
Abstract
More than two decades of intense research has provided a detailed understanding of hepatitis C virus (HCV), which chronically infects 2% of the world's population. This effort has paved the way for the development of antiviral compounds to spare patients from life-threatening liver disease. An exciting new era in HCV therapy dawned with the recent approval of two viral protease inhibitors, used in combination with pegylated interferon-α and ribavirin; however, this is just the beginning. Multiple classes of antivirals with distinct targets promise highly efficient combinations, and interferon-free regimens with short treatment duration and fewer side effects are the future of HCV therapy. Ongoing and future trials will determine the best antiviral combinations and whether the current seemingly rich pipeline is sufficient for successful treatment of all patients in the face of major challenges, such as HCV diversity, viral resistance, the influence of host genetics, advanced liver disease and other co-morbidities.
Similar content being viewed by others

Main
HCV constitutes a significant health burden worldwide, with an estimated 130–170 million people chronically infected. Severe liver disease, including advanced fibrosis, cirrhosis and hepatocellular carcinoma, is often a complication of long-term HCV infection, making HCV the most common indication for liver transplantation in developed countries (discussed by Thomas in this issue1). The previous standard-of-care HCV therapy consisted of pegylated interferon-α (peg-IFN-α) and ribavirin for up to 48 weeks, which leads to a virologic cure for about 50% of adherent patients. IFN-α elicits a general antiviral state in cells, whereas several mechanisms have been suggested for the activity of ribavirin. These include favoring of T helper type 1 immune responses, induction of IFN-stimulated genes (ISGs), inhibition of inosine monophosphate dehydrogenase (leading to GTP depletion), direct inhibition of the HCV polymerase or mutagenesis of newly synthesized viral RNA2. Severe side effects are one of the most frequent causes of treatment discontinuation, and they include flu-like and neuropsychiatric symptoms, autoimmune diseases and hemolytic anemia3. Since 2011, a triple combination adding one of two direct-acting antivirals (DAAs) protease inhibitors has been approved for HCV genotype 1 infections, increasing cure rates to around 70% (refs. 4,5). However, added severe side effects, resistance and drug-drug interactions are still issues6, and the quest for the holy grail of HCV treatment, an all-oral highly effective IFN-free regimen, continues (Fig. 1). HCV prevention in high-risk groups has seen only limited improvement, and there is no vaccine available or on the near horizon (discussed by Liang in this issue7).

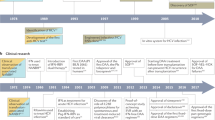
Major developments in natural history, virology and model systems, direct-acting antivirals (DAA) development, host-targeting agents (HTA) development and clinical implementations are indicated. Immediate implications of breakthroughs in basic research for inhibitor development and patient therapy are indicated with horizontal arrows.
HCV was discovered in 1989 (ref. 8) and was found to be the major cause of non-A, non-B post-transfusion hepatitis9. This breakthrough quickly led to serologic- and nucleic acid–based diagnostics for blood product screening10. In contrast, it has been a struggle to establish research tools and cell culture systems for HCV (Fig. 1). The only true HCV animal model is the chimpanzee, which has been crucial in studies of HCV immunity and pathogenesis11. Small animals are not naturally infected by HCV, which has encouraged development of human-liver chimeric12 and genetically modified13 HCV-permissive mice. Establishment of cell culture systems has been a painfully slow process, but these now include selectable replicon systems14, retrovirus-based pseudotyped particles15,16 and complete viral replication systems17,18,19, which have been essential for dissecting the viral lifecycle, identifying promising targets and developing antiviral compounds (Box 1).
HCV is a positive-stranded RNA virus in the Flaviviridae family, which also includes classical flaviviruses such as yellow fever and dengue. Only the enigmatic GB-virus B and the recently identified nonprimate, rodent and bat hepaciviruses (NPHV20, RHV21 and BHV22) are grouped with HCV in the Hepacivirus genus. The HCV 9.6-kb genome contains 5′ and 3′ untranslated regions (UTRs) flanking a long open reading frame (ORF) that is translated via an internal ribosome entry site (IRES)23. The resulting polyprotein is processed to yield structural (core, E1 and E2) and nonstructural (p7, NS2, NS3, NS4A, NS4B, NS5A and NS5B) proteins (Fig. 2)24. Membrane-associated viral replication takes place in the cytoplasm, and assembly and release through secretory pathways are coupled to lipoprotein biogenesis25. Approved DAAs inhibit the cleavage of the polyprotein into mature nonstructural proteins by the viral NS3-NS4A serine protease, and other antivirals in development target RNA replication. Progress in understanding HCV biology has been key for creating a remarkably rich pipeline of antiviral compounds in various stages of preclinical and clinical development (Table 1).

The HCV RNA genome (top) contains one long ORF (blue) flanked by 5′ and 3′ UTRs (red). The binding of two copies of miR-122 (green) to the 5′ UTR is highlighted in the inset. IRES-mediated translation of the ORF leads to a polyprotein (bottom) that is co- and post-translationally processed into ten viral proteins. The maturation process of the core protein involves a cellular signal peptide peptidase cleavage of a C-terminal signal peptide (white triangle) and cleavage from E1 by the cellular signal peptidase123, which also cleaves E1, E2 and p7 from the polyprotein (gray triangles). In an autocleavage mechanism requiring two identical molecules to make up the composite active site, the NS2-NS3 protease cleaves itself (red triangle)72. The NS3 protease located in the first one-third of NS3 (ref. 165), assisted by its membrane-bound cofactor, NS4A166, cleaves the remaining proteins NS3, NS4A, NS4B, NS5A and NS5B (green triangles). Glycosylation of the envelope proteins (black dots) and the functions of the individual HCV proteins are indicated.
HCV genotype and host genetics affect antiviral drug response
Genetic heterogeneity looms large for HCV compared to, for example, hepatitis B virus (HBV) and HIV; HCV isolates have been grouped into seven genotypes (1–7, ∼30% sequence divergence) and a number of subtypes (a, b, and so on, ∼20% sequence divergence)24. Distinct differences are observed in geographic distribution, with genotype 1 dominating in the Americas (70% of cases), Japan (75%) and Europe (50–70%); genotypes 2 and 3 are also prevalent in these regions. Genotypes 3 and 6 are widespread in South and Southeast Asia, and genotypes 4 and 5 are most common in Africa but spreading to Europe. Genotype 7 was recently found in a few patients from Central Africa, but thus far it is not of major clinical importance. Disease association is largely similar across genotypes; however, a higher risk of hepatic steatosis26 and of progressive liver disease27 is associated with genotype 3. For reasons that still remain obscure, IFN-based regimens lead to a sustained viral response (SVR) of nearly 80% for genotype 2– and genotype 3–infected patients but only about 50% for genotype 1 and genotype 4 infections3; genotypes 5 and 6 have intermediate response rates. However, selection of true viral resistance to peg-IFN-α and ribavirin has not been observed. This genotype variability remains important in the dawning era of DAAs. The first-generation NS3-NS4A protease inhibitors telaprevir and boceprevir, owing to genotype-dependent efficacy, were approved for treatment of only genotype 1. Selective targeting of genotype 1 has been driven by its high prevalence in developed countries and the use of genotype 1b replicon systems as the industry standard for drug development.
Several host factors influence spontaneous clearance of HCV and the outcome of therapy, including HLA type, ethnicity, gender, age and obesity. Further, high baseline levels of ISGs predict an unfavorable outcome of HCV therapy28. Single nucleotide polymorphisms in the gene encoding IFN-λ3 (formerly known as IL28B) and the recently discovered IFN-λ4 have now become important predictors of treatment outcome (discussed by Horner and Gale in this issue29, Fig. 1).
The viral life cycle and points of intervention
The HCV particle.
Much remains to be understood on the composition and structure of infectious HCV particles. Enveloped HCV virions are 50–80 nm in diameter, with E1 and E2 glycoprotein heterodimers embedded in the lipid bilayer surrounding a nucleocapsid composed of core protein and the single-stranded RNA genome30,31 (Fig. 3). HCV virions, existing as lipoviroparticles (LVPs), are not icosahedral, and because of their association with low-density and very-low-density lipoproteins (LDL and VLDL) in the infected host32,33, they are pleomorphic with heterogeneous and low buoyant density, which varies depending upon growth conditions34. This Trojan horse strategy may help shield the virus from neutralization25. Apolipoprotein E (apoE) and apoC are associated with both in vivo– and cell culture–derived particles, whereas association with apoB is less pronounced in cell culture35.

Points of intervention in the HCV life cycle are marked with numbered circles, and types of inhibitors of the individual steps are indicated in the legend. Interaction of extracellular HCV LVPs (1) with cellular surface receptors initiates the entry process (2), which can also occur from direct cell-to-cell transmission. After pH-dependent fusion and uncoating, the incoming HCV genome is translated and the resulting polyprotein processed (bottom inset and Fig. 2, (3)). Replication takes place in ER-derived membrane spherules (membranous web, bottom right inset, (4)), the architecture of which remains to be fully defined76. The spatiotemporal contribution of miR-122 binding to the HCV genome is not yet fully understood, and miR-122 presence is indicated with '?'. In the assembly and release process (top right inset, (5)), core protein is transferred from cLDs to form nucleocapsids that, assisted by NS5A, are loaded with RNA. Replicase proteins supposedly bind HCV RNA during transfer from replication to packaging, the intracellular sites of which might converge. It is not clear whether the RNA is transiently located on the cLD. The p7, NS2 and NS3-NS4A proteins are also involved in coordination of assembly. HCV virion morphogenesis is coupled to the VLDL pathway, and particles are produced as LVPs. Particles released from cell culture have less ApoB association and resemble the ApoB-deficient particle illustrated. LVPs also associate with ApoC, which for simplicity is not shown here. EphA2, ephrin receptor type A2; GAG, glycosaminoglycans; PL, phospholipids; TG, triglycerides. In the translation inset, NS5A is shown as a dimer, though several other HCV proteins also form homo- or heterodimers or oligomers. For comprehensive discussion and illustration of HCV assembly and release and of membrane-associated HCV protein structures, see refs. 25,167, respectively.
In theory, selective lysis of infectious particles could help prevent the observed universal re-infection of liver allografts after transplantation or could be used as a supplement to other antivirals. Peptides exhibiting virocidal activity for HCV and other viruses have been reported, including a peptide derived from the HCV NS5A protein36. However, issues of in vivo delivery, efficacy and safety have not been explored. Hence, virocidal compounds have not emerged as serious antiviral players.
HCV-neutralizing antibodies, an obvious objective for vaccination strategies (see Liang in this issue7), could also play a part in therapy, for example by passive administration to prevent re-infection after liver transplantation. However, relative to the recent progress for HIV37, understanding of HCV neutralization and HCV-neutralizing antibodies is still in its infancy. Antibodies derived from patients or experimentally infected chimpanzees can neutralize HCV. But limited cross-genotype neutralization38,39 and continuous viral escape40 pose serious challenges. For example, the E2 glycoprotein contains hypervariable regions containing immunodominant neutralization epitopes thought to function as immunological decoys to shield more conserved neutralization epitopes, possibly by facilitating association with host lipids41,42. ApoE-specific antibodies can neutralize HCV infection35 and are well tolerated at least in mouse models. However, dosing and safety are concerns when targeting a protein that is also an abundant host serum component. Few clinical studies have examined the efficacy of neutralizing antibodies, and the results thus far have been disappointing43. However, efforts are under way to identify and characterize more potent, cross-reactive neutralizing antibodies44,45. Given HCV's diversity and ability to persist in the face of an evolving neutralizing response, a cocktail of such antibodies is likely to be required for effective control in the context of established chronic infection or liver transplantation.
Entry and uncoating.
A growing number of cellular molecules are involved in HCV entry into hepatocytes (Fig. 3). The LDL receptor and glycosaminoglycans are thought to mediate initial low-affinity cell binding46,47, before E1-E2 interaction with the co-receptors SR-BI48 and CD81 (ref. 49). Claudin-1 (CLDN1) and occludin (OCLN) are also required for entry50,51. New in vitro systems mimicking the polarized nature of liver cells52 are helping to examine migration of HCV-receptor complexes to tight junctions, where these entry factors are normally found. CLDN6 and CLDN9 can replace CLDN1 for HCV entry, but they are expressed only at low levels in the liver53. Species differences in CD81 and OCLN, for example, restrict host tropism51. The additional factors epidermal growth factor receptor (EGFR) and ephrin receptor type A2 are required for HCV entry and possibly modulate interactions between CD81 and CLDN1 (ref. 54). Virion-associated cholesterol seems to be involved at a late stage of HCV entry, at or before fusion, through interaction with the NPC1L1 cholesterol absorption receptor55. Uptake occurs through clathrin-mediated endocytosis56, and fusion requires a low pH compartment, which is probably encountered in endosomes57,58. HCV E2 is primed by CD81 for activation in acidic environments59. Although its structure has not been solved, it has been predicted to be a class II fusion protein60. However, the structure of the related pestivirus E2 ectodomain was recently solved, revealing a novel fold and organization, which led to speculation that E1 may actually be the fusogen61,62. These entry processes eventually lead to release of the HCV genome into the cytoplasm, where primary translation can occur.
In addition to infection with cell-free virus, direct cell-to-cell transmission probably also occurs in the liver, which may provide a means to avoid neutralization63. However, most host entry factors overlap between the two routes, making them potential targets for intervention. In human-liver chimeric mice, antibodies targeting CD81 or SR-BI protect mice from infection, and also from viral dissemination in the case of SR-BI–specific antibodies64,65. Small-molecule host-targeting agents (HTAs) are also being investigated66. ITX 5061, which inhibits the uptake of high-density lipoprotein (HDL) through SR-BI and uptake of HCV particles67, is currently in phase 2 clinical trials (Table 1). Mutations in E2 hypervariable region 1 can confer ITX 5061 resistance, but the trade-off for such variants may be increased susceptibility to neutralization68. Erlotinib, a clinically approved inhibitor of EGFR, and the NPC1L1 inhibitor ezetimibe have been shown to impair HCV infection in human-liver chimeric mice54,55, but it is yet unclear whether a sufficient therapeutic window exists in humans. The late stages of HCV uptake, including fusion with host membranes and viral uncoating, are poorly understood; however, peptidomimetics inhibiting fusion have been described69. Although there are scant clinical data to date and concerns about safety, targeting host entry factors with antibodies or small-molecule inhibitors could block the spread of DAA-resistant variants and disturb infection dynamics necessary to maintain chronic liver infection.
Translation and polyprotein processing.
The intracellular replicative phase of the HCV life cycle has provided a wealth of antiviral targets, including key viral enzymes required for protein production and RNA amplification. The HCV genome is highly structured with essential RNA elements in the 5′ and 3′ UTRs as well as in the coding region (Fig. 2)24. Endoplasmic reticulum (ER)-associated translation is initiated by an IRES located in the HCV 5′ UTR23. The resulting HCV polyprotein is co- and post-translationally cleaved by cellular proteases (signalase and signal peptide peptidase) and the viral NS2-NS3 and NS3-NS4A proteases to release ten HCV proteins (Fig. 2).
Interestingly, the NS3-NS4A serine protease also cleaves the MAVS and TRIF adaptor proteins, blocking IFN synthesis initiated by retinoic acid–inducible gene-I (RIG-I) and Toll-like receptor 3 (see Horner and Gale in this issue29). Inhibiting NS3-NS4A thus has an attractive dual function, making this protease a prime target for antiviral development. The linear NS3-NS4A protease inhibitors telaprevir and boceprevir were the first DAAs approved in triple combination with peg-IFN-α and ribavirin for treatment of HCV genotype 1 infection (Fig. 1). In phase 3 studies, adding telaprevir or boceprevir increased SVR rates from ∼50% to ∼70% (refs. 4,5). A number of second-wave, primarily macrocyclic, NS3-NS4A protease inhibitors are in advanced clinical development and show more potent antiviral activity with superior tolerability and pharmacokinetics (Table 1)6. These include ongoing phase 3 studies with simeprevir (TMC435), faldaprevir (BI201335), vaniprevir (MK-7009), asunaprevir (BMS-650032) and ABT-450. In spite of their promise, these first-generation protease inhibitors still have suboptimal resistance profiles and genotype coverage6,70. However, true second-generation protease inhibitors with high potency, broad genotype coverage and superior resistance profiles, such as MK-5172 and neceprevir (ACH-2684), are already in the clinical development pipeline71.
Less attention has been given to cellular proteins involved in IRES-mediated translation23 and the NS2-NS3 protease66. For NS2-NS3, biochemical assays were challenging to develop, and a crystal structure for the post-cleavage form was not solved until 2006 (ref. 72). However, NS2-NS3 cleavage is essential for formation of the HCV RNA replicase, with mature NS2 also required for infectious virus production73,74. Thus NS2, at least in concept, could make an attractive antiviral target.
HCV RNA replication.
RNA replication is believed to take place in association with ER-derived membrane spherules induced by NS4B and NS5A75,76, which in aggregate are termed the membranous web (Fig. 3). The RNA-dependent RNA polymerase, NS5B, is the workhorse of the HCV replicase complex, with a classical right-hand structure consisting of finger, palm and thumb domains77. The NS3 protein contains a superfamily 2 DExH/D-box helicase domain, capable of nucleic-acid binding and 3′ to 5′ translocation coupled to hydrolysis of ATP78. Although its exact role is unknown, this activity could be important for separation of nascent and template RNA strands, unwinding of local RNA secondary structures or displacement of RNA-binding proteins. The NS5A phosphoprotein consists of three loosely defined domains79, with a high-resolution structure available only for domain I, revealing a dimer with a basic channel possibly involved in RNA binding80,81. NS5A domains I and II are essential for RNA replication79,82, whereas domain III participates in virus assembly83,84. The phosphorylation state of NS5A regulates the balance between RNA replication and downstream processes85.
Owing to its pivotal role in viral RNA synthesis, the NS5B polymerase has been a prime target for antiviral development. Perhaps the most promising of new inhibitor classes are nucleoside or nucleotide inhibitors that target the NS5B polymerase active site, acting as RNA chain terminators. Given the high conservation of the active site, these inhibitors have pan-genotype coverage. Although these inhibitors have a low genetic barrier to resistance, requiring only a single amino acid substitution, such resistance mutants have low fitness, resulting in an overall high barrier to resistance70. Clinical studies of these drugs in combination with peg-IFN-α and ribavirin look promising, in particular for sofosbuvir (GS-7977), which had tolerable safety profiles and ∼90% SVR86,87 (Table 1). However, this class of inhibitors, particularly the purine analogs, has been plagued with severe adverse events and safety concerns, possibly as a result of mitochondrial toxicity88. Clinical development of several initially promising compounds, including valopicitabine (NM283), R1626, BMS-986094, PSI-938 and IDX-184, has been discontinued. Nonetheless, it is predicted that this class of compounds will emerge as the cornerstone of all-oral, pan-genotype combinatorial regimens.
Non-nucleoside inhibitors, which target one of four allosteric sites on NS5B (thumb and palm domains), typically have narrow genotype coverage and a low barrier to resistance, owing to the high fitness of escape mutants. However, compounds such as ABT-333, ABT-072, BI207127, BMS-791325, lumibuvir (VX-222) and setrobuvir (RG7790) may be useful when combined with other antivirals and are now being evaluated primarily in combination trials (Table 1)6. As for the nucleosides, several allosteric polymerase inhibitors have also been halted for safety issues. Although not likely to be a component of pan-genotype regimens, potent genotype- or even subtype-specific inhibitors of this class may still prove useful for treating targeted patient populations.
NS5A, because of its lack of enzymatic activity, emerged as a viable target only recently after the advent of cell-based screening platforms (Fig. 1). Despite its late emergence as a drug target, NS5A inhibitors are now perhaps the most potent antivirals ever discovered, with low- or even subpicomolar activity in cell-based assays89,90. In fact, the extremely rapid rate of HCV decline in single-dose escalation trials for daclatasvir (BMS-790052) necessitated revising estimates of the circulating virus half-life from 2.5 h to 45 min (ref. 91). On the basis of the location of resistance mutations, current compounds seem to target NS5A domain I89,90; however, the exact mechanism of action of these inhibitors is the subject of ongoing research. Despite their high potency and reasonably broad genotype coverage, this class of compounds has a relatively low barrier to resistance with high fitness for escape mutants90. Daclatasvir, the most advanced compound, is currently in phase 3 studies of patients with genotype 1 HCV, with promising results92 (Table 1). However, it is likely that NS5A inhibitors will ultimately be used in combination with other antiviral classes, and encouraging results have been obtained, for example, with daclatasvir and sofosbuvir93.
Although the NS3 helicase activity would seem to be an attractive target, the large number of cellular ATP-binding motor proteins has posed specificity challenges that have not yet been overcome. Nonetheless, a better understanding of NS3's role in RNA replication, snapshots during unwinding steps that reveal unique structural intermediates94 and improved antiviral screening methods may yet yield effective and specific helicase-targeted drugs66. Several classes of inhibitors targeting NS4B have been identified95, including clemizole, an approved antihistamine drug, which blocks an RNA-binding activity of NS4B96. None of the NS4B-targeted compounds have progressed to advanced clinical development. Silibinin, an approved therapeutic successfully used to prevent re-infection of the allograft after liver transplantation and for treatment of IFN nonresponders, may target NS4B, given its resistance profile and ability to interfere with formation of replication sites97.
Although too numerous to detail here, a number of host factors influencing translation, replication, assembly and release have been identified using RNAi-screening and mass spectrometry interactome approaches (Fig. 3 and Table 1, reviewed in ref. 52). The front-runner is cyclophilin A (CypA), a peptidyl-prolyl cis-trans isomerase required for HCV replication. This was discovered in a cell-based screen testing approved compounds for anti-HCV activity98. Cyclosporin A demonstrated potent antiviral activity, as did nonimmunosuppressive analogs with even higher antiviral potency such as alisporivir (DEBIO-025) and SCY-635, both of which then moved quickly into clinical development99,100. CypA, which binds NS5A, is believed to catalyze conformational changes necessary for HCV RNA replication101. Resistance to CypA inhibitors maps to NS5A in a region with overlapping CypA and NS5B binding sites (ref. 102 and references therein). A recent study suggests that CypA inhibitors may also enhance innate antiviral immunity by inducing type I and III IFNs and ISGs103. In patients, alisporivir enhances the efficacy of IFN-based regimens and demonstrates reasonable cross-genotype coverage with a high barrier to resistance104. However, owing to several cases of acute pancreatitis among trial participants, this inhibitor is now on clinical hold. Although this is a setback for this first-in-class HTA, CypA inhibitors may well re-emerge in the context of IFN-free regimens.
Another intriguing aspect of HCV biology is its addiction to the abundant liver-specific host microRNA miR-122 (ref. 105). Near its 5′ end, the HCV RNA genome harbors two conserved miR-122 seed sites, with additional miR-122–HCV genome nonseed interactions required for efficient HCV replication106 (Fig. 2). miR-122 binding has a stimulatory effect on translation107. More recently, miR-122–argonaute 2 complexes were shown to protect uncapped HCV RNA from 5′–3′ degradation by exonuclease Xrn1 (ref. 108). It has also been suggested that miR-122–HCV RNA complexes might shield the 5′ end of the HCV genome from recognition by innate immunity–inducing pattern recognition receptors, such as RIG-I109. Studies in chimpanzees provided proof of concept for targeting miR-122, by sequestering the miRNA using the miR-122 locked nucleic acid (LNA) antagonist miravirsen (SPC3649)110, a strategy that is effective against all HCV genotypes111. Potential drawbacks are that miravirsen is administered by injection, and miR-122 regulates hundreds of hepatocyte mRNAs, which might cause unwanted side effects. Treatment with miravirsen led to transient reduction in serum cholesterol110, and miR-122–knockout mice developed hepatitis and hepatocellular carcinomas112,113, which, interestingly, resembled HCV-related disease in humans. Despite potential drawbacks, a short-term phase 2 study of miravirsen indicated a tolerable safety profile, significant antiviral activity and a high barrier to resistance114.
Studying HCV's intimate association with hepatocytes, membranes and lipid metabolism has uncovered additional host-targeted therapeutic angles. Cholesterol and fatty acid biosynthesis as well as geranylgeranylation are important in HCV replication, possibly for forming membrane-associated RNA replication complexes115,116. Statins targeting HMG-CoA reductase inhibit HCV replication in cell culture115,116, although at doses beyond those used in the clinic to control serum cholesterol. As might be expected from this, statin monotherapy does not significantly reduce HCV RNA levels but does show a modest increase in response rates to IFN-based therapy117. An unexpected hit from several siRNA screens is the requirement for phosphatidylinositol-4 kinase III-α (PI4KIIIα) in HCV replication118. This enzyme is hijacked by HCV NS5A, which stimulates the production of phosphatidylinositol-4-phosphate119,120. Exactly how this is required for HCV RNA replication is still under investigation. Interestingly, 4-anilino quinazolines, originally thought to target NS5A based on mapping resistance mutations, turn out to be inhibitors of PI4KIIIα (ref. 121). Whether or not PI4KIIIα is a viable target is still a matter of debate. In mice, this enzyme is essential, and even conditional ablation of PI4KIIIα leads to severe pathology122. However, partial PI4KIIIα inhibition, liver targeting or short treatment duration might still provide a therapeutic window useful for HCV treatment.
Virus assembly and release.
Although late stages in the virus life cycle should also provide attractive targets for intervention, progress is lagging, as experimental systems for studying assembly and release are recent developments (Fig. 1 and Box 1). Virus assembly and release is a tightly regulated process coupled to host cell lipid synthesis (Fig. 3)25. After cleavage, first by signal peptidase and subsequently by signal peptide peptidase, the mature core protein relocates from ER membranes to cytoplasmic lipid droplets (cLDs)123,124, assisted by diacylglycerol acyltransferase-1 (DGAT1)125. The current model for nucleocapsid formation involves interaction of core with NS5A, either on cLDs, whereto NS5A is also directed by DGAT1 via its N-terminal amphipathic α-helix, or after translocation from the mobile cLDs to the ER83,126,127. Delivery of the HCV genome RNA to sites of nucleocapid assembly is still poorly understood but is probably facilitated by the close juxtaposition of sites of RNA replication and virion assembly25 and by NS2-coordinated virion assembly through interactions with the glycoproteins, p7, NS3 and NS5A73,74. A series of signal and stop-transfer sequences orchestrate ER translocation of the E1 and E2 glycoproteins, which assume a type I membrane protein topology. After folding, heterodimer formation and addition of N-linked sugars, the E1-E2 glycans are then trimmed by glycosidases I and II (ref. 128).
HCV protein-protein interactions or cellular machinery necessary for these steps could be potential antiviral targets. Compounds targeting DGAT1 do inhibit HCV125 and are already in clinical development for obesity-associated disease. In addition, recent evidence suggests that potent NS5A inhibitors, such as daclatasvir, act by inhibiting both RNA replication and virion assembly and that this is responsible for the strikingly rapid decline in viral load observed after administration of a single dose91. Iminosugar derivatives targeting ER glycosidases have demonstrated antiviral potential in vitro129,130. The glycosidase I inhibitor celgosivir (MX-3253) showed a synergistic effect when combined with peg-IFN-α and ribavirin in phase 2 trials131, but development has subsequently been halted. p7 oligomers are believed to function as ion channels that prevent acidification of intracellular compartments and virus inactivation during egress through the secretory pathway132. Given this functional similarity of p7 to the influenza M2 viroporin, amantadine-like compounds have been tested for their inhibitory activity. The effects in cell culture are moderate and isolate dependent129,130,132, and the clinical benefit of adding amantadine to peg-IFN-α and ribavirin, if any, is unclear3. Though improved SVR rates were achieved by combining peg-IFN-α and ribavirin with a new p7 inhibitor, BIT225, in a small clinical study133, larger studies are needed to evaluate its potential contribution.
During later stages of assembly, HCV co-opts the VLDL pathway134,135, a strategy that may enhance hepatotropism and contribute to persistence. At lipid-rich microdomains of the ER, the nucleocapsid is thought to be transferred to luminal lipid droplets (luLDs), which are precursors of VLDL particles136. Nucleocapsid-containing luLDs fuse with apoB-containing pre-VLDL particles to form LVPs, which also acquire apoE and apoC25,35,135 and exit through the Golgi137. The intraluminal ER microsomal transfer protein (MTP) is responsible for transfer of triglycerides and phospholipids to nascent luLDs and has been implicated in HCV assembly135. However, other cell culture studies found less apparent LVP-apoB association and MTP dependence138, possibly as a result of inefficient VLDL production in cell culture. This correlates with the higher density of cell culture–produced HCV, which is still infectious but of lower specific infectivity34. MTP inhibitors being evaluated in the clinic for dyslipidemia possess antiviral activity in vitro134,135, but their in vivo efficacy against HCV has yet to be demonstrated.
The future of HCV therapy: high hopes and challenges
Since type I IFN was tested and shown to be effective for treating non-A, non-B hepatitis, the standard of care steadily improved from single-digit success rates to overall virologic cure rates of around 50% with peg-IFN-α and ribavirin. These remarkable advances were achieved empirically, through testing in the clinic, without understanding of how these drugs exerted their anti-HCV effects. Now, after more than two decades of intense effort in academia, industry and clinical investigation, a deeper understanding of the HCV lifecycle has ushered in a new era in HCV treatment, beginning with the approval of the first two DAAs in 2011 (Fig. 1).
So where does the field stand in terms of clinical development? Initial strategies have examined combination of one or more DAAs with peg-IFN-α and ribavirin. This might include one DAA with a high barrier to resistance or two DAAs with lower resistance barriers. Benefits include the direct action of a DAA with the proven and probably independent antiviral activities of peg-IFN-α and ribavirin. This might shorten treatment, particularly for patients with a favorable IFN-λ genotype139. Responses are generally superior for HCV genotype 1b versus 1a, for the reasons discussed earlier. However, this strategy also has the potential of increasing the already harsh side effects (as seen for telaprevir and boceprevir) and, in some regimens, is less effective in previous IFN nonresponders or unusable in patients with contraindications to IFN. Phase 3 studies combining the second-wave protease inhibitors simeprevir or faldaprevir with peg-IFN-α and ribavirin improved SVR rates to ∼80% with milder side effects140,141,142. Even higher SVR rates of ∼90% were achieved for the nucleotide inhibitor sofosbuvir with peg-IFN-α and ribavirin86. Very high SVR rates were also observed with two DAAs (quadruple therapy), either protease and NS5A inhibitors (asunaprevir plus daclatasvir143 or vedroprevir (GS-9451) plus ledipasvir (GS-5885)144), or protease and nucleoside polymerase inhibitors (danoprevir plus mericitabine (RG7128)145). However, multiple-DAA regimens are now being pursued largely without IFN. Although there is ongoing speculation about the imminent demise of IFN-α–based therapies, IFN-λ is still being evaluated as a replacement given its similar antiviral properties and more desirable side-effect profile146.
Until recently, it was debated whether the holy grail, an IFN-free DAA and/or HTA regimen, would be sufficient to achieve a virologic cure. Might the far-reaching, systemic and antiviral action of IFN be required to eliminate the virus? In cell culture this is not the case, and in seminal chimpanzee147 and human-liver chimeric mouse148 experiments, combining two DAAs led to SVR. In a subsequent phase 2 pilot study, combination treatment with protease and NS5A inhibitors (asunaprevir and daclatasvir) for the first time demonstrated IFN-free SVR in HCV genotype 1–infected previous nonresponders149.
The HCV field is now in the midst of an explosion of IFN-free clinical studies combining one, two or three antiviral agents, with or without ribavirin. Combinations of three DAAs—protease, NS5A and non-nucleoside polymerase inhibitors with or without ribavirin— achieved close to 100% SVR in relatively large groups of HCV genotype 1–infected patients (for example ritonavir-boosted ABT-450, ABT-267 and ABT-333 (ref. 150), which included previous nonresponders, or asunaprevir, daclatasvir and BMS-791325 (ref. 151)). Several two-DAA combinations with or without ribavirin also showed remarkable results with SVR rates of at least 90%, for example sofosbuvir in combination with one of the NS5A inhibitors ledipasvir152 and daclatasvir153, a combination also proven to be efficacious for previous telaprevir or boceprevir failures93. Similar or slightly lower SVR rates were achieved by combining a protease inhibitor with a non-nucleotide inhibitor (for example, ritonavir-boosted ABT-450 and ABT-333 (ref. 154), telaprevir and VX-222 (ref. 155) or faldaprevir and BI-207127 (ref. 156)), the latter with a stronger bias toward HCV genotype 1b versus 1a. In phase 3 studies with sofosbuvir and ribavirin, impressive SVR rates of 97% were achieved for genotype 2, but only 56% for genotype 3, and with cirrhosis having more impact for poor response in genotype 3 (ref. 86). With response rates ranging from 59% to 84% for genotype 1, and even as low as 10% for previous nonresponders157, longer treatment duration or combination with additional drugs is likely to be necessary, at least for patients infected with an HCV genotype other than genotype 2. Ribavirin continues to have a role in some DAA regimens, for example, eliminating ribavirin from the sofosbuvir regimen significantly lowered SVR rates157. Interestingly, even in the absence of exogenous IFN, favorable IFN-λ genotype and low ISG baseline levels still positively correlate with DAA treatment response, suggesting a continuing role for innate immunity in viral clearance158. However, regimens that combine compounds of sufficient potency and high resistance barrier seem to be able to overcome the influence of the host determinants on treatment response87,150,153.
Broad genotype coverage is still a challenge, in particular for non-nucleoside polymerase inhibitors, but also for first-generation NS3-NS4A protease inhibitors, and NS5A inhibitors (Table 1). Some DAA combinations, anchored by nucleotide polymerase inhibitors given their cross-genotype efficacy, have been tested in patients with non–genotype 1 HCV. As mentioned above, sofosbuvir and ribavirin alone gave high SVR rates for genotype 2 but not genotype 3 patients86,87, and daclatasvir combined with sofosbuvir and ribavirin improved the already high peg-IFN-α plus ribavirin SVR for genotypes 2 and 3 (ref. 153), although a small number of failures have been reported for genotype 3 with this regimen and only small numbers of patients have been studied. Thus, it will be important to continue development of IFN-free regimens for all HCV genotypes, including genotype 3, which might be more difficult to treat with certain DAAs. Replicon and infectious cell culture systems for other genotypes159,160,161 should aid the development of second-generation pan-genotype protease and NS5A inhibitors to address this need (Table 1).
HCV's intrinsically high error rate churns out an estimated 1012 variants per day in a single infected individual. The emergence of DAA resistance is an obvious concern, and it has been seen in cell culture and in patients70. However, unlike the case with HIV, which requires life-long antiviral treatment, HCV therapy can result in eradication of detectable virus; in the vast majority of cases where SVR is achieved, relapse is not observed. Resistance can be minimized by using compounds with a high resistance barrier, such as nucleoside or nucleotide polymerase inhibitors, or by combining several classes of DAAs with nonoverlapping resistance profiles. A relatively high prevalence of pre-existing resistance mutations to certain protease inhibitors has been observed70,140, and the more fit resistance variants selected by certain DAAs, such as NS5A inhibitors, persist long after cessation of treatment162. Monitoring resistance in treatment failures is likely to become an important consideration for choosing which DAA classes to use for retreatment.
HTAs provide yet another angle, with the possible advantage of limiting virus-acquired resistance and increasing therapeutic options to avoid drug-drug interactions. Although development of the CypA inhibitor alisporivir in combination with peg-IFN-α and ribavirin is currently on clinical hold, combining alisporivir with ribavirin in a recent study of patients with genotype 2 and genotype 3 HCV gave high SVR rates of ∼90% and improved safety compared to peg-IFN-α and ribavirin163. Thus, alisporivir could well have a part in future IFN-free regimens. Recent efficacy and safety data for the miR-122 antagonist miravirsen are encouraging, even when it was administered as a monotherapy114. Although CypA inhibitors and small-molecule entry inhibitors can be orally formulated, receptor-blocking antibodies and miravirsen will require injections. However, infrequent administration of miravirsen, perhaps even once monthly, could make injections less of a problem and perhaps even favor patient compliance compared to oral DAAs that will require at least daily dosing114. Toxicity from targeting normal cellular functions is a concern for HTAs but may be manageable, particularly as treatment regimens shorten110,114.
Where does the field go from here and how close is it to the 'ideal' regimen: an all-oral, IFN-free combination cocktail with pan-genotype coverage, minimal side effects and high virologic cure rates in all patient groups? With dozens of compounds already in clinical development, and many more at preclinical stages, expectations for achieving this goal for HCV are justifiably high. However, this will not happen overnight, and the widespread excitement associated with initial trials is often tempered after more experience in the clinic. It is plausible that the most efficient IFN-free future regimens, combining nucleotide polymerase inhibitors with, for example, ribavirin and NS5A inhibitors, will overcome even immunologic limitations imposed by prior IFN nonresponse. In this scenario, IFNL3 and IFNL4 genotyping might become less relevant for most patient groups. However, individualized therapy may still be necessary for special patient groups, such as people with HCV and HIV co-infections, in whom drug-drug interactions can be problematic, and for patients with advanced fibrosis and cirrhosis. For many DAAs and HTAs, although their targets are known, how they actually work is still incompletely understood, and this would be useful knowledge for deciding which combinations might be most effective91. It is still not possible to culture and phenotype clinical isolates or predict antiviral resistance and, until recently, the ability to study HCV biology and assess inhibitor efficacy in primary human hepatocytes was limited160,161,164. Fortunately, in vitro systems or animal models can be used in drug combination studies to evaluate synergy and potential complications of drug-drug interactions. Testing combinations in the clinic presents an exciting opportunity to evaluate the value of in vitro systems; thus far, predictions of resistance and genotype coverage have largely held true in patients70,89,90. However, finding optimal combinations is not straightforward. Initial clinical studies are driven by corporate market share decisions rather than finding the best combination from the broader pipeline. The latter often occurs later, after approval, in an ad hoc manner or in the context of more organized clinical investigations.
In closing, it is worth remembering that even if SVR rates >90% are reached, the identification of infected patients, global access to drugs and implementation of therapy will be major challenges. Assuming it were possible to identify and treat every infected person, an overall failure rate of even 5% will still leave nearly 10 million people without treatment options. This is hardly a niche population or an orphan disease scenario. Hence, it is still important to keep up the momentum and invest in hepatitis C research and clinical development until options that work are available for all.
References
Thomas, D.L. Nat. Med. 19, 850–858 (2013).
Feld, J.J. & Hoofnagle, J.H. Mechanism of action of interferon and ribavirin in treatment of hepatitis C. Nature 436, 967–972 (2005).
Manns, M.P., Wedemeyer, H. & Cornberg, M. Treating viral hepatitis C: Efficacy, side effects, and complications. Gut 55, 1350–1359 (2006).
Jacobson, I.M. et al. Telaprevir for previously untreated chronic hepatitis C virus infection. N. Engl. J. Med. 364, 2405–2416 (2011).
Poordad, F. et al. Boceprevir for untreated chronic HCV genotype 1 infection. N. Engl. J. Med. 364, 1195–1206 (2011).
Sarrazin, C., Hezode, C., Zeuzem, S. & Pawlotsky, J.M. Antiviral strategies in hepatitis C virus infection. J. Hepatol. 56 (suppl. 1), S88–S100 (2012).
Liang, T.J. Nat. Med. 19, 869–878 (2013).
Choo, Q.L. et al. Isolation of a cDNA clone derived from a blood-borne non-A, non-B viral hepatitis genome. Science 244, 359–362 (1989).
Alter, H.J. et al. Detection of antibody to hepatitis C virus in prospectively followed transfusion recipients with acute and chronic non-A, non-B hepatitis. N. Engl. J. Med. 321, 1494–1500 (1989).
Kuo, G. et al. An assay for circulating antibodies to a major etiologic virus of human non-A, non-B hepatitis. Science 244, 362–364 (1989).
Bukh, J. Animal models for the study of hepatitis C virus infection and related liver disease. Gastroenterology 142, 1279–1287.e3 (2012).
Mercer, D.F. et al. Hepatitis C virus replication in mice with chimeric human livers. Nat. Med. 7, 927–933 (2001).
Dorner, M. et al. A genetically humanized mouse model for hepatitis C virus infection. Nature 474, 208–211 (2011).
Lohmann, V. et al. Replication of subgenomic hepatitis C virus RNAs in a hepatoma cell line. Science 285, 110–113 (1999).
Bartosch, B., Dubuisson, J. & Cosset, F.L. Infectious hepatitis C virus pseudo-particles containing functional E1-E2 envelope protein complexes. J. Exp. Med. 197, 633–642 (2003).
Hsu, M. et al. Hepatitis C virus glycoproteins mediate pH-dependent cell entry of pseudotyped retroviral particles. Proc. Natl. Acad. Sci. USA 100, 7271–7276 (2003).
Wakita, T. et al. Production of infectious hepatitis C virus in tissue culture from a cloned viral genome. Nat. Med. 11, 791–796 (2005).
Lindenbach, B.D. et al. Complete replication of hepatitis C virus in cell culture. Science 309, 623–626 (2005).
Zhong, J. et al. Robust hepatitis C virus infection in vitro. Proc. Natl. Acad. Sci. USA 102, 9294–9299 (2005).
Kapoor, A. et al. Characterization of a canine homolog of hepatitis C virus. Proc. Natl. Acad. Sci. USA 108, 11608–11613 (2011).
Kapoor, A. et al. Identification of rodent homologs of hepatitis C virus and pegiviruses. MBio 4, e00216–e00213 (2013).
Quan, P.L. et al. Bats are a major natural reservoir for hepaciviruses and pegiviruses. Proc. Natl. Acad. Sci. USA 110, 8194–8199 (2013).
Hoffman, B. & Liu, Q. Hepatitis C viral protein translation: Mechanisms and implications in developing antivirals. Liver Int. 31, 1449–1467 (2011).
Gottwein, J.M. & Bukh, J. Cutting the gordian knot-development and biological relevance of hepatitis C virus cell culture systems. Adv. Virus Res. 71, 51–133 (2008).
Bartenschlager, R., Penin, F., Lohmann, V. & Andre, P. Assembly of infectious hepatitis C virus particles. Trends Microbiol. 19, 95–103 (2011).
Negro, F. Steatosis and insulin resistance in response to treatment of chronic hepatitis C. J. Viral Hepat. 19 (suppl. 1), 42–47 (2012).
van der Meer, A.J. et al. Association between sustained virological response and all-cause mortality among patients with chronic hepatitis C and advanced hepatic fibrosis. J. Am. Med. Assoc. 308, 2584–2593 (2012).
Kau, A., Vermehren, J. & Sarrazin, C. Treatment predictors of a sustained virologic response in hepatitis B and C. J. Hepatol. 49, 634–651 (2008).
Horner, S.M. & Gale, M. Jr. Nat. Med. 19, 879–888 (2013).
Gastaminza, P. et al. Ultrastructural and biophysical characterization of hepatitis C virus particles produced in cell culture. J. Virol. 84, 10999–11009 (2010).
Bassendine, M.F. et al. Hcv and the hepatic lipid pathway as a potential treatment target. J. Hepatol. 55, 1428–1440 (2011).
André, P. et al. Characterization of low- and very-low-density hepatitis C virus RNA-containing particles. J. Virol. 76, 6919–6928 (2002).
Merz, A. et al. Biochemical and morphological properties of hepatitis C virus particles and determination of their lipidome. J. Biol. Chem. 286, 3018–3032 (2011).
Lindenbach, B.D. et al. Cell culture-grown hepatitis C virus is infectious in vivo and can be recultured in vitro. Proc. Natl. Acad. Sci. USA 103, 3805–3809 (2006).
Chang, K.S., Jiang, J., Cai, Z. & Luo, G. Human apolipoprotein E is required for infectivity and production of hepatitis C virus in cell culture. J. Virol. 81, 13783–13793 (2007).
Cheng, G. et al. A virocidal amphipathic α-helical peptide that inhibits hepatitis C virus infection in vitro. Proc. Natl. Acad. Sci. USA 105, 3088–3093 (2008).
Burton, D.R., Poignard, P., Stanfield, R.L. & Wilson, I.A. Broadly neutralizing antibodies present new prospects to counter highly antigenically diverse viruses. Science 337, 183–186 (2012).
Gottwein, J.M. et al. Development and characterization of hepatitis C virus genotype 1–7 cell culture systems: Role of CD81 and scavenger receptor class B type I and effect of antiviral drugs. Hepatology 49, 364–377 (2009).
Meuleman, P. et al. In vivo evaluation of the cross-genotype neutralizing activity of polyclonal antibodies against hepatitis C virus. Hepatology 53, 755–762 (2011).
von Hahn, T. et al. Hepatitis C virus continuously escapes from neutralizing antibody and T-cell responses during chronic infection in vivo. Gastroenterology 132, 667–678 (2007).
Farci, P. et al. Prevention of hepatitis C virus infection in chimpanzees by hyperimmune serum against the hypervariable region 1 of the envelope 2 protein. Proc. Natl. Acad. Sci. USA 93, 15394–15399 (1996).
Prentoe, J. et al. Hypervariable region 1 differentially impacts viability of hepatitis C virus strains of genotypes 1 to 6 and impairs virus neutralization. J. Virol. 85, 2224–2234 (2011).
Galun, E. et al. Clinical evaluation (phase I) of a human monoclonal antibody against hepatitis C virus: Safety and antiviral activity. J. Hepatol. 46, 37–44 (2007).
Law, M. et al. Broadly neutralizing antibodies protect against hepatitis C virus quasispecies challenge. Nat. Med. 14, 25–27 (2008).
Giang, E. et al. Human broadly neutralizing antibodies to the envelope glycoprotein complex of hepatitis C virus. Proc. Natl. Acad. Sci. USA 109, 6205–6210 (2012).
Barth, H. et al. Cellular binding of hepatitis C virus envelope glycoprotein e2 requires cell surface heparan sulfate. J. Biol. Chem. 278, 41003–41012 (2003).
Agnello, V., Abel, G., Elfahal, M., Knight, G.B. & Zhang, Q.X. Hepatitis C virus and other flaviviridae viruses enter cells via low density lipoprotein receptor. Proc. Natl. Acad. Sci. USA 96, 12766–12771 (1999).
Scarselli, E. et al. The human scavenger receptor class B type I is a novel candidate receptor for the hepatitis C virus. EMBO J. 21, 5017–5025 (2002).
Pileri, P. et al. Binding of hepatitis C virus to CD81. Science 282, 938–941 (1998).
Evans, M.J. et al. Claudin-1 is a hepatitis C virus co-receptor required for a late step in entry. Nature 446, 801–805 (2007).
Ploss, A. et al. Human occludin is a hepatitis C virus entry factor required for infection of mouse cells. Nature 457, 882–886 (2009).
Shulla, A. & Randall, G. Hepatitis C virus-host interactions, replication, and viral assembly. Curr. Opin. Virol. 2, 725–732 (2012).
Zheng, A. et al. Claudin-6 and claudin-9 function as additional coreceptors for hepatitis C virus. J. Virol. 81, 12465–12471 (2007).
Lupberger, J. et al. EGFR and EPHA2 are host factors for hepatitis C virus entry and possible targets for antiviral therapy. Nat. Med. 17, 589–595 (2011).
Sainz, B. Jr. et al. Identification of the Niemann-Pick C1-like 1 cholesterol absorption receptor as a new hepatitis C virus entry factor. Nat. Med. 18, 281–285 (2012).
Blanchard, E. et al. Hepatitis C virus entry depends on clathrin-mediated endocytosis. J. Virol. 80, 6964–6972 (2006).
Tscherne, D.M. et al. Time- and temperature-dependent activation of hepatitis C virus for low-pH-triggered entry. J. Virol. 80, 1734–1741 (2006).
Koutsoudakis, G. et al. Characterization of the early steps of hepatitis C virus infection by using luciferase reporter viruses. J. Virol. 80, 5308–5320 (2006).
Sharma, N.R. et al. Hepatitis C virus is primed by CD81 protein for low pH-dependent fusion. J. Biol. Chem. 286, 30361–30376 (2011).
Krey, T. et al. The disulfide bonds in glycoprotein E2 of hepatitis C virus reveal the tertiary organization of the molecule. PLoS Pathog. 6, e1000762 (2010).
El Omari, K., Iourin, O., Harlos, K., Grimes, J.M. & Stuart, D.I. Structure of a pestivirus envelope glycoprotein E2 clarifies its role in cell entry. Cell Rep. 3, 30–35 (2013).
Li, Y., Wang, J., Kanai, R. & Modis, Y. Crystal structure of glycoprotein E2 from bovine viral diarrhea virus. Proc. Natl. Acad. Sci. USA 110, 6805–6810 (2013).
Timpe, J.M. et al. Hepatitis C virus cell-cell transmission in hepatoma cells in the presence of neutralizing antibodies. Hepatology 47, 17–24 (2008).
Meuleman, P. et al. Anti-cd81 antibodies can prevent a hepatitis C virus infection in vivo. Hepatology 48, 1761–1768 (2008).
Meuleman, P. et al. A human monoclonal antibody targeting scavenger receptor class B type I precludes hepatitis C virus infection and viral spread in vitro and in vivo. Hepatology 55, 364–372 (2012).
Lemon, S.M. et al. Development of novel therapies for hepatitis C. Antiviral Res. 86, 79–92 (2010).
Syder, A.J. et al. Small molecule scavenger receptor BI antagonists are potent HCV entry inhibitors. J. Hepatol. 54, 48–55 (2011).
Zhu, H. et al. Evaluation of ITC 5061, a scavenger receptor B1 antagonist: Resistance selection and activity in combination with other hepatitis C virus antivirals. J. Infect. Dis. 205, 656–662 (2012).
Liu, R. et al. A peptide derived from hepatitis C virus E2 envelope protein inhibits a post-binding step in HCV entry. Antiviral Res. 86, 172–179 (2010).
Vermehren, J. & Sarrazin, C. The role of resistance in HCV treatment. Best Pract. Res. Clin. Gastroenterol. 26, 487–503 (2012).
Clark, V.C., Peter, J.A. & Nelson, D.R. New therapeutic strategies in HCV: Second-generation protease inhibitors. Liver Int. 33 (suppl. 1), 80–84 (2013).
Lorenz, I.C., Marcotrigiano, J., Dentzer, T.G. & Rice, C.M. Structure of the catalytic domain of the hepatitis C virus NS2–3 protease. Nature 442, 831–835 (2006).
Jirasko, V. et al. Structural and functional studies of nonstructural protein 2 of the hepatitis C virus reveal its key role as organizer of virion assembly. PLoS Pathog. 6, e1001233 (2010).
Popescu, C.I. et al. Ns2 protein of hepatitis C virus interacts with structural and non-structural proteins towards virus assembly. PLoS Pathog. 7, e1001278 (2011).
Egger, D. et al. Expression of hepatitis C virus proteins induces distinct membrane alterations including a candidate viral replication complex. J. Virol. 76, 5974–5984 (2002).
Romero-Brey, I. et al. Three-dimensional architecture and biogenesis of membrane structures associated with hepatitis C virus replication. PLoS Pathog. 8, e1003056 (2012).
Lesburg, C.A. et al. Crystal structure of the RNA-dependent RNA polymerase from hepatitis C virus reveals a fully encircled active site. Nat. Struct. Biol. 6, 937–943 (1999).
Raney, K.D., Sharma, S.D., Moustafa, I.M. & Cameron, C.E. Hepatitis C virus non-structural protein 3 (HCV NS3): A multifunctional antiviral target. J. Biol. Chem. 285, 22725–22731 (2010).
Tellinghuisen, T.L., Marcotrigiano, J., Gorbalenya, A.E. & Rice, C.M. The NS5A protein of hepatitis C virus is a zinc metalloprotein. J. Biol. Chem. 279, 48576–48587 (2004).
Tellinghuisen, T.L., Marcotrigiano, J. & Rice, C.M. Structure of the zinc-binding domain of an essential component of the hepatitis C virus replicase. Nature 435, 374–379 (2005).
Love, R.A., Brodsky, O., Hickey, M.J., Wells, P.A. & Cronin, C.N. Crystal structure of a novel dimeric form of NS5A domain I protein from hepatitis C virus. J. Virol. 83, 4395–4403 (2009).
Tellinghuisen, T.L., Foss, K.L., Treadaway, J.C. & Rice, C.M. Identification of residues required for RNA replication in domains II and III of the hepatitis C virus NS5A protein. J. Virol. 82, 1073–1083 (2008).
Appel, N. et al. Essential role of domain III of nonstructural protein 5a for hepatitis C virus infectious particle assembly. PLoS Pathog. 4, e1000035 (2008).
Tellinghuisen, T.L., Foss, K.L. & Treadaway, J. Regulation of hepatitis C virion production via phosphorylation of the NS5A protein. PLoS Pathog. 4, e1000032 (2008).
Neddermann, P. et al. Reduction of hepatitis C virus NS5A hyperphosphorylation by selective inhibition of cellular kinases activates viral RNA replication in cell culture. J. Virol. 78, 13306–13314 (2004).
Lawitz, E. et al. Sofosbuvir for previously untreated chronic hepatitis C infection. N. Engl. J. Med. 368, 1878–1887 (2013).
Jacobson, I.M. et al. Sofosbuvir for hepatitis C genotype 2 or 3 in patients without treatment options. N. Engl. J. Med. 368, 1867–1877 (2013).
Arnold, J.J. et al. Sensitivity of mitochondrial transcription and resistance of RNA polymerase II dependent nuclear transcription to antiviral ribonucleosides. PLoS Pathog. 8, e1003030 (2012).
Gao, M. et al. Chemical genetics strategy identifies an HCV NS5A inhibitor with a potent clinical effect. Nature 465, 96–100 (2010).
Scheel, T.K., Gottwein, J.M., Mikkelsen, L.S., Jensen, T.B. & Bukh, J. Recombinant HCV variants with NS5A from genotypes 1–7 have different sensitivities to an NS5A inhibitor but not interferon-α. Gastroenterology 140, 1032–1042 (2011).
Guedj, J. et al. Modeling shows that the NS5A inhibitor daclatasvir has two modes of action and yields a shorter estimate of the hepatitis C virus half-life. Proc. Natl. Acad. Sci. USA 110, 3991–3996 (2013).
Pol, S. et al. Daclatasvir for previously untreated chronic hepatitis C genotype-1 infection: A randomised, parallel-group, double-blind, placebo-controlled, dose-finding, phase 2a trial. Lancet Infect. Dis. 12, 671–677 (2012).
Sulkowski, M. et al. Sustained virologic response with daclatasvir plus sofosbuvir ± ribavirin (RBV) in chronic HCV genotype (GT) 1-infected patients who previously failed telaprevir (TVR) or boceprevir (BOC). J. Hepatol. 58, S570 (2013).
Gu, M. & Rice, C.M. Three conformational snapshots of the hepatitis C virus ns3 helicase reveal a ratchet translocation mechanism. Proc. Natl. Acad. Sci. USA 107, 521–528 (2010).
Rai, R. & Deval, J. New opportunities in anti-hepatitis C virus drug discovery: Targeting ns4b. Antiviral Res. 90, 93–101 (2011).
Einav, S. et al. Discovery of a hepatitis C target and its pharmacological inhibitors by microfluidic affinity analysis. Nat. Biotechnol. 26, 1019–1027 (2008).
Esser-Nobis, K. et al. Analysis of hepatitis C virus resistance to silibinin in vitro and in vivo points to a novel mechanism involving nonstructural protein 4b. Hepatology 57, 953–963 (2013).
Watashi, K., Hijikata, M., Hosaka, M., Yamaji, M. & Shimotohno, K. Cyclosporin a suppresses replication of hepatitis C virus genome in cultured hepatocytes. Hepatology 38, 1282–1288 (2003).
Paeshuyse, J. et al. The non-immunosuppressive cyclosporin debio-025 is a potent inhibitor of hepatitis C virus replication in vitro. Hepatology 43, 761–770 (2006).
Hopkins, S. et al. Scy-635, a novel nonimmunosuppressive analog of cyclosporine that exhibits potent inhibition of hepatitis C virus RNA replication in vitro. Antimicrob. Agents Chemother. 54, 660–672 (2010).
Yang, F. et al. Cyclophilin A is an essential cofactor for hepatitis C virus infection and the principal mediator of cyclosporine resistance in vitro. J. Virol. 82, 5269–5278 (2008).
Rosnoblet, C. et al. Hepatitis C virus NS5B and host cyclophilin a share a common binding site on NS5A. J. Biol. Chem. 287, 44249–44260 (2012).
Hopkins, S. et al. The cyclophilin inhibitor scy-635 suppresses viral replication and induces endogenous interferons in patients with chronic HCV genotype 1 infection. J. Hepatol. 57, 47–54 (2012).
Flisiak, R. et al. The cyclophilin inhibitor debio 025 combined with peg IFN-α2a significantly reduces viral load in treatment-naive hepatitis C patients. Hepatology 49, 1460–1468 (2009).
Jopling, C.L., Yi, M., Lancaster, A.M., Lemon, S.M. & Sarnow, P. Modulation of hepatitis C virus RNA abundance by a liver-specific microRNA. Science 309, 1577–1581 (2005).
Machlin, E.S., Sarnow, P. & Sagan, S.M. Masking the 5′ terminal nucleotides of the hepatitis C virus genome by an unconventional microRNA-target RNA complex. Proc. Natl. Acad. Sci. USA 108, 3193–3198 (2011).
Jangra, R.K., Yi, M. & Lemon, S.M. Regulation of hepatitis C virus translation and infectious virus production by the microRNA miR-122. J. Virol. 84, 6615–6625 (2010).
Li, Y., Masaki, T., Yamane, D., McGivern, D.R. & Lemon, S.M. Competing and noncompeting activities of miR-122 and the 5′ exonuclease xrn1 in regulation of hepatitis C virus replication. Proc. Natl. Acad. Sci. USA 110, 1881–1886 (2013).
Shimakami, T. et al. Stabilization of hepatitis C virus RNA by an ago2-miR-122 complex. Proc. Natl. Acad. Sci. USA 109, 941–946 (2012).
Lanford, R.E. et al. Therapeutic silencing of microRNA-122 in primates with chronic hepatitis C virus infection. Science 327, 198–201 (2010).
Li, Y.P., Gottwein, J.M., Scheel, T.K., Jensen, T.B. & Bukh, J. MicroRNA-122 antagonism against hepatitis C virus genotypes 1–6 and reduced efficacy by host RNA insertion or mutations in the HCV 5′ UTR. Proc. Natl. Acad. Sci. USA 108, 4991–4996 (2011).
Hsu, S.H. et al. Essential metabolic, anti-inflammatory, and anti-tumorigenic functions of miR-122 in liver. J. Clin. Invest. 122, 2871–2883 (2012).
Tsai, W.C. et al. MicroRNA-122 plays a critical role in liver homeostasis and hepatocarcinogenesis. J. Clin. Invest. 122, 2884–2897 (2012).
Janssen, H.L. et al. Treatment of HCV infection by targeting microRNA. N. Engl. J. Med. 368, 1685–1694 (2013).
Ye, J. et al. Disruption of hepatitis C virus RNA replication through inhibition of host protein geranylgeranylation. Proc. Natl. Acad. Sci. USA 100, 15865–15870 (2003).
Kapadia, S.B. & Chisari, F.V. Hepatitis C virus RNA replication is regulated by host geranylgeranylation and fatty acids. Proc. Natl. Acad. Sci. USA 102, 2561–2566 (2005).
Harrison, S.A. et al. Serum cholesterol and statin use predict virological response to peginterferon and ribavirin therapy. Hepatology 52, 864–874 (2010).
Bishé, B., Syed, G. & Siddiqui, A. Phosphoinositides in the hepatitis C virus life cycle. Viruses 4, 2340–2358 (2012).
Berger, K.L. et al. Roles for endocytic trafficking and phosphatidylinositol 4-kinase III -α in hepatitis C virus replication. Proc. Natl. Acad. Sci. USA 106, 7577–7582 (2009).
Reiss, S. et al. Recruitment and activation of a lipid kinase by hepatitis C virus NS5A is essential for integrity of the membranous replication compartment. Cell Host Microbe 9, 32–45 (2011).
Bianco, A. et al. Metabolism of phosphatidylinositol 4-kinase III-α-dependent PI4P is subverted by HCV and is targeted by a 4-anilino quinazoline with antiviral activity. PLoS Pathog. 8, e1002576 (2012).
Vaillancourt, F.H. et al. Evaluation of phosphatidylinositol-4-kinase III-α as a hepatitis C virus drug target. J. Virol. 86, 11595–11607 (2012).
McLauchlan, J., Lemberg, M.K., Hope, G. & Martoglio, B. Intramembrane proteolysis promotes trafficking of hepatitis C virus core protein to lipid droplets. EMBO J. 21, 3980–3988 (2002).
Boulant, S. et al. Structural determinants that target the hepatitis C virus core protein to lipid droplets. J. Biol. Chem. 281, 22236–22247 (2006).
Herker, E. et al. Efficient hepatitis C virus particle formation requires diacylglycerol acyltransferase-1. Nat. Med. 16, 1295–1298 (2010).
Miyanari, Y. et al. The lipid droplet is an important organelle for hepatitis C virus production. Nat. Cell Biol. 9, 1089–1097 (2007).
Shavinskaya, A., Boulant, S., Penin, F., McLauchlan, J. & Bartenschlager, R. The lipid droplet binding domain of hepatitis C virus core protein is a major determinant for efficient virus assembly. J. Biol. Chem. 282, 37158–37169 (2007).
Lavie, M., Goffard, A. & Dubuisson, J. Assembly of a functional HCV glycoprotein heterodimer. Curr. Issues Mol. Biol. 9, 71–86 (2007).
Steinmann, E. et al. Antiviral effects of amantadine and iminosugar derivatives against hepatitis C virus. Hepatology 46, 330–338 (2007).
Griffin, S. et al. Genotype-dependent sensitivity of hepatitis C virus to inhibitors of the p7 ion channel. Hepatology 48, 1779–1790 (2008).
Durantel, D. Celgosivir, an α-glucosidase I inhibitor for the potential treatment of HCV infection. Curr. Opin. Investig. Drugs 10, 860–870 (2009).
Wozniak, A.L. et al. Intracellular proton conductance of the hepatitis C virus p7 protein and its contribution to infectious virus production. PLoS Pathog. 6, e1001087 (2010).
Tanwandee, T. et al. High sustained viral response with a HCV p7 inhibitor, bit225: Antiviral activity and tolerability of bit225 plus pegylated interferon alfa 2b and weight-based ribavirin for 28 days in HCV treatment-naive patients. Hepatology 56, 1530 (2012).
Huang, H. et al. Hepatitis C virus production by human hepatocytes dependent on assembly and secretion of very low-density lipoproteins. Proc. Natl. Acad. Sci. USA 104, 5848–5853 (2007).
Gastaminza, P. et al. Cellular determinants of hepatitis C virus assembly, maturation, degradation, and secretion. J. Virol. 82, 2120–2129 (2008).
Counihan, N.A., Rawlinson, S.M. & Lindenbach, B.D. Trafficking of hepatitis C virus core protein during virus particle assembly. PLoS Pathog. 7, e1002302 (2011).
Coller, K.E. et al. Molecular determinants and dynamics of hepatitis C virus secretion. PLoS Pathog. 8, e1002466 (2012).
Jiang, J. & Luo, G. Apolipoprotein E but not B is required for the formation of infectious hepatitis C virus particles. J. Virol. 83, 12680–12691 (2009).
Hunt, D. & Pockros, P. What are the promising new therapies in the field of chronic hepatitis C after the first-generation direct-acting antivirals? Curr. Gastroenterol. Rep. 15, 303 (2013).
Jacobson, I. et al. Simeprevir (TMC435) with peginterferon/ribavirin for chronic HCV genotype-1 infection in treatment-naïve patients: Results from QUEST-1, a phase III trial. J. Hepatol. 58, S574 (2013).
Manns, M. et al. Simeprevir (TMC435) with peginterferon/ribavirin for treatment of chronic HCV genotype-1 infection in treatment-naïve patients: Results from QUEST-2, a phase III trial. J. Hepatol. 58, S568 (2013).
Ferenci, P. et al. Faldaprevir plus pegylated interferon alfa-2a and ribavirin in chronic HCV genotype-1 treatment-naïve patients: Final results from startverso1, a randomised, double-blind, placebo-controlled phase III trial. J. Hepatol. 58, S569 (2013).
Lok, A.S. et al. Sustained virologic response in chronic HCV genotype (GT) 1-infected null responders with combination of daclatasvir (DCV; NS5A inhibitor) and asunaprevir (ASV; ns3 inhibitor) with or without peginterferon alfa-2a/ribavirin (PEG/RBV). Hepatology 56, 230A–231A (2012).
Thompson, A.J. et al. Six weeks of a NS5A inhibitor (GS-5885), protease inhibitor (GS-9451) plus peginterferon/ribavirin (PR) achieves high SVR4 rates in genotype 1 IL28B CC treatment naive HCV patients: Interim results of a prospective, randomized trial. Hepatology 56, 556A–557A (2012).
Feld, J.J. et al. Up to 100% svr4 rates with ritonavir-boosted danoprevir (DNVR), mericitabine (MCB) and ribavirin (R) ± peginterferon alfa-2a (40kd) (P) in HCV genotype 1-infected partial and null responders: Results from the matterhorn study. Hepatology 56, 231A–232A (2012).
Muir, A.J. et al. Peginterferon lambda-1a (lambda) compared to peginterferon alfa-2a (alfa) in treatment-naive patients with HCV genotypes (GT) 1 or 4: Svr24 results from emerge phase 2b. Hepatology 56, 299A (2012).
Olsen, D.B. et al. Sustained viral response in a hepatitis C virus-infected chimpanzee via a combination of direct-acting antiviral agents. Antimicrob. Agents Chemother. 55, 937–939 (2011).
Ohara, E. et al. Elimination of hepatitis C virus by short term NS3–4A and NS5B inhibitor combination therapy in human hepatocyte chimeric mice. J. Hepatol. 54, 872–878 (2011).
Lok, A.S. et al. Preliminary study of two antiviral agents for hepatitis C genotype 1. N. Engl. J. Med. 366, 216–224 (2012).
Kowdley, K. et al. Safety and efficacy of interferon-free regimens of ABT-450/R, ABT-267, ABT-333 ± ribavirin in patients with chronic HCV gt1 infection: Results from the aviator study. J. Hepatol. 58, S2 (2013).
Everson, G. et al. Interim analysis of an interferon (IFN)- and ribavirin (RBV)-free regimen of daclatasvir (DCV), asunaprevir (ASV), and BMS-791325 in treatment-naive, hepatitis C virus genotype 1–infected patients. J. Hepatol. 58, S573 (2013).
Gane, E. et al. All-oral sofosbuvir-based 12-week regimens for the treatment of chronic HCV infection: The electron study. J. Hepatol. 58, S6–S7 (2013).
Sulkowski, M. et al. High rate of sustained virologic response with the all-oral combination of daclatasvir (NS5A inhibitor) plus sofosbuvir (nucleotide NS5B inhibitor) with or without ribavirin, in treatment-naive patients chronically infected with HCV GT 1, 2, or 3. Hepatology 56, 1516–1517 (2012).
Poordad, F. et al. Exploratory study of oral combination antiviral therapy for hepatitis C. N. Engl. J. Med. 368, 45–53 (2013).
Jacobson, I.M. et al. VX-222, telaprevir and ribavirin in treatment-naive patients with genotype 1 chronic hepatitis C: Results of the zenith study interferon-free regimen. Hepatology 56, 308A (2012).
Zeuzem, S. et al. An analysis of response rates by fibrosis stage in patients treated with faldaprevir, BI 207127 and ribavirin in the SOUND-C2 study. J. Hepatol. 58, S498 (2013).
Gane, E.J. et al. Nucleotide polymerase inhibitor sofosbuvir plus ribavirin for hepatitis C. N. Engl. J. Med. 368, 34–44 (2013).
Zeuzem, S. et al. Interferon (IFN)-free combination treatment with the HCV NS3/4A protease inhibitor bi 201335 and the non-nucleoside NS5B inhibitor BI 207127 +/− ribavirin (R): Final results of sound-c2 and predictors of response. Hepatology 56, 308A–309A (2012).
Saeed, M. et al. Efficient replication of genotype 3a and 4a HCV replicons in human hepatoma cells. Antimicrob. Agents Chemother. 56, 5365–5373 (2012).
Li, Y.P. et al. Highly efficient full-length hepatitis C virus genotype 1 (strain TN) infectious culture system. Proc. Natl. Acad. Sci. USA 109, 19757–19762 (2012).
Li, Y.P. et al. Robust full-length hepatitis C virus genotype 2a and 2b infectious cultures using mutations identified by a systematic approach applicable to patient strains. Proc. Natl. Acad. Sci. USA 109, E1101–E1110 (2012).
McPhee, F. et al. Resistance analysis of hepatitis C virus genotype 1 prior treatment null responders receiving daclatasvir and asunaprevir. Hepatology published online, http://dx.doi.org/10.1002/hep.26388 (2013).
Pawlotsky, J.M. et al. Alisporivir plus ribavirin achieves high rates of sustained HCV clearance (SVR24) as interferon (IFN)-free or IFN-add-on regimen in treatment-naive patients with HCV GT2 or GT3: Final results from vital-1 study. Hepatology 56, 309A–310A (2012).
Marukian, S. et al. Hepatitis C virus induces interferon-λ and interferon-stimulated genes in primary liver cultures. Hepatology 54, 1913–1923 (2011).
Love, R.A. et al. The crystal structure of hepatitis C virus NS3 proteinase reveals a trypsin-like fold and a structural zinc binding site. Cell 87, 331–342 (1996).
Wölk, B. et al. Subcellular localization, stability, and trans-cleavage competence of the hepatitis C virus NS3-NS4A complex expressed in tetracycline-regulated cell lines. J. Virol. 74, 2293–2304 (2000).
Moradpour, D., Penin, F. & Rice, C.M. Replication of hepatitis C virus. Nat. Rev. Microbiol. 5, 453–463 (2007).
Kolykhalov, A.A. et al. Transmission of hepatitis C by intrahepatic inoculation with transcribed RNA. Science 277, 570–574 (1997).
Yanagi, M., Purcell, R.H., Emerson, S.U. & Bukh, J. Transcripts from a single full-length cDNA clone of hepatitis C virus are infectious when directly transfected into the liver of a chimpanzee. Proc. Natl. Acad. Sci. USA 94, 8738–8743 (1997).
Gottwein, J.M. et al. Novel infectious cDNA clones of hepatitis C virus genotype 3a (strain S52) and 4a (strain ED43): Genetic analyses and in vivo pathogenesis studies. J. Virol. 84, 5277–5293 (2010).
Bitzegeio, J. et al. Adaptation of hepatitis C virus to mouse CD81 permits infection of mouse cells in the absence of human entry factors. PLoS Pathog. 6, e1000978 (2010).
Blight, K.J., Kolykhalov, A.A. & Rice, C.M. Efficient initiation of HCV RNA replication in cell culture. Science 290, 1972–1974 (2000).
Bukh, J. et al. Mutations that permit efficient replication of hepatitis C virus RNA in Huh-7 cells prevent productive replication in chimpanzees. Proc. Natl. Acad. Sci. USA 99, 14416–14421 (2002).
Kaul, A., Woerz, I., Meuleman, P., Leroux-Roels, G. & Bartenschlager, R. Cell culture adaptation of hepatitis C virus and in vivo viability of an adapted variant. J. Virol. 81, 13168–13179 (2007).
Pietschmann, T. et al. Construction and characterization of infectious intragenotypic and intergenotypic hepatitis C virus chimeras. Proc. Natl. Acad. Sci. USA 103, 7408–7413 (2006).
Imhof, I. & Simmonds, P. Development of an intergenotypic hepatitis C virus (HCV) cell culture method to assess antiviral susceptibilities and resistance development of HCV NS3 protease genes from HCV genotypes 1 to 6. J. Virol. 84, 4597–4610 (2010).
Gastaminza, P., Whitten-Bauer, C. & Chisari, F.V. Unbiased probing of the entire hepatitis C virus life cycle identifies clinical compounds that target multiple aspects of the infection. Proc. Natl. Acad. Sci. USA 107, 291–296 (2010).
Chockalingam, K., Simeon, R.L., Rice, C.M. & Chen, Z. A cell protection screen reveals potent inhibitors of multiple stages of the hepatitis C virus life cycle. Proc. Natl. Acad. Sci. USA 107, 3764–3769 (2010).
Acknowledgements
We are grateful to I.M. Jacobson for critical reading of the manuscript. Our research is supported by grants from the US Public Health Service, the National Institutes of Health (NIH), National Institute of Allergy and Infectious Diseases (AI099284, AI072613, AI075099, AI091707, AI090055), Office of the Director through the NIH Roadmap for Medical Research (DK085713), US National Cancer Institute (CA057973), The Rockefeller University Center for Clinical and Translational Science (UL1RR024143), the Center for Basic and Translational Research on Disorders of the Digestive System through the generosity of the Leona M. and Harry B. Helmsley Charitable Trust, the Greenberg Medical Research Institute and the Starr Foundation. T.K.H.S. is supported by a postdoctoral fellowship and a Sapere Aude Research Talent Award from The Danish Council for Independent Research. We apologize to the many HCV investigators whose work could not be cited owing to space restrictions.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
C.M.R. declares an equity interest in Apath, which holds commercial rights to HCV-related technology, and has in addition served as a consultant or scientific advisor to Genentech, GlaxoSmithKline, iTherX, Merck & Co. Inc. and Novartis. C.M.R. and T.K.H.S. both hold patent rights to HCV-related technology.
Rights and permissions
About this article
Cite this article
Scheel, T., Rice, C. Understanding the hepatitis C virus life cycle paves the way for highly effective therapies. Nat Med 19, 837–849 (2013). https://doi.org/10.1038/nm.3248
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/nm.3248
This article is cited by
-
Mitochondrial DNA copy number in Hepatitis C virus-related chronic liver disease: impact of direct-acting antiviral therapy
Scientific Reports (2023)
-
A single mutation in the E2 glycoprotein of hepatitis C virus broadens the claudin specificity for its infection
Scientific Reports (2022)
-
Computational Design and Analysis of a Poly-Epitope Fusion Protein: A New Vaccine Candidate for Hepatitis and Poliovirus
International Journal of Peptide Research and Therapeutics (2020)
-
Whole-genome sequencing of human Pegivirus variant from an Egyptian patient co-infected with hepatitis C virus: a case report
Virology Journal (2019)
-
Impact of Direct Acting Antivirals on Survival in Patients with Chronic Hepatitis C and Hepatocellular Carcinoma
Scientific Reports (2019)
