
Abstract
The avascularity of cardiac valves is abrogated in several valvular heart diseases (VHDs). This study investigated the molecular mechanisms underlying valvular avascularity and its correlation with VHD. Chondromodulin-I, an antiangiogenic factor isolated from cartilage, is abundantly expressed in cardiac valves. Gene targeting of chondromodulin-I resulted in enhanced Vegf-A expression, angiogenesis, lipid deposition and calcification in the cardiac valves of aged mice. Echocardiography showed aortic valve thickening, calcification and turbulent flow, indicative of early changes in aortic stenosis. Conditioned medium obtained from cultured valvular interstitial cells strongly inhibited tube formation and mobilization of endothelial cells and induced their apoptosis; these effects were partially inhibited by chondromodulin-I small interfering RNA. In human VHD, including cases associated with infective endocarditis, rheumatic heart disease and atherosclerosis, VEGF-A expression, neovascularization and calcification were observed in areas of chondromodulin-I downregulation. These findings provide evidence that chondromodulin-I has a pivotal role in maintaining valvular normal function by preventing angiogenesis that may lead to VHD.
Similar content being viewed by others


Main
A balance of angiogenic and antiangiogenic factors is critical to normal development and organ homeostasis. Although the heart is a vascular-rich organ, cardiac valves are avascular and oxygen supply is via diffusion from the blood stream1. Under pathological conditions such as atherosclerosis, rheumatic valvular heart disease (RHD) or infective endocarditis, cardiac valves express angiogenic factors leading to neovascularization2,3. The contribution of antiangiogenic factors to the maintenance of avascularity in cardiac valves is unknown.
The mesenchymal cells of cardiac valve tissue, known as valvular interstitial cells (VICs), have an incomplete basal lamina, are sparsely distributed, and have direct and extensive contacts with collagen fibers, elastin microfibrils and proteoglycans of the extracellular matrix4,5,6. Cartilage is another avascular tissue, and transcription factors considered essential for chondrogenesis during endochondral ossification, including Sox9 (ref. 7), NFATc8, Cbfa1 (ref. 9) and MSX2 (ref. 10), and growth factors such as BMP2 (ref. 11) and TGFß2 (ref. 12), are also expressed in developing cardiac valves. Sox9, in conjunction with Sox5 and Sox6, can induce the expression of genes specific to cartilage, including chondromodulin-1 (encoded by Chm1, also known as Lect1), even in nonchondrogenic cells13, and are essential to cardiac valve development7. Chm-I is a 121–amino acid residue glycoprotein derived from a 335–amino acid residue type II transmembrane precursor located primarily in avascular tissue of the eye and cartilage14,15. After translation, the Chm-I precursor is cleaved by furin proteases16, and secreted Chm-I accumulates in the interstitial space of the cartilage matrix17. The inhibition of endothelial cell proliferation and tube morphogenesis by Chm-I provided the first evidence of the antiangiogenic activity of this protein17,18,19,20.
The aim of this study was to determine the role of antiangiogenic factors in valvular heart disease (VHD). We show that CHM-I is expressed strongly in normal cardiac valves, but at markedly reduced levels in human VHD and Apoe−/− mice. We also provide direct evidence that Chm-I has a role as an antiangiogenic factor by showing that loss of Chm-I leads to neovascularization, as well as unusual calcification and infiltration of inflammatory cells into the matrix of cardiac valves. These data provide new insight into the mechanisms underlying maintenance of avascularity in cardiac valves and the disruption of this process in pathological conditions. Understanding these mechanisms may form the basis of new therapeutic regimens for the treatment of VHD.
Results
Cardiac valves express chondromodulin-I
RT-PCR for Chm1 showed a faint level of expression in the heart, cartilage and eye (Supplementary Fig. 1 online). Chm1 expression was strong in cardiac valves, but absent from the atrium and ventricle. Expression of Chm1 first appeared in the heart at embryonic day (E) 9.5 and persisted in the adult. Quantitative PCR showed that Chm1 expression was 800 times higher in cardiac valves than in the atrium or ventricle. Western-blot analysis identified the 25-kDa mature glycosylated form of Chm-I in rat and human cardiac valves, which was also present in cartilage.
Immunohistochemical analysis
Immunohistochemistry (Fig. 1 and Supplementary Fig. 2 online) showed localization of Chm-I to all four cardiac valves. Analysis of serial sections showed that Chm-I and Vegf-A expression did not overlap. Chm-I was detected throughout the VICs and extracellular matrix, but not in the outer endothelial cell layer.

(a) Chm-I was expressed in all four valves in the adult. Positive signals are shown in brown. The left two micrographs are views along the short axis; the right two micrographs are views along the long axis. AV, aortic valve; IVS, interventricular septum; LV, left ventricle; MV, mitral valve; PA, pulmonary artery; PV, pulmonary valve. (b–e) Immunofluorescence staining for Chm-I and Vegf-A in the adult mouse heart. The boxed region in b is shown in c and e; the boxed region in c is shown in d. (f) Serial sections of the mitral valve immunostained for Chm-I, vWF, vimentin and actinin. (g) Immunofluorescence staining for Chm-I and Vegf-A in various developmental stages. A, atrium; AVC, atrioventricular cushion; EC, endothelial cell; LA, left atrium; OFT, outflow tract; RV, right ventricle; TV, tricuspid valve.
During development, cardiac valve precursor cells from the atrioventricular cushions and outflow tract express Chm-I from E9.5. In E10.0 embryos, Chm-I is expressed in the cardiac jelly covering the trabeculating cardiomyocytes of the left ventricle, the outer curvature of the right ventricle and the outflow tract. Chm-I expression in the ventricles decreased gradually as development progressed, and by mid-embryogenesis, both transcripts and protein were absent. The non-overlapping localization of Chm-I and Vegf-A was apparent at all development stages.
Chm1 disruption induces angiogenesis in cardiac valves
Cardiac valves of 8-week-old Chm1−/− mice were histologically similar to those of wild-type mice. By old age (90.2 ± 7.4 weeks), the aortic valves of Chm1−/− mice were significantly more bulky and the density of VICs more sparse than in age-matched wild-type mice (P < 0.05; Fig. 2). The mean thickness of the aortic valve gradually increased after 32 weeks and reached a thickness 1.8 times greater than that of wild-type mice at 90 weeks. No significant morphological changes were observed in the mitral or other valves (P < 0.05 at 90 weeks). Cardiac valves of Chm1−/− mice had newly developed capillary-like structures and were Chm-I–negative and Vegf-A–positive. Vegf-A expression was not observed in valves from young adults but was upregulated in aged animals. Cells forming capillary-like structures were positive for von Willebrand factor (vWF)–specific antibody, confirming that they were endothelial cells. Calcium deposits in the cardiac valves of Chm1−/− mice were detected with von Kossa staining, and the presence of inflammatory cells was confirmed by Mac-1 staining. New vessel formation and invasion of inflammatory cells are indicative of sclerotic processes. Sudan IV staining showed that affected valves were laden with lipid. Deposits of calcium and lipid were relatively large at the laminae ventricularis compared with the other laminae. Chm1+/+ littermates did not show these characteristics. Capillary structures in three leaflets of the aortic valve were counted in each 20-μm section. The capillary number in Chm1−/− mice was 13.8 times higher than that in Chm1+/+ mice (P < 0.01). The aortic valves showed strong expression of Vegf-A, enhanced de novo capillary-like structures, increased calcium and lipid deposits, and Mac-1–positive staining. Changes in these characteristics were not notable in other valves.

(a,c) Hematoxylin and eosin staining of aortic valves in aged Chm1−/− and age-matched wild-type mice at low magnification. Chm1−/− mice had bulky leaflets. Chm1−/− mice were confirmed negative for Chm-I immunostaining. Vegf-A staining in the aortic valve showed a strong positive signal only in Chm1−/− mice. Only Chm1−/− mice exhibited lipid deposits (arrowheads) by Sudan IV staining. (b,d) Hematoxylin and eosin staining at high magnification. Capillary-like structures (arrowheads) were observed only in aged Chm1−/− mice. The cell density decreased at the neovascular area. Immunofluorescence staining for vWF and Mac-1 are shown; positive signals were observed only in Chm1−/− mice. Von Kossa staining showed calcium deposits in Chm1−/− mice (arrowheads). The boxed regions in a and c are shown in b and d, respectively. The cell density (e) and the number of vWF-positive cells (f) in three leaflets of the aortic valve were counted for every 20-μm section stained for immunofluorescence in which at least one leaflet was visible. The cell density and the number of vWF-positive cells were determined at 90 weeks of age for Chm1−/− (n = 12) and Chm1+/+ (n = 12) mice. (g) The mean thickness of the aortic valves was investigated at 8, 20, 32, 64 and 90 weeks of age. §P < 0.05; *P < 0.01.
Early phase aortic stenosis in Chm1−/− mice
Echocardiography revealed bright echogenic aortic valves that moved backward and forward with slight acoustic shadowing, suggestive of thickening or calcification (Fig. 3). A color Doppler study showed a mosaic turbulent jet distal to the aortic valve. No echogenic object or turbulent jet was observed in Chm1+/+ mice. An M-mode scan showed marked thickening of the aortic valve. There were no significant differences between Chm1+/+ and Chm1−/− mice in left ventricular wall thickness, left ventricular end-diastolic and end-systolic dimensions, ejection fraction, brightness of the mitral valve, or turbulent flow in the area of the mitral valve. The area trace of the aortic valve gradually increased from 32 weeks (P < 0.01) and had increased approximately 3.8-fold by 90 weeks (P < 0.001).

(a,b) Top, two-dimensional echocardiography showed an abnormal bright echogenic mass in the aortic valve leaflets in Chm1−/− mice that were not detected in the aortic valves of Chm1+/+ mice. Middle, a color Doppler study showed a mosaic turbulent jet distal to the aortic valve in Chm1−/− mice that was not detected in the aortic valve of Chm1+/+ mouse hearts. Inset shows the aortic valve level short-axis view of each long-axis figure. Bottom, M-mode scanning of the aortic valve area. Arrowheads indicate the thickened and calcified aortic valve leaflets in Chm1−/− mice. Mice were 90 weeks of age. (c) Thickness of the interventricular septum. (d) Thickness of the left ventricular posterior wall. (e) Left ventricular end-diastolic dimension (LVEDD). (f) Left ventricular end-systolic dimension (LVESD). (g) Ejection fraction (EF). (h) Time course of the area trace of the aortic valve. WT, wild-type; young KO, young adult knockout mice at 8 and 20 weeks of age; aged KO, young adult knockout mice at 90 weeks of age; Ao, aorta; AV, aortic valve; IVS, interventricular septum; LA, left atrium; LV, left ventricle; LVFW, left ventricular free wall; PW, posterior wall; RV, right ventricle. §P < 0.05; *P < 0.01; NS, not significant.
Analysis of sclerotic cardiac valves in Apoe−/− mice
The expression of antiangiogenic Chm-I and angiogenic Vegf-A was examined in the cardiac valves of aged Apoe−/− mice (87.3 ± 5.3 weeks old, n = 10) as a model of atherosclerosis with abnormal lipid deposits and calcification in cardiac valves. Chm-I was absent from calcified regions (Fig. 4) in aortic and mitral valves containing Vegf-A positive cells. Age-matched wild-type mice (88.5 ± 4.8 weeks old, n = 8) showed the expected physiological Chm-I–positive and Vegf-A–negative expression pattern and no sclerotic changes or calcification. In situ hybridization for Chm1 showed no signal in the sclerotic aortic valve leaflet in Apoe−/− mice, whereas age-matched wild-type mice showed positive signals. The Chm-I–positive area in aged Apoe−/− mice was less than 48.0% the size of that in age-matched wild-type mice (P < 0.01). The Vegf-A–positive area and the microvessel density in aged Apoe−/− mice were significantly greater compared with age-matched wild-type mice (P < 0.01).

(a) Left, immunohistochemistry of Chm-I and Vegf-A in a sclerotic lesion (arrowheads) of the mitral valve (MV) of an Apoe−/− mouse heart. Boxed area is enlarged in the middle panel. Right, an adjacent section was immunostained using the Vegf-A–specific antibody. (b) In situ hybridization of Chm1 in Apoe−/− and age-matched Apoe+/+ mouse aortic valves (AV). (c,d) Immunofluorescence staining of Chm-I and Vegf-A in Apoe−/− (c) and age-matched Apoe+/+ mice (d). Vegf-A was markedly upregulated in the sclerotic valve area where Chm-I was downregulated in Apoe−/− mice. Arrowheads indicate the valvular area in which Chm-I was downregulated and Vegf-A was upregulated in Apoe−/− mice. (e,f) Quantitative analysis of the percentage of the Chm-I–positive area and the Vegf-A–positive area in the cardiac valves of Apoe+/+ and Apoe−/− mice. (g) Quantitative analysis of the microvessel density in the cardiac valves of Apoe+/+ and Apoe−/− mice. *P < 0.01.
VIC-derived Chm-I directly suppresses angiogenesis
We next investigated whether VICs produce Chm-I and, if so, whether VIC-derived Chm-I can affect proliferation or tube morphogenesis of human coronary artery endothelial cells (HCAECs). Primary VICs were obtained from explant cultures of rat cardiac valves (Fig. 5). Explanted cells comprise a heterogeneous population of cobblestone and spindle-type cells characteristic of VICs after 3 d (ref. 21) and formed an orthogonal pattern of overgrowth5. The cells were negative for the acetyl-LDL–DiI conjugate, consistent with cardiac valves being composed of VICs. Immunostaining showed Chm-I in the cytoplasm of VICs and the absence of Chm-I in NIH3T3 cells. RT-PCR and western-blot analysis confirmed that VICs secreted Chm-I into the culture media. Furthermore, Chm-I secretion was inhibited by treatment of VICs with specific small interfering RNA (siRNA).

(a) Rat VICs after post-explant culture. At 7 d, VICs showed a cobblestone-like (Cb) or spindle-like (Sp) appearance. At 14 d, VICs exhibited a fibroblast-like appearance. Ex, explant of cardiac valve. VICs were negative for acetyl-LDL–DiI staining; HCAECs are shown as a positive control (inset). Immunofluorescence staining of Chm-I in VICs and NIH3T3 cells is shown; nuclei were stained with TOTO-3. Upper graph, RT-PCR for Chm1 in VICs; Lower graph, western-blot analysis of Chm-I expression in VIC-CM. Lane 1, VIC; lane 2, VIC + siRNA specific to Chm1; lane 3, VIC + control siRNA. (b) Tube formation assay. VIC conditioned medium (CM) inhibited endothelial cell tube formation on Matrigel. Representative micrographs of tube formation of HCAECs are shown. Tube formation was significantly suppressed by VIC-CM but not by NIH3T3-CM. Treatment of VICs with siRNA specific to Chm1 but not control siRNA reduced the VIC-CM–induced suppression. The graph shows quantitative analysis of tube lengths. (c) Cell migration assay. VICs abrogated HCAEC chemotaxis in vitro. HCAECs were inhibited from migrating when cocultured with VICs, but not when cultured without cells (control) or with NIH3T3 cells. Treatment of VICs with siRNA specific to Chm1 but not control siRNA reduced the suppression of chemotaxis. The graph shows quantitative analysis of cell migration. (d) Induction of apoptosis by VIC-CM. HCAECs cultured with NIH3T3-CM (negative control, upper panels) or VIC-CM (middle panels) were treated with FITC-conjugated Annexin V. Treatment of VICs with siRNA specific to Chm1 reduced the number of Annexin V–positive cells (lower panels). Arrowheads show Annexin V–positive cells. The graph shows quantitative analysis of Annexin V staining. *P < 0.01; **P < 0.001; NS, not significant.
HCAECs formed capillary-like tube structures on Matrigel after 6 h (ref. 22). Capillary-like structures were less prominent following treatment with conditioned medium from VICs (VIC-CM) compared with mock medium or conditioned medium from control NIH3T3 cells (NIH3T3-CM). HCAECs cultured with conditioned medium from siRNA-treated VICs regained their ability to form capillary-like structures. The total length of capillary-like structures was evaluated using NIH image software. Quantitative analysis of the length of tube formation compared with controls showed that VIC-CM inhibited tube formation by 75.9% (P < 0.001) and that Chm1-specific siRNA led to recovery of tube formation capacity by 62.9% (P < 0.001).
Chm-I also inhibited the migration capacity of HCAECs. In a modified Boyden chamber assay, HCAECs cocultured with VICs lost their migratory capacity compared with NIH3T3 cells. Treatment of VICs with Chm1-specific siRNA led to HCAECs partially regaining migratory capacity. Control siRNA had no effect on VIC-mediated inhibition of migration. Quantitative analysis showed that VICs decreased the number of migrated cells by 82.1% (P < 0.001), and that siRNA specific to Chm1 recovered the migratory capacity by 25.7% (P < 0.01). These results imply that Chm-I has a pivotal role as a chemoattractant-inhibitor in cardiac valves.
Morphological changes in HCAECs following treatment with VIC-CM were suggestive of apoptosis. To determine whether Chm-I induces apoptosis, we stained HCAECs treated with VIC-CM with Annexin V and propidium iodide. Positive staining for Annexin V–fluorescein isothiocyanate (FITC) showed induction of apoptosis, and fluorescent cells were both propidium iodide–positive (early apoptotic labeling pattern) and –negative (late apoptotic labeling pattern). HCAECs treated with NIH3T3-CM were Annexin V–FITC–negative. Annexin V–FITC–positive fluorescent cells increased by 8.0-fold in VIC-CM–treated cells (P < 0.01). VIC-CM treated with siRNA specific to Chm1 strongly attenuated VIC-mediated apoptosis (P < 0.01). The antiangiogenic activity of Chm-I was confirmed by examining the effects of purified recombinant human CHM-I on tube formation and migration in vitro. The recombinant human CHM-I inhibited tube formation and migration in HCAECs and induced apoptosis in a dose-dependent manner (Supplementary Fig. 3 online). To test whether Chm-I has hyperplastic or cell survival effects on VICs, cell survival was assessed using the MTT assay with or without siRNA specific to Chm1. We found no significant difference in survival between VICs treated with siRNA specific to Chm1 and untreated VICs or VICs treated with control siRNA. This result indicates that Chm-I is not a survival factor for VICs (Supplementary Fig. 4 online). These findings imply that Chm-I protects cardiac valves from endothelial infiltration and vascular formation.
Pathological analysis of human cardiac valves
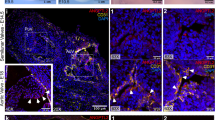
Hematoxylin and eosin staining and immunostaining of autopsy or surgical specimens were used to determine the expression profile of CHM-I and VEGF-A in cardiac valves of patients with VHD (Fig. 6). Blood vessels were absent from cardiac valves of patients without VHD. In normal valves, CHM-I was detected in the laminae fibrosa, spongiosa and ventricularis layers, but not in endothelial cells, whereas VEGF-A was absent from all cell layers. By comparison, numerous vessels were observed in cardiac valves of patients with atherosclerosis, RHD and infective endocarditis. CHM-I was markedly downregulated in regions of new vessel formation that strongly expressed VEGF-A. In normal cardiac valves, or areas without neovascular formation in infective endocarditis and RHD valves, cells stained with vimentin and the interstitial space stained strongly with CHM-I. This result suggests the cells were VICs. By comparison, in regions of neovascularization in diseased valves, the vWF-positive cells adjacent to newly developed vessels were endothelial cells and were at a higher cell density than in normal cardiac valves. Coimmunostaining showed localization of CBFA-1 to the neovascular area of diseased valves in which CHM-I was downregulated, but the absence of CBFA-1 in normal cardiac valves or regions of diseased valves lacking neovascularization (Supplementary Fig. 5 online).

(a) Samples from autopsies with no VHD. HE, hematoxylin and eosin staining. CHM-I was strongly expressed, whereas VEGF-A was not expressed. (b–d) Representative micrographs of immunohistochemistry for surgical samples of various VHDs. Prominent angiogenesis was found in infective endocarditis (IE, b), RHD (c) and atherosclerotic VHD (d). (e) The aortic valve in annuloaortic ectasia (AAE) showed no angiogenesis and stained well for CHM-I, but not VEGF-A. In b, double immunostaining for CHM-I (brown) and vimentin (purple) is shown in the neovascular and non-neovascular areas in the fourth and fifth micrographs from the left, respectively. The level of CHM-I was markedly diminished in the large area with strong VEGF-A expression and new vessel formation. AV, aortic valve; MV, mitral valve. (f) Computer-assisted quantitative analysis of vessel number, calcification area, percentage CHM-I–positive area, and percentage VEGF-A–positive area. *P < 0.01; §P < 0.05. NS, not significant.
Matrix metalloproteinases (MMPs) are important in repair and remodeling of damaged tissue, and in early atherosclerosis. We thus investigated the colocalization of MMPs and CHM-I in various VHDs (Supplementary Fig. 6 online). In the atherosclerotic valve, MMP-1, MMP-2, MMP-9 and MMP-13 showed positive signals, whereas in the infective endocarditis and RHD valves, only MMP-1, MMP-2 and MMP-13 showed positive signals. By comparison, MMP-1 and MMP-2 showed weak signals in valves with annuloaortic ectasia and no signal in the individuals without VHD. Furthermore, expression of MMPs and CHM-I was mutually exclusive. The cells in the MMP-positive area stained positive for vimentin, desmin (data not shown), and α-smooth muscle actin (Supplementary Fig. 7 online). In the atherosclerotic valve, but not in infective endocarditis and RHD valves, some cells were positive for CD11b and CD14 (data not shown). MMP expression in atherosclerotic valves was different from that in valves from patients without VHD, and the interstitial space did not stain positive for CHM-I as in normal valves. These results suggest that the cells staining positive for CD11b and CD14 are not VICs but infiltrating macrophages or activated myofibroblasts. This pathological expression profile was not observed in the cardiac valves of patients with annuloaortic ectasia or rupture of the chordae tendineae of the mitral valve. Computer image analysis showed that vessel number (7.7 ± 1.8, P < 0.01; 7.1 ± 1.3, P < 0.01; 6.0 ± 1.9, P < 0.01; versus 0 ± 0 in control), calcification area (1.6 ± 0.6%, P < 0.05; 4.1 ± 0.7%, P < 0.01; 5.0 ± 0.5%, P < 0.01; versus 0 ± 0% in control), and the percentage of VEGF-A–positive cells (22.9 ± 6.0%, P < 0. 01; 31.1 ± 9.4%, P < 0.01; 18.5 ± 6.2%, P < 0. 01; versus 1.5 ± 0.3% in control) were markedly increased in infective endocarditis, RHD and atherosclerosis, respectively, whereas the percentages of CHM-I–positive cells were markedly decreased (10.0 ± 3.2%, P < 0.01; 4.9 ± 2.2%, P < 0.01; 25.8 ± 6.1%, P < 0.01; versus 72.2 ± 6.1% in control). These findings show that development of angiogenesis and sclerotic change in diseased cardiac valves correlate with downregulation of CHM-I and upregulation of MMP expression levels.
Discussion
Despite the clinical importance of heart disease and intense study in this field, little is known of the mechanisms underlying the degeneration of cardiac valves. The present study addresses this issue by demonstrating the key role of the potent antiangiogenic factor Chm-I in the prevention of VHD by maintaining the avascular nature of cardiac valves.
Chm-I is expressed in the cartilage, eye and thymus of various species including human23, rabbit19, mouse24, chick25,26, cow14 and zebra fish27. The present study showed that Chm-I expression persisted in normal cardiac valves throughout life but was downregulated in diseased cardiac valves, suggesting that Chm-I has an important role in maintenance of cardiac valve function. The conceptual framework for this role of Chm-I is shown in Supplementary Figure 8 online. Expression of endostatin, an antiangiogenic factor, is enhanced in aortic valves under pathological, but not normal conditions28. The present study is the first report, to our knowledge, of an antiangiogenic factor expressed in cardiac valves under physiological conditions to suppress neovascularization. As cardiac valves are flow-regulating tissues in a dynamic chambered pump, they are subjected to mechanical stress and damage of the endothelial cell lining on the outer layer of the valve. Chm-I may protect cardiac valves from inflammation and vascularization resulting from mechanical damage. To confirm this hypothesis, we analyzed Chm-I expression profiles in cardiac valves in normal and diseased states, in various in vitro functional assays, in Chm1−/− mice and in Apoe−/− mice as an in vivo model of cardiac valve disease. The mutually exclusive expression pattern of Chm-I and Vegf-A can be explained by several mechanisms. First, an upstream signal may control the genetic switch between angiogenic and antiangiogenic factors. In support of this mechanism, genetic rescue in Cbfa1-knockout mice showed that Cbfa1, a critical transcription factor mediating endochondral ossification in cartilage, induced a coordinated change in upregulation of Vegf-A and downregulation of Chm-I in chondrocytes29. Cbfa1 expression was shown to be upregulated in atherosclerotic aortic valves9. It is possible that upregulated Cbfa1 represses Chm-I and activates Vegf-A. Second, it is possible that loss of VICs may be induced by acute inflammation of cardiac valves in RHD or infective endocarditis; excessive mechanical stress imposed on valves, such as the bicuspid aortic valves; hypertension; or the chronic inflammation found in atherosclerosis. A decrease in VIC number may lead to a reduction in the level of Chm-I produced by diseased valves, and then to the infiltration of Vegf-A–expressing cells. It is also possible that a combination of the above two mechanisms may explain the downregulation of Chm-I and the upregulation of Vegf-A.
Suppression of the antiangiogenic activity of VIC-CM by Chm1-specific siRNA suggests that Chm-I is a critical antiangiogenic factor in cardiac valves. The incomplete suppression of antiangiogenic activity implies that VICs may secrete other antiangiogenic factors, similar to those identified in the eye. In the eye, the antiangiogenic factors endostatin and pigment epithelium–derived factor are expressed in addition to Chm-I30.
The echocardiographic findings of early changes in aortic stenosis, neoangiogenesis, infiltration of inflammatory cells, lipid deposition and calcification were observed only in the cardiac valves of aged Chm1−/− mice. The expression of other antiangiogenic factors may delay development of cardiac valve defects such that the phenotype is not apparent during embryogenesis or in the young adult. It is likely that deformation of cardiac valves in aged Chm1−/− mice results from ordinary mechanical stress in the absence of the protective antiangiogenic activity of Chm-I. The aortic valve receives the greatest mechanical stress of the four heart valves, which may explain the pathological changes observed in Chm1−/− mice. The molecular mechanism underlying the influence of mechanical stress on intracellular signaling needs further investigation.
Previous investigations showed that (i) the expression of angiogenic factors and neoangiogenesis occurs in VHD2,30,31,32,33; (ii) atherosclerotic plaque progression is associated with angiogenesis34,35; (iii) the early lesion of degenerative VHD is an active inflammatory process with similarities to atherosclerosis32,36,37,38,39,40; and (iv) endothelial cells in VHD have significantly increased angiogenic potency28. These findings suggest that angiogenesis of cardiac valves results in the progression of sclerogenic change. Further supporting this, it was reported that although VICs in normal valves stained positively for vimentin but not alpha-actin or desmin, VICs in myxomatous valves contained both vimentin and alpha-actin or desmin (characteristics of myofibroblasts) and strongly expressed SMemb, MMPs, cathepsins and IL-1β, which stained weakly in controls. It was concluded that VICs in myxomatous valves have features of activated myofibroblasts and express excessive levels of catabolic enzymes41. After an investigation of the expression of MMPs and tissue inhibitor of metalloproteases (TIMPs) in aortic valve disease, it was reported that levels of MMP-3, MMP-9 and TIMP-1 were increased in aortic stenosis42. Taken together, the present findings and previous investigations support the hypothesis that antiangiogenic factors protect cardiac valves from angiogenesis and the development of dystrophic changes. By comparison, angiogenic factors and MMPs promote dystrophic changes. Further studies to support this hypothesis include investigations to determine the protective effect of Chm1 overexpression. The present findings support preclinical investigations of Chm-I, or proteins with a similar function, as therapeutic agents in preventing VHD.
Methods
RT-PCR.
Total RNA was isolated using Trizol reagent (Gibco-BRL) and treated with DNase I (Roche). RT-PCR was carried out as described previously43 using the following primers: Chm1, 5′-CTTAAGCCCATGTATCCAAA-3′ (forward), 3′-CCAGTGGTTCACAGATCTTC-5′ (reverse); Gapdh, 5′-TTCAACGGCACAGTCAAGG-3′ (forward), 3′-CATGGACTGTGGTCATGAG-5′ (reverse).
In situ hybridization.
Paraffin-embedded sections were treated with proteinase K, and in situ hybridization was carried out as described previously43. Template DNA was an 879-bp cDNA encoding mouse Chm1 cloned into the pCR II-TOPO vector. Color was developed with 0.2 mg ml−1 of 3,3′-diaminobenzidine tetrahydrochloride in 50 mmol liter−1 Tris HCl, pH 7.6, containing 0.003% hydrogen peroxide. The sections were counterstained with hematoxylin and eosin and were observed under microscopy.
Immunohistochemical and immunofluorescence staining.
Adult mouse hearts were perfused from the apex with phosphate-buffered saline, perfusion-fixed with 4% paraformaldehyde in phosphate-buffered saline and used for immunostaining as described previously15. The hearts were dissected, immersion-fixed overnight at 4 °C in 4% paraformaldehyde and then embedded in paraffin. Before application of the primary antibodies, paraffin was removed from the sections in xylene and the sections were heated in a microwave oven in 10 mmol citric acid monohydrate (DAKO), pH 6.0, for 3 min. After sections were rinsed in phosphate-buffered saline, they were incubated overnight at 4 °C with 5% normal rabbit serum and affinity-purified rabbit polyclonal antibody to Chm-I, rabbit polyclonal antibody to Vegf-A (1:200 dilution; Santa Cruz Biotechnology), vWF (1:200 dilution; Lab vision Corporation), Mac-1 (1:200 dilution; BD PharMingen, Inc.), Cbfa1 (1:50 dilution; R&D systems), MMP-1 (ref. 44; Daiichi fine chemical), MMP-2 (ref. 44; Daiichi fine chemical), MMP-3 (ref. 44; Daiichi fine chemical), MMP-9 (ref. 44; Daiichi fine chemical), MMP-13 (ref. 44; 1:100 dilution; Biogenesis), α smooth muscle actin (1:400 dilution; Sigma) or CD11b (1:200 dilution; BD PharMingen). Immunohistochemical signals were detected by applying 0.05% 3,3′-diaminobenzidine tetrahydrochloride (Sigma-Aldrich) containing 0.01% hydrogen peroxide in 0.05 M Tris-buffered saline (pH 7.6) as a chromogenic substrate. The sections were then counterstained with hematoxylin and eosin, dehydrated in a graded ethanol series, and mounted in Permount (Fisher Scientific)23.
For immunofluorescence studies, the sections were incubated with secondary antibodies conjugated with Alexa 488 or Alexa 594 (Molecular Probes). Nuclei were stained with TOTO-3 (Molecular Probes). Slides were observed under a confocal laser-scanning microscope (LSM 510 META; Carl Zeiss). Optical sections were obtained at 1024 × 1024 pixels resolution, and analyzed using LSM software (Carl Zeiss). We substituted nonimmune rabbit serum for primary antibodies as a negative control for each immunostaining experiment. Histological examinations were done at 8 (n = 10), 20 (n = 7), 32 (n = 7), 64 (n = 7) and 90 weeks (n = 12) in wild-type and Chm1−/− mice. Quantitative analysis of the stained area was carried out by converting images to monochrome with optimum saturation and counting the black pixels using NIH Image software.
Animals.
Wild-type ICR mice and Wistar rats were purchased from Japan CLEA. Apoe−/− mice, deficient in apolipoprotein E, were used as an atherosclerosis animal model. These mice are also known to show stage-related aortic stenosis45. These mice were purchased from the Jackson Laboratory and maintained on a normal diet. The genetic backgrounds of the Chm1−/− and age-matched control mice have been described previously46. TT2 embryonic stem cell clones47 harboring the targeted Chm1 coding region were identified by Southern-blot analysis and were aggregated with CD-1 single eight-cell embryos to generate chimeras as described previously46. Chimeras were crossed with C57BL/6 female mice to achieve germline transmission of the targeted allele. Offspring were genotyped either by Southern blotting or by PCR. Heterozygous Chm1+/− mice were backcrossed twice to C57/BL6 mice and were maintained. Homozygous Chm1−/− strains, kept until 90.2 ± 7.4 weeks old, were used for experimental procedures. Homozygous Chm1+/+ littermates of the Chm1−/− strain were used as controls for experimental procedures to ensure that controls were age-matched and had the same genetic background as the knockout mice. All experimental procedures and protocols were approved by the Animal Care and Use Committee of Keio University, Japan.
Echocardiography.
Transthoracic echocardiography was carried out with the Sonos 1000 echocardiogram (Hewlett-Packard) equipped with a 10-MHz linear-array transducer. The heart was imaged in two-dimensional and color Doppler mode in the long- and short-axis view at the level of the aortic valve48. M-mode scanning along the long axis view was examined, and the thickness of the interventricular septum and left ventricular free wall, left ventricular end-diastolic dimension, left ventricular end-systolic dimension, and ejection fraction were measured. The area trace of the aortic valve was also examined at each time point. An echocardiographic examination was done at 8 (n = 10), 20 (n = 10), 32 (n = 10), 64 (n = 10) and 90 weeks (n = 12) in wild-type and Chm1−/− mice.
Isolation of adult rat aortic VICs.
The hearts were dissected from anesthetized 5-week-old Wistar rats. The leaflet of the aortic valves was rapidly removed, chopped under a stereomicroscope, and used for the explant culture as previously described5. Pieces that measured 5 × 5 mm were cut from the tissue, placed in 12-well collagen coated dishes (Iwaki), and grown in M199 medium (Sigma-Aldrich) containing 10% fetal bovine serum. Conditioned medium was obtained from confluent VICs 3 d after changing the medium and used in further analyses.
Human samples.
Samples comprising 24 aortic and 11 mitral valves (from 35 men; mean age, 59.9 ± 17.1 y) were collected from patients undergoing valve replacement due to valvular stenosis or regurgitation. Samples were fixed immediately after removal in formaldehyde and then embedded in paraffin. For controls, six micro- and macroscopically normal, noncalcified, smooth and pliable aortic and mitral valves were collected from three autopsied patients (mean age, 53.2 ± 19.2 years). The use of autopsied and surgical specimens of human tissue was approved by the institutional review board of Keio University.
Statistical analysis.
Values are presented as the mean ± SEM. Statistical significance was evaluated using the unpaired student's t-test for comparisons between two mean values. Multiple comparisons between more than three groups were carried out using an ANOVA test. A value of P < 0.05 was considered significant.
Other methods.
Other methods are described in the Supplementary Methods online.
GenBank accession numbers.
Mouse Chm1, NM_010701; rat Chm1, NM_030854.
Note: Supplementary information is available on the Nature Medicine website.
References
Hammon, J.W., Jr ., O'Sullivan, M.J., Oury, J. & Fosburg, R.G. Allograft cardiac valves. A view through the scanning electron microscope. J. Thorac. Cardiovasc. Surg. 68, 352–360 (1974).
Soini, Y., Salo, T. & Satta, J. Angiogenesis is involved in the pathogenesis of nonrheumatic aortic valve stenosis. Hum. Pathol. 34, 756–763 (2003).
Yamauchi, R. et al. Upregulation of SR-PSOX/CXCL16 and recruitment of CD8+ T cells in cardiac valves during inflammatory valvular heart disease. Arterioscler. Thromb. Vasc. Biol. 24, 282–287 (2004).
Filip, D.A., Radu, A. & Simionescu, M. Interstitial cells of the heart valves possess characteristics similar to smooth muscle cells. Circ. Res. 59, 310–320 (1986).
Lester, W., Rosenthal, A., Granton, B. & Gotlieb, A.I. Porcine mitral valve interstitial cells in culture. Lab. Invest. 59, 710–719 (1988).
Gotlieb, A.I., Rosenthal, A. & Kazemian, P. Fibroblast growth factor 2 regulation of mitral valve interstitial cell repair in vitro. J. Thorac. Cardiovasc. Surg. 124, 591–597 (2002).
Akiyama, H. et al. Essential role of Sox9 in the pathway that controls formation of cardiac valves and septa. Proc. Natl. Acad. Sci. USA 101, 6502–6507 (2004).
Ranger, A.M. et al. The transcription factor NF-ATc is essential for cardiac valve formation. Nature 392, 186–190 (1998).
Rajamannan, N.M. et al. Human aortic valve calcification is associated with an osteoblast phenotype. Circulation 107, 2181–2184 (2003).
Chan-Thomas, P.S., Thompson, R.P., Robert, B., Yacoub, M.H. & Barton, P.J. Expression of homeobox genes Msx-1 (Hox-7) and Msx-2 (Hox-8) during cardiac development in the chick. Dev. Dyn. 197, 203–216 (1993).
Sugi, Y., Yamamura, H., Okagawa, H. & Markwald, R.R. Bone morphogenetic protein-2 can mediate myocardial regulation of atrioventricular cushion mesenchymal cell formation in mice. Dev. Biol. 269, 505–518 (2004).
Camenisch, T.D. et al. Temporal and distinct TGFbeta ligand requirements during mouse and avian endocardial cushion morphogenesis. Dev. Biol. 248, 170–181 (2002).
Ikeda, T. et al. The combination of SOX5, SOX6, and SOX9 (the SOX trio) provides signals sufficient for induction of permanent cartilage. Arthritis Rheum. 50, 3561–3573 (2004).
Hiraki, Y. [Molecular cloning of a novel cartilage-specific functional matrix, chondromodulin-I, and its role in endochondral bone formation]. Seikagaku 63, 1449–1454 (1991).
Funaki, H. et al. Expression and localization of angiogenic inhibitory factor, chondromodulin-I, in adult rat eye. Invest. Ophthalmol. Vis. Sci. 42, 1193–1200 (2001).
Azizan, A., Holaday, N. & Neame, P.J. Post-translational processing of bovine chondromodulin-I. J. Biol. Chem. 276, 23632–23638 (2001).
Hiraki, Y. et al. Identification of chondromodulin I as a novel endothelial cell growth inhibitor. Purification and its localization in the avascular zone of epiphyseal cartilage. J. Biol. Chem. 272, 32419–32426 (1997).
Inoue, H., Kondo, J., Koike, T., Shukunami, C. & Hiraki, Y. Identification of an autocrine chondrocyte colony-stimulating factor: chondromodulin-I stimulates the colony formation of growth plate chondrocytes in agarose culture. Biochem. Biophys. Res. Commun. 241, 395–400 (1997).
Shukunami, C. & Hiraki, Y. Expression of cartilage-specific functional matrix chondromodulin-I mRNA in rabbit growth plate chondrocytes and its responsiveness to growth stimuli in vitro. Biochem. Biophys. Res. Commun. 249, 885–890 (1998).
Hiraki, Y., Kono, T., Sato, M., Shukunami, C. & Kondo, J. Inhibition of DNA synthesis and tube morphogenesis of cultured vascular endothelial cells by chondromodulin-I. FEBS Lett. 415, 321–324 (1997).
Zacks, S. et al. Characterization of Cobblestone mitral valve interstitial cells. Arch. Pathol. Lab. Med. 115, 774–779 (1991).
Oshima, Y. et al. Expression and localization of tenomodulin, a transmembrane type chondromodulin-I-related angiogenesis inhibitor, in mouse eyes. Invest. Ophthalmol. Vis. Sci. 44, 1814–1823 (2003).
Hiraki, Y. et al. Molecular cloning of human chondromodulin-I, a cartilage-derived growth modulating factor, and its expression in Chinese hamster ovary cells. Eur. J. Biochem. 260, 869–878 (1999).
Shukunami, C., Iyama, K., Inoue, H. & Hiraki, Y. Spatiotemporal pattern of the mouse chondromodulin-I gene expression and its regulatory role in vascular invasion into cartilage during endochondral bone formation. Int. J. Dev. Biol. 43, 39–49 (1999).
Shukunami, C., Yamamoto, S., Tanabe, T. & Hiraki, Y. Generation of multiple transcripts from the chicken chondromodulin-I gene and their expression during embryonic development. FEBS Lett. 456, 165–170 (1999).
Dietz, U.H., Ziegelmeier, G., Bittner, K., Bruckner, P. & Balling, R. Spatio-temporal distribution of chondromodulin-I mRNA in the chicken embryo: expression during cartilage development and formation of the heart and eye. Dev. Dyn. 216, 233–243 (1999).
Sachdev, S.W. et al. Sequence analysis of zebrafish chondromodulin-1 and expression profile in the notochord and chondrogenic regions during cartilage morphogenesis. Mech. Dev. 105, 157–162 (2001).
Chalajour, F. et al. Angiogenic activation of valvular endothelial cells in aortic valve stenosis. Exp. Cell Res. 298, 455–464 (2004).
Takeda, S., Bonnamy, J.P., Owen, M.J., Ducy, P. & Karsenty, G. Continuous expression of Cbfa1 in nonhypertrophic chondrocytes uncovers its ability to induce hypertrophic chondrocyte differentiation and partially rescues Cbfa1-deficient mice. Genes Dev. 15, 467–481 (2001).
Dawson, D.W. et al. Pigment epithelium-derived factor: a potent inhibitor of angiogenesis. Science 285, 245–248 (1999).
Shworak, N.W. Angiogenic modulators in valve development and disease: does valvular disease recapitulate developmental signaling pathways? Curr. Opin. Cardiol. 19, 140–146 (2004).
Mohler, E.R., III . et al. Bone formation and inflammation in cardiac valves. Circulation 103, 1522–1528 (2001).
Mazzone, A. et al. Neoangiogenesis, T-lymphocyte infiltration, and heat shock protein-60 are biological hallmarks of an immunomediated inflammatory process in end-stage calcified aortic valve stenosis. J. Am. Coll. Cardiol. 43, 1670–1676 (2004).
O'Brien, K.D., McDonald, T.O., Chait, A., Allen, M.D. & Alpers, C.E. Neovascular expression of E-selectin, intercellular adhesion molecule-1, and vascular cell adhesion molecule-1 in human atherosclerosis and their relation to intimal leukocyte content. Circulation 93, 672–682 (1996).
Moulton, K.S. et al. Inhibition of plaque neovascularization reduces macrophage accumulation and progression of advanced atherosclerosis. Proc. Natl. Acad. Sci. USA 100, 4736–4741 (2003).
Otto, C.M., Kuusisto, J., Reichenbach, D.D., Gown, A.M. & O'Brien, K.D. Characterization of the early lesion of 'degenerative' valvular aortic stenosis. Histological and immunohistochemical studies. Circulation 90, 844–853 (1994).
O'Brien, K.D. et al. Osteopontin is expressed in human aortic valvular lesions. Circulation 92, 2163–2168 (1995).
Agmon, Y. et al. Aortic valve sclerosis and aortic atherosclerosis: different manifestations of the same disease? Insights from a population-based study. J. Am. Coll. Cardiol. 38, 827–834 (2001).
O'Brien, K.D. et al. Association of angiotensin-converting enzyme with low-density lipoprotein in aortic valvular lesions and in human plasma. Circulation 106, 2224–2230 (2002).
Pohle, K. et al. Association of cardiovascular risk factors to aortic valve calcification as quantified by electron beam computed tomography. Mayo Clin. Proc. 79, 1242–1246 (2004).
Rabkin, E. et al. Activated interstitial myofibroblasts express catabolic enzymes and mediate matrix remodeling in myxomatous heart valves. Circulation 104, 2525–2532 (2001).
Fondard, O. et al. Extracellular matrix remodelling in human aortic valve disease: the role of matrix metalloproteinases and their tissue inhibitors. Eur. Heart J. 26, 1333–1341 (2005).
Enomoto, H. et al. Vascular endothelial growth factor isoforms and their receptors are expressed in human osteoarthritic cartilage. Am. J. Pathol. 162, 171–181 (2003).
Fujimoto, N. & Iwata, K. Use of EIA to measure MMPs and TIMPs. Methods Mol. Biol. 151, 347–358 (2001).
Tanaka, K. et al. Age-associated aortic stenosis in apolipoprotein E-deficient mice. J. Am. Coll. Cardiol. 46, 134–141 (2005).
Nakamichi, Y. et al. Chondromodulin I is a bone remodeling factor. Mol. Cell. Biol. 23, 636–644 (2003).
Kyuwa, S., Xiao, Y., Toyoda, Y. & Sato, E. Characterization of embryonic stem-like cell lines derived from embryoid bodies. Exp. Anim. 46, 11–16 (1997).
Litwin, S.E., Katz, S.E., Morgan, J.P. & Douglas, P.S. Serial echocardiographic assessment of left ventricular geometry and function after large myocardial infarction in the rat. Circulation 89, 345–354 (1994).
Acknowledgements
We thank Y. Nishizaki and S. Kondo for technical support. This study was supported in part by research grants from the Ministry of Education, Science and Culture, Japan, and by the Program for Promotion of Fundamental Studies in Health Science of the National Institute of Biomedical Innovation.
Author information
Authors and Affiliations
Contributions
This study was designed by K.F.; experiments were performed by M.Y., S.Y., K.M., K.K. and N.K.; the Chm1 knockout mouse and antibody to Chm-I were made by C.S. and Y.H.; surgical specimens were collected by M.M., H.S. and R.Y.; the MMP experiment was performed by T.S. and Y.O.; specimens from Apoe knockout mice were distributed by M.S.; S.O. contributed to the writing of the paper.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Supplementary Fig. 1
Temporal and spatial expression of Chm1 in rodent and human heart (PDF 2002 kb)
Supplementary Fig. 2
Immunofluorescent staining for Chm-I in middle- and late-stage embryonic mouse heart. (PDF 287 kb)
Supplementary Fig. 3
Antiangiogenic effect of recombinant human CHM-I on HCAECs in vitro. (PDF 224 kb)
Supplementary Fig. 4
MTT assay for VICs. (PDF 982 kb)
Supplementary Fig. 5
Cbfa1-positive cells were detected in vascular areas in VHD. (PDF 189 kb)
Supplementary Fig. 6
Immunohistochemistry of the cardiac valves for MMP-1, -2, -3, -9 and -13 in humans with various VHDs. (PDF 450 kb)
Supplementary Fig. 7
Activated myofibroblasts were detected in vascular areas in VHD. (PDF 279 kb)
Supplementary Fig. 8
The conceptual framework of the role of Chm-I in cardiac valves. (PDF 1790 kb)
Rights and permissions
About this article
Cite this article
Yoshioka, M., Yuasa, S., Matsumura, K. et al. Chondromodulin-I maintains cardiac valvular function by preventing angiogenesis. Nat Med 12, 1151–1159 (2006). https://doi.org/10.1038/nm1476
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/nm1476
This article is cited by
-
Epigenome-wide association study in peripheral white blood cells involving insulin resistance
Scientific Reports (2019)
-
Chondromodulin-1 in health, osteoarthritis, cancer, and heart disease
Cellular and Molecular Life Sciences (2019)
-
Scleraxis is a transcriptional activator that regulates the expression of Tenomodulin, a marker of mature tenocytes and ligamentocytes
Scientific Reports (2018)
-
Calcific Aortic Valve Disease: a Developmental Biology Perspective
Current Cardiology Reports (2018)
-
Proteomic Alterations Associated with Biomechanical Dysfunction are Early Processes in the Emilin1 Deficient Mouse Model of Aortic Valve Disease
Annals of Biomedical Engineering (2017)
