activated macrophages on fibrogenic activities of human fibroblasts. Cell. Immunol. 204, 19–28 (2000).
Sun, L. et al. New concepts of IL-10–induced lung fibrosis: fibrocyte recruitment and M2 activation in a CCL2/CCR2 axis. Am. J. Physiol. Lung Cell Mol. Physiol. 300, L341–L353 (2011).
Herbert, D.R. et al. Alternative macrophage activation is essential for survival during schistosomiasis and downmodulates T helper 1 responses and immunopathology. Immunity 20, 623–635 (2004).
Pesce, J.T. et al. Arginase-1–expressing macrophages suppress TH2 cytokine-driven inflammation and fibrosis. PLoS Pathog. 5, e1000371 (2009).
Murray, P.J. & Wynn, T.A. Protective and pathogenic functions of macrophage subsets. Nat. Rev. Immunol. 11, 723–737 (2011).
Ricote, M., Li, A.C., Willson, T.M., Kelly, C.J. & Glass, C.K. The peroxisome proliferator–activated receptor-γ is a negative regulator of macrophage activation. Nature 391, 79–82 (1998).
Odegaard, J.I. et al. Macrophage-specific PPAR-γ controls alternative activation and improves insulin resistance. Nature 447, 1116–1120 (2007).
Kulkarni, A.A. et al. PPAR-γ ligands repress TGF-β–induced myofibroblast differentiation by targeting the PI3K/Akt pathway: implications for therapy of fibrosis. PLoS ONE 6, e15909 (2011).
Iglarz, M. et al. Peroxisome proliferator–activated receptor-α and receptor-γ activators prevent cardiac fibrosis in mineralocorticoid-dependent hypertension. Hypertension 42, 737–743 (2003).
Yang, L., Stimpson, S.A., Chen, L., Wallace Harrington, W. & Rockey, D.C. Effectiveness of the PPAR-γ agonist, GW570, in liver fibrosis. Inflamm. Res. 59, 1061–1071 (2010).
Kawai, T. et al. PPAR-γ agonist attenuates renal interstitial fibrosis and inflammation through reduction of TGF-β. Lab. Invest. 89, 47–58 (2009).
Aoki, Y. et al. Pioglitazone, a peroxisome proliferator–activated receptor-γ ligand, suppresses bleomycin-induced acute lung injury and fibrosis. Respiration 77, 311–319 (2009).
Levick, S.P. et al. Cardiac mast cells mediate left ventricular fibrosis in the hypertensive rat heart. Hypertension 53, 1041–1047 (2009).
Reiman, R.M. et al. Interleukin-5 (IL-5) augments the progression of liver fibrosis by regulating IL-13 activity. Infect. Immun. 74, 1471–1479 (2006).
Minshall, E.M. et al. Eosinophil-associated TGF-β1 mRNA expression and airways fibrosis in bronchial asthma. Am. J. Respir. Cell Mol. Biol. 17, 326–333 (1997).
Humbles, A.A. et al. A critical role for eosinophils in allergic airways remodeling. Science 305, 1776–1779 (2004).
Levi-Schaffer, F. et al. Human eosinophils regulate human lung- and skin-derived fibroblast properties in vitro: a role for transforming growth factor-β (TGF-β). Proc. Natl. Acad. Sci. USA 96, 9660–9665 (1999).
Peterson, M.W., Monick, M. & Hunninghake, G.W. Prognostic role of eosinophils in pulmonary fibrosis. Chest 92, 51–56 (1987).
Gilbert, H.S. Myelofibrosis revisited: characterization and classification of myelofibrosis in the setting of myeloproliferative disease. Prog. Clin. Biol. Res. 154, 3–17 (1984).
Wilson, M.S. et al. Bleomycin and IL-1β–mediated pulmonary fibrosis is IL-17A dependent. J. Exp. Med. 207, 535–552 (2010).
Faust, S.M. et al. Role of T cell TGF-β signaling and IL-17 in allograft acceptance and fibrosis associated with chronic rejection. J. Immunol. 183, 7297–7306 (2009).
Fan, L. et al. Neutralizing IL-17 prevents obliterative bronchiolitis in murine orthotopic lung transplantation. Am. J. Transplant. 11, 911–922 (2011).
Feng, W. et al. IL-17 induces myocardial fibrosis and enhances RANKL/OPG and MMP/TIMP signaling in isoproterenol-induced heart failure. Exp. Mol. Pathol. 87, 212–218 (2009).
Wang, L., Chen, S.J. & Xu, K.S. IL-17 expression is correlated with hepatitis B–related liver diseases and fibrosis. Int. J. Mol. Med. 27, 385–392 (2011).
Laan, M. et al. Neutrophil recruitment by human IL-17 via CXC chemokine release in the airways. J. Immunol. 162, 2347–2352 (1999).
Zhu, F. et al. IL-17 induces apoptosis of vascular endothelial cells—a potential mechanism for human acute coronary syndrome. Clin. Immunol. 141, 152–160 (2011).
Kinder, B.W. et al. Baseline BAL neutrophilia predicts early mortality in idiopathic pulmonary fibrosis. Chest 133, 226–232 (2008).
Gasse, P. et al. IL-1 and IL-23 mediate early IL-17A production in pulmonary inflammation leading to late fibrosis. PLoS ONE 6, e23185 (2011).
Cortez, D.M. et al. IL-17 stimulates MMP-1 expression in primary human cardiac fibroblasts via p38 MAPK- and ERK1/2-dependent C/EBP-β, NF-κB and AP-1 activation. Am. J. Physiol. Heart Circ. Physiol. 293, H3356–H3365 (2007).
Wynn, T.A. et al. An IL-12–based vaccination method for preventing fibrosis induced by schistosome infection. Nature 376, 594–596 (1995).
Wynn, T.A. Fibrotic disease and the TH1/TH2 paradigm. Nat. Rev. Immunol. 4, 583–594 (2004).
Ong, C., Wong, C., Roberts, C.R., Teh, H.S. & Jirik, F.R. Anti–IL-4 treatment prevents dermal collagen deposition in the tight-skin mouse model of scleroderma. Eur. J. Immunol. 28, 2619–2629 (1998).
Chiaramonte, M.G., Donaldson, D.D., Cheever, A.W. & Wynn, T.A. An IL-13 inhibitor blocks the development of hepatic fibrosis during a T helper type 2–dominated inflammatory response. J. Clin. Invest. 104, 777–785 (1999).
Yang, G. et al. Anti–IL-13 monoclonal antibody inhibits airway hyper-responsiveness, inflammation and airway remodeling. Cytokine 28, 224–232 (2004).
Murray, L.A. et al. Hyper-responsiveness of IPF/UIP fibroblasts: interplay between TGF-β1, IL-13 and CCL2. Int. J. Biochem. Cell Biol. 40, 2174–2182 (2008).
Kolodsick, J.E. et al. Protection from fluorescein isothiocyanate–induced fibrosis in IL-13–deficient, but not IL-4–deficient, mice results from impaired collagen synthesis by fibroblasts. J. Immunol. 172, 4068–4076 (2004).
Fuschiotti, P. Role of IL-13 in systemic sclerosis. Cytokine 56, 544–549 (2011).
Oh, M.H. et al. IL-13 induces skin fibrosis in atopic dermatitis by thymic stromal lymphopoietin. J. Immunol. 186, 7232–7242 (2011).
Han, G., Zhang, H., Xie, C.H. & Zhou, Y.F. TH2-like immune response in radiation-induced lung fibrosis. Oncol. Rep. 26, 383–388 (2011).
Heller, F., Fuss, I.J., Nieuwenhuis, E.E., Blumberg, R.S. & Strober, W. Oxazolone colitis, a TH2 colitis model resembling ulcerative colitis, is mediated by IL-13–producing NKT cells. Immunity 17, 629–638 (2002).
Weng, H.L. et al. The etiology of liver damage imparts cytokines transforming growth factor-β1 or interleukin-13 as driving forces in fibrogenesis. Hepatology 50, 230–243 (2009).
Shimamura, T. et al. Novel role of IL-13 in fibrosis induced by nonalcoholic steatohepatitis and its amelioration by IL-13R–directed cytotoxin in a rat model. J. Immunol. 181, 4656–4665 (2008).
Lee, C.G. et al. Interleukin-13 induces tissue fibrosis by selectively stimulating and activating transforming growth factor-β1. J. Exp. Med. 194, 809–822 (2001).
Liu, Y. et al. IL-13 induces connective tissue growth factor in rat hepatic stellate cells via TGF-β–independent Smad signaling. J. Immunol. 187, 2814–2823 (2011).
Kaviratne, M. et al. IL-13 activates a mechanism of tissue fibrosis that is completely TGF-β independent. J. Immunol. 173, 4020–4029 (2004).
Kuperman, D.A. et al. Direct effects of interleukin-13 on epithelial cells cause airway hyperreactivity and mucus overproduction in asthma. Nat. Med. 8, 885–889 (2002).
Lee, J.H. et al. Interleukin-13 induces dramatically different transcriptional programs in three human airway cell types. Am. J. Respir. Cell Mol. Biol. 25, 474–485 (2001).
Wilson, M.S. et al. Colitis and intestinal inflammation in IL10−/− mice results from IL-13Rα2–mediated attenuation of IL-13 activity. Gastroenterology 140, 254–264 e2 (2011).
Ramalingam, T.R. et al. Unique functions of the type II interleukin-4 receptor identified in mice lacking the interleukin-13 receptor-α1 chain. Nat. Immunol. 9, 25–33 (2008).
Chiaramonte, M.G. et al. Regulation and function of the interleukin-13 receptor-α2 during a T helper cell type 2–dominant immune response. J. Exp. Med. 197, 687–701 (2003).
Mentink-Kane, M.M. et al. Accelerated and progressive and lethal liver fibrosis in mice that lack interleukin (IL)-10, IL-12p40, and IL-13Rα2. Gastroenterology 141, 2200–2209 (2011).
Mentink-Kane, M.M. & Wynn, T.A. Opposing roles for IL-13 and IL-13 receptor-α2 in health and disease. Immunol. Rev. 202, 191–202 (2004).
Baroni, G.S. et al. Interferon-γ decreases hepatic stellate cell activation and extracellular matrix deposition in rat liver fibrosis. Hepatology 23, 1189–1199 (1996).
Giri, S.N., Hyde, D.M. & Marafino, B.J. Jr. Ameliorating effect of murine interferon-γ on bleomycin-induced lung collagen fibrosis in mice. Biochem. Med. Metab. Biol. 36, 194–197 (1986).
Oldroyd, S.D., Thomas, G.L., Gabbiani, G. & El Nahas, A.M. Interferon-γ inhibits experimental renal fibrosis. Kidney Int. 56, 2116–2127 (1999).
Kim, J.H. et al. Natural killer T (NKT) cells attenuate bleomycin-induced pulmonary fibrosis by producing interferon-γ. Am. J. Pathol. 167, 1231–1241 (2005).
Jeong, W.I., Park, O. & Gao, B. Abrogation of the antifibrotic effects of natural killer cells/interferon-γ contributes to alcohol acceleration of liver fibrosis. Gastroenterology 134, 248–258 (2008).
Ulloa, L., Doody, J. & Massague, J. Inhibition of transforming growth factor-β/SMAD signalling by the interferon-γ/STAT pathway. Nature 397, 710–713 (1999).
Gurujeyalakshmi, G. & Giri, S.N. Molecular mechanisms of antifibrotic effect of interferon-γ in bleomycin mouse model of lung fibrosis: downregulation of TGF-β and procollagen I and III gene expression. Exp. Lung Res. 21, 791–808 (1995).
Shao, D.D., Suresh, R., Vakil, V., Gomer, R.H. & Pilling, D. Pivotal advance: TH1 cytokines inhibit, and TH2 cytokines promote fibrocyte differentiation. J. Leukoc. Biol. 83, 1323–1333 (2008).
Keane, M.P., Belperio, J.A., Burdick, M.D. & Strieter, R.M. IL-12 attenuates bleomycin-induced pulmonary fibrosis. Am. J. Physiol. Lung Cell Mol. Physiol. 281, L92–L97 (2001).
King, T.E. Jr. et al. Effect of interferon-γ-1b on survival in patients with idiopathic pulmonary fibrosis (INSPIRE): a multicentre, randomised, placebo-controlled trial. Lancet 374, 222–228 (2009).
Kotsianidis, I. et al. Global impairment of CD4+CD25+FOXP3+ regulatory T cells in idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 179, 1121–1130 (2009).
Vetrone, S.A. et al. Osteopontin promotes fibrosis in dystrophic mouse muscle by modulating immune cell subsets and intramuscular TGF-β. J. Clin. Invest. 119, 1583–1594 (2009).
Kanellakis, P., Dinh, T.N., Agrotis, A. & Bobik, A. CD4+CD25+Foxp3+ regulatory T cells suppress cardiac fibrosis in the hypertensive heart. J. Hypertens. 29, 1820–1828 (2011).
Zhang, J.L. et al. CD3 mAb treatment ameliorated the severity of the cGVHD-induced lupus nephritis in mice by upregulation of Foxp3+ regulatory T cells in the target tissue: kidney. Transpl. Immunol. 24, 17–25 (2010).
Claassen, M.A., de Knegt, R.J., Tilanus, H.W., Janssen, H.L. & Boonstra, A. Abundant numbers of regulatory T cells localize to the liver of chronic hepatitis C–infected patients and limit the extent of fibrosis. J. Hepatol. 52, 315–321 (2010).
Estes, J.D. et al. Simian immunodeficiency virus-induced lymphatic tissue fibrosis is mediated by transforming growth factor-β1–positive regulatory T cells and begins in early infection. J. Infect. Dis. 195, 551–561 (2007).
Liu, F. et al. CD4+CD25+Foxp3+ regulatory T cells depletion may attenuate the development of silica-induced lung fibrosis in mice. PLoS ONE 5, e15404 (2010).
Baarsma, H.A. et al. Activation of Wnt/β-catenin signaling in pulmonary fibroblasts by TGF-β is increased in chronic obstructive pulmonary disease. PLoS ONE 6, e25450 (2011).
Wei, J. et al. Canonical Wnt signaling induces skin fibrosis and subcutaneous lipoatrophy: a novel mouse model for scleroderma? Arthritis Rheum. 63, 1707–1717 (2011).
Surendran, K., Schiavi, S. & Hruska, K.A. Wnt-dependent β-catenin signaling is activated after unilateral ureteral obstruction, and recombinant secreted frizzled-related protein 4 alters the progression of renal fibrosis. J. Am. Soc. Nephrol. 16, 2373–2384 (2005).
Wang, D., Dai, C., Li, Y. & Liu, Y. Canonical Wnt/β-catenin signaling mediates transforming growth factor-β1–driven podocyte injury and proteinuria. Kidney Int. 80, 1159–1169 (2011).
Jiang, F., Parsons, C.J. & Stefanovic, B. Gene-expression profile of quiescent and activated rat hepatic stellate cells implicates Wnt signaling pathway in activation. J. Hepatol. 45, 401–409 (2006).
Königshoff, M. et al. WNT1-inducible signaling protein-1 mediates pulmonary fibrosis in mice and is upregulated in humans with idiopathic pulmonary fibrosis. J. Clin. Invest. 119, 772–787 (2009).
Hinz, B., Celetta, G., Tomasek, J.J., Gabbiani, G. & Chaponnier, C. α-smooth muscle actin expression upregulates fibroblast contractile activity. Mol. Biol. Cell 12, 2730–2741 (2001).
Heise, R.L., Stober, V., Cheluvaraju, C., Hollingsworth, J.W. & Garantziotis, S. Mechanical stretch induces epithelial-mesenchymal transition in alveolar epithelia via hyaluronan activation of innate immunity. J. Biol. Chem. 286, 17435–17444 (2011).
Chen, J.H., Chen, W.L., Sider, K.L., Yip, C.Y. & Simmons, C.A. β-catenin mediates mechanically regulated, transforming growth factor-β1–induced myofibroblast differentiation of aortic valve interstitial cells. Arterioscler. Thromb. Vasc. Biol. 31, 590–597 (2011).
Balestrini, J.L., Chaudhry, S., Sarrazy, V., Koehler, A. & Hinz, B. The mechanical memory of lung myofibroblasts. Integr. Biol. 4, 410–421 (2012).
Hinz, B. Tissue stiffness, latent TGF-β1 activation, and mechanical signal transduction: implications for the pathogenesis and treatment of fibrosis. Curr. Rheumatol. Rep. 11, 120–126 (2009).
Bechtel, W. et al. Methylation determines fibroblast activation and fibrogenesis in the kidney. Nat. Med. 16, 544–550 (2010).
Coward, W.R., Watts, K., Feghali-Bostwick, C.A., Jenkins, G. & Pang, L. Repression of IP-10 by interactions between histone deacetylation and hypermethylation in idiopathic pulmonary fibrosis. Mol. Cell Biol. 30, 2874–2886 (2010).
Sanders, Y.Y. et al. Thy-1 promoter hypermethylation: a novel epigenetic pathogenic mechanism in pulmonary fibrosis. Am. J. Respir. Cell Mol. Biol. 39, 610–618 (2008).
Malhi, H. & Gores, G.J. Molecular mechanisms of lipotoxicity in nonalcoholic fatty liver disease. Semin. Liver Dis. 28, 360–369 (2008).
Korfei, M. et al. Epithelial endoplasmic reticulum stress and apoptosis in sporadic idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 178, 838–846 (2008).
Lawson, W.E. et al. Endoplasmic reticulum stress in alveolar epithelial cells is prominent in IPF: association with altered surfactant protein processing and herpesvirus infection. Am. J. Physiol. Lung Cell Mol. Physiol. 294, L1119–L1126 (2008).
Calado, R.T. & Young, N.S. Telomere diseases. N. Engl. J. Med. 361, 2353–2365 (2009).
Tsakiri, K.D. et al. Adult-onset pulmonary fibrosis caused by mutations in telomerase. Proc. Natl. Acad. Sci. USA 104, 7552–7557 (2007).
Cronkhite, J.T. et al. Telomere shortening in familial and sporadic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 178, 729–737 (2008).
Alder, J.K. et al. Short telomeres are a risk factor for idiopathic pulmonary fibrosis. Proc. Natl. Acad. Sci. USA 105, 13051–13056 (2008).
Armanios, M.Y. et al. Telomerase mutations in families with idiopathic pulmonary fibrosis. N. Engl. J. Med. 356, 1317–1326 (2007).
Lee, J. et al. Lung alveolar integrity is compromised by telomere shortening in telomerase-null mice. Am. J. Physiol. Lung Cell Mol. Physiol. 296, L57–L70 (2009).
Liu, T. et al. Telomerase regulation of myofibroblast differentiation. Am. J. Respir. Cell Mol. Biol. 34, 625–633 (2006).
Li, H., Xu, D., Li, J., Berndt, M.C. & Liu, J.P. Transforming growth factor-β suppresses human telomerase reverse transcriptase (hTERT) by Smad3 interactions with c-Myc and the hTERT gene. J. Biol. Chem. 281, 25588–25600 (2006).
Proctor, C.J. & Kirkwood, T.B. Modelling telomere shortening and the role of oxidative stress. Mech. Ageing Dev. 123, 351–363 (2002).
Liu, T. et al. Telomerase activity is required for bleomycin-induced pulmonary fibrosis in mice. J. Clin. Invest. 117, 3800–3809 (2007).
Liu, G. et al. miR-21 mediates fibrogenic activation of pulmonary fibroblasts and lung fibrosis. J. Exp. Med. 207, 1589–1597 (2010).
Thum, T. et al. MicroRNA-21 contributes to myocardial disease by stimulating MAP kinase signalling in fibroblasts. Nature 456, 980–984 (2008).
Chan, J.A., Krichevsky, A.M. & Kosik, K.S. MicroRNA-21 is an antiapoptotic factor in human glioblastoma cells. Cancer Res. 65, 6029–6033 (2005).
Patrick, D.M. et al. Stress-dependent cardiac remodeling occurs in the absence of microRNA-21 in mice. J. Clin. Invest. 120, 3912–3916 (2010).
Chung, A.C., Huang, X.R., Meng, X. & Lan, H.Y. miR-192 mediates TGF-β/Smad3-driven renal fibrosis. J. Am. Soc. Nephrol. 21, 1317–1325 (2010).
Kato, M. et al. TGF-β activates Akt kinase through a microRNA-dependent amplifying circuit targeting PTEN. Nat. Cell Biol. 11, 881–889 (2009).
Maurer, B. et al. MicroRNA-29, a key regulator of collagen expression in systemic sclerosis. Arthritis Rheum. 62, 1733–1743 (2010).
Roderburg, C. et al. Micro-RNA profiling reveals a role for miR-29 in human and murine liver fibrosis. Hepatology 53, 209–218 (2011).
van Rooij, E. et al. Dysregulation of microRNAs after myocardial infarction reveals a role of miR-29 in cardiac fibrosis. Proc. Natl. Acad. Sci. USA 105, 13027–13032 (2008).
Ogawa, T. et al. Suppression of type I collagen production by microRNA-29b in cultured human stellate cells. Biochem. Biophys. Res. Commun. 391, 316–321 (2010).
Pandit, K.V. et al. Inhibition and role of let-7d in idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 182, 220–229 (2010).
Duisters, R.F. et al. miR-133 and miR-30 regulate connective tissue growth factor: implications for a role of microRNAs in myocardial matrix remodeling. Circ. Res. 104, 170–178 (2009).
Venugopal, S.K. et al. Liver fibrosis causes downregulation of miRNA-150 and miRNA-194 in hepatic stellate cells, and their overexpression causes decreased stellate cell activation. Am. J. Physiol. Gastrointest. Liver Physiol. 298, G101–G106 (2010).
Wang, B. et al. miR-200a Prevents renal fibrogenesis through repression of TGF-β2 expression. Diabetes 60, 280–287 (2011).
Richeldi, L. & du Bois, R.M. Pirfenidone in idiopathic pulmonary fibrosis: the CAPACITY program. Expert Rev. Respir. Med. 5, 473–481 (2011).
Nakanishi, H., Sugiura, T., Streisand, J.B., Lonning, S.M. & Roberts, J.D. Jr. TGF-β–neutralizing antibodies improve pulmonary alveologenesis and vasculogenesis in the injured newborn lung. Am. J. Physiol. Lung Cell Mol. Physiol. 293, L151–L161 (2007).
Munger, J.S. et al. The integrin αvβ6 binds and activates latent TGF-β1: a mechanism for regulating pulmonary inflammation and fibrosis. Cell 96, 319–328 (1999).
Hahm, K. et al. αvβ6 integrin regulates renal fibrosis and inflammation in Alport mouse. Am. J. Pathol. 170, 110–125 (2007).
Hemavathy, K. & Wang, J.C. Epigenetic modifications: new therapeutic targets in primary myelofibrosis. Curr. Stem Cell Res. Ther. 4, 281–286 (2009).
Tedstone, J.L., Richards, S.M., Garman, R.D. & Ruzek, M.C. Ultrasound imaging accurately detects skin thickening in a mouse scleroderma model. Ultrasound Med. Biol. 34, 1239–1247 (2008).
Kono, M. et al. Plasma CCN2 (connective tissue growth factor; CTGF) is a potential biomarker in idiopathic pulmonary fibrosis (IPF). Clin. Chim. Acta 412, 2211–2215 (2011).
Pradère, J.P. et al. LPA1 receptor activation promotes renal interstitial fibrosis. J. Am. Soc. Nephrol. 18, 3110–3118 (2007).
Tager, A.M. et al. The lysophosphatidic acid receptor LPA1 links pulmonary fibrosis to lung injury by mediating fibroblast recruitment and vascular leak. Nat. Med. 14, 45–54 (2008).
Zeisberg, M. et al. BMP-7 counteracts TGF-β1–induced epithelial-to-mesenchymal transition and reverses chronic renal injury. Nat. Med. 9, 964–968 (2003).
Sidharta, P.N., van Giersbergen, P.L., Halabi, A. & Dingemanse, J. Macitentan: entry-into-humans study with a new endothelin receptor antagonist. Eur. J. Clin. Pharmacol. 67, 977–984 (2011).
Barry-Hamilton, V. et al. Allosteric inhibition of lysyl oxidase–like-2 impedes the development of a pathologic microenvironment. Nat. Med. 16, 1009–1017 (2010).
Myllyharju, J. Prolyl 4-hydroxylases, key enzymes in the synthesis of collagens and regulation of the response to hypoxia, and their roles as treatment targets. Ann. Med. 40, 402–417 (2008).
Fineschi, S. et al. In vivo investigations on antifibrotic potential of proteasome inhibition in lung and skin fibrosis. Am. J. Respir. Cell Mol. Biol. 39, 458–465 (2008).
Anan, A. et al. Proteasome inhibition induces hepatic stellate cell apoptosis. Hepatology 43, 335–344 (2006).
Kralovics, R. et al. A gain-of-function mutation of JAK2 in myeloproliferative disorders. N. Engl. J. Med. 352, 1779–1790 (2005).
Daniels, C.E. et al. Imatinib treatment for idiopathic pulmonary fibrosis: randomized placebo-controlled trial results. Am. J. Respir. Crit. Care Med. 181, 604–610 (2010).
Kay, J. & High, W.A. Imatinib mesylate treatment of nephrogenic systemic fibrosis. Arthritis Rheum. 58, 2543–2548 (2008).
Richeldi, L. et al. Efficacy of a tyrosine kinase inhibitor in idiopathic pulmonary fibrosis. N. Engl. J. Med. 365, 1079–1087 (2011).
Verstovsek, S. Therapeutic potential of Janus-activated kinase-2 inhibitors for the management of myelofibrosis. Clin. Cancer Res. 16, 1988–1996 (2010).
Pockros, P.J. et al. Final results of a double-blind, placebo-controlled trial of the antifibrotic efficacy of interferon-γ1b in chronic hepatitis C patients with advanced fibrosis or cirrhosis. Hepatology 45, 569–578 (2007).
Noble, P.W., Richeldi, L. & Kaminski, N. End of an ERA: lessons from negative clinical trials in idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 184, 4–5 (2011).
Park, S.W. et al. Interleukin-13 and its receptors in idiopathic interstitial pneumonia: clinical implications for lung function. J. Korean Med. Sci. 24, 614–620 (2009).
Corren, J. et al. Lebrikizumab treatment in adults with asthma. N. Engl. J. Med. 365, 1088–1098 (2011).
Kraft, M. Asthma phenotypes and interleukin-13—moving closer to personalized medicine. N. Engl. J. Med. 365, 1141–1144 (2011).
Ekert, J.E. et al. Chemokine (C-C motif) ligand-2 mediates direct and indirect fibrotic responses in human and murine cultured fibrocytes. Fibrogenesis Tissue Repair 4, 23 (2011).
Duffield, J.S. & Lupher, M.L. Jr. PRM-151 (recombinant human serum amyloid P/pentraxin 2) for the treatment of fibrosis. Drug News Perspect. 23, 305–315 (2010).
Castaño, A.P. et al. Serum amyloid P inhibits fibrosis through FcγR-dependent monocyte-macrophage regulation in vivo. Sci. Transl. Med. 1, 5ra13 (2009).
McKinsey, T.A. Therapeutic potential for HDAC inhibitors in the heart. Annu. Rev. Pharmacol. Toxicol. 52, 303–319 (2012).
Dart, M.L. et al. Interleukin-17–dependent autoimmunity to collagen type V in atherosclerosis. Circ. Res. 107, 1106–1116 (2010).
Acknowledgements
We would like to sincerely thank the past and present members of our laboratory for their guidance, comments and support. The Wynn laboratory is supported by the intramural research program of the US National Institutes of Health, National Institute of Allergy and Infectious Diseases and has benefited from cooperative research-and-development agreements with Pfizer, MedImmune, Amgen, Regeneron, Centocor and Genentech.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
T.A.W. holds patents on IL-13 as a target for fibrotic disease.
Rights and permissions
About this article
Cite this article
Wynn, T., Ramalingam, T. Mechanisms of fibrosis: therapeutic translation for fibrotic disease. Nat Med 18, 1028–1040 (2012). https://doi.org/10.1038/nm.2807
Published:
Issue Date:
DOI: https://doi.org/10.1038/nm.2807
This article is cited by
-
The occurrence and development mechanisms of esophageal stricture: state of the art review
Journal of Translational Medicine (2024)
-
Modelling and targeting mechanical forces in organ fibrosis
Nature Reviews Bioengineering (2024)
-
Progressive alteration of murine bladder elasticity in actinic cystitis detected by Brillouin microscopy
Scientific Reports (2024)
-
Ketotifen directly modifies the fibrotic response of human skin fibroblasts
Scientific Reports (2024)
-
Molecular mechanisms of cellular dysfunction in testes from men with non-obstructive azoospermia
Nature Reviews Urology (2024)
