
Abstract
Acute oxidative stress induced by ischemia-reperfusion or inflammation causes serious damage to tissues, and persistent oxidative stress is accepted as one of the causes of many common diseases including cancer. We show here that hydrogen (H2) has potential as an antioxidant in preventive and therapeutic applications. We induced acute oxidative stress in cultured cells by three independent methods. H2 selectively reduced the hydroxyl radical, the most cytotoxic of reactive oxygen species (ROS), and effectively protected cells; however, H2 did not react with other ROS, which possess physiological roles. We used an acute rat model in which oxidative stress damage was induced in the brain by focal ischemia and reperfusion. The inhalation of H2 gas markedly suppressed brain injury by buffering the effects of oxidative stress. Thus H2 can be used as an effective antioxidant therapy; owing to its ability to rapidly diffuse across membranes, it can reach and react with cytotoxic ROS and thus protect against oxidative damage.
Similar content being viewed by others


Main
Oxidative stress arises from the strong cellular oxidizing potential of excess reactive oxygen species (ROS), or free radicals1,2,3,4,5. Most of the superoxide anion radical (O2−•) produced is generated in mitochondria by electron leakage from the electron transport chain and the Krebs cycle6. O2−• is also produced by metabolic oxidases, including NADPH oxidase and xanthine oxidase7. Superoxide dismutase converts O2−• into hydrogen peroxide (H2O2)8, which is detoxified into H2O by either glutathione peroxidase or catalase. Excess O2−• reduces transition metal ions such as Fe3+ and Cu2+ (ref. 2), the reduced forms of which in turn can react with H2O2 to produce hydroxyl radicals (•OH) by the Fenton reaction. •OH is the strongest of the oxidant species and reacts indiscriminately with nucleic acids, lipids and proteins. There is no known detoxification system for •OH; therefore, scavenging •OH is a critical antioxidant process9.
Despite their cytotoxic effects, O2−• and H2O2 play important physiological roles at low concentrations: they function as regulatory signaling molecules that are involved in numerous signal transduction cascades and also regulate biological processes such as apoptosis, cell proliferation and differentiation7,10. At higher concentrations, H2O2 is converted into hypochlorous acid by myeloperoxidase; hypochlorous acid defends against bacterial invasion5. Nitric oxide (NO•), another ROS, functions as a neurotransmitter and is essential for the dilation of blood vessels11. Thus, cytotoxic radicals such as •OH must be neutralized without compromising the essential biological activities of other, physiologically beneficial, ROS. Here we demonstrate that molecular hydrogen (dihydrogen, H2) can alleviate •OH-induced cytotoxicity without affecting the other ROS, and propose that H2 has potential as an antioxidant for preventive and therapeutic applications.
Results
H2 selectively reduces •OH in cultured cells
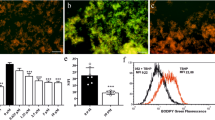
H2 reduces the •OH that is produced by radiolysis or photolysis of water12; however, whether H2 can effectively neutralize •OH in living cells has not been directly investigated. As the cellular damage produced by spontaneous generation of •OH is not sufficient to be detectable, we induced O2−• production in PC12 cultured cells. To do this, we treated the cells with a mitochondrial respiratory complex III inhibitor, antimycin A (ref. 13); following such treatment, O2−• in these cells is rapidly converted into H2O2. The addition of antimycin A increased levels of O2−• and H2O2, as judged by the fluorescence signals emitted by the oxidized forms of MitoSOX (Fig. 1a) and 2′,7′-dichlorodihydrofluorescein (H2DCF) (Supplementary Fig. 1 online), respectively. We dissolved H2 and O2 into medium as described in the Methods, and confirmed the prolonged (24 h long) maintenance of H2 levels (Supplementary Fig. 2 online). H2 dissolved in culture medium did not decrease MitoSOX and DCF signals in the cells (Fig. 1a,b and Supplementary Fig. 1). Additionally, H2 did not decrease the steady-state level of NO• (Supplementary Fig. 1). In contrast, H2 treatment significantly decreased levels of •OH, as assessed by the fluorescence signal emitted by the oxidized form of 2-[6-(4′-hydroxy)phenoxy-3H-xanthen-3-on-9-yl] benzoate (HPF) (refs. 14,15 and Fig. 1c,d). When we exposed the cells to antimycin A (30 μg/ml) in the absence of H2, the HPF signals increased in both the nuclear region and the cytoplasm, probably because H2O2 diffused from the mitochondria to produce •OH. Notably, H2 decreased •OH levels even in the nuclear region (Fig. 1c).

(a,b) PC12 cells were incubated in medium with or without 0.6 mM H2, and exposed to antimycin A (30 μg/ml for 30 min) in order to induce O2−• production. They were then treated with 0.5 μM MitoSOX. Representative fluorescence images of MitoSOX-treated cells were obtained by laser-scanning confocal microscopy (Olympus FV300). MitoSOX fluorescence was quantified from 100 cells of each independent experiment (n = 5). (c) Representative laser-scanning confocal images of the fluorescence of the •OH marker HPF were taken 30 min after the addition of antimycin A. Arrows and arrowheads indicate the increase and decrease, respectively, in HPF signals in the nuclear region. (d) HPF fluorescence in cells treated with antimycin A with or without 0.6 mM H2 was quantified from 100 cells (n = 4). **P < 0.01, ***P < 0.001. (e) At 30 min after adding antimycin A (10 μg/ml) with or without H2 (0.6 mM), cells were incubated with 1 μM MTGreen and 100 nM TMRM for 10 min and then imaged. The two images were superimposed (merge). (f) Cells were pretreated with 4.5 g/l 2-deoxyl-D-glucose (an inhibitor of glycolysis) and 1 mM pyruvate, and relative cellular ATP levels were quantified after exposure to 30 μg/ml antimycin A. ATP levels of cells not treated with antimycin A were set at 100% (n = 3). *P < 0.05, **P < 0.01. Scale bars: 100 μm in a; 50 μm in c; 20 μm in e. Histograms show mean ± s.d.
After antimycin A treatment, H2 prevented the decline of the mitochondrial membrane potential, as detected by fluorescence of tetramethylrhodamine methyl ester (TMRM), which depends upon the mitochondrial membrane potential, whereas fluorescence levels of MitoTracker Green (MTGreen), which are independent of the membrane potential, were unchanged (Fig. 1e). This suggested that H2 protected mitochondria from •OH. H2-treated cells looked normal, whereas H2-untreated cells were shrunken and had abnormal round shapes (Fig. 1e). Along with this protective effect, H2 also prevented a decrease in the cellular levels of ATP synthesized in mitochondria (Fig. 1f). The fact that H2 protected mitochondria and nuclear DNA provided evidence that H2 penetrated most membranes and diffused into organelles.
H2 dissolved in medium protects cultured cells against •OH
We placed PC12 cells in culture medium containing H2 and O2, and, at the same time, induced oxidative stress by adding antimycin A. At 24 h after the induction of ROS with antimycin A, we observed that H2 seemed to protect nuclear DNA from oxidation, as shown by decreased levels of oxidized guanine (8-OH-G) (Fig. 2a,b and ref. 16). Moreover, H2 also decreased levels of 4-hydroxyl-2-nonenal (HNE), an end-product of lipid peroxides (Fig. 2c,d and ref. 17), indicating that it protected lipids from peroxidation. Further, H2 dissolved in medium protected cells from cell death in a dose-dependent manner (Fig. 2e,f). When we removed H2 from medium that had been saturated with H2, the protective effect disappeared (Fig. 2f), suggesting that the observed effect was not due to a reaction of H2 with the medium. Moreover, we confirmed that H2 protected cellular viability by using two methods: a modified MTT assay (WST-1 assay) and measurement of cellular lactate dehydrogenase (LDH) leakage from damaged cells (Supplementary Fig. 3 online). To exclude the possibility that the protective effect of H2 was due to a reaction with antimycin A, we induced ROS by adding menadione, an inhibitor that acts on mitochondrial complex I, and observed that H2 protected cells in this system as well (Supplementary Fig. 3).

(a–d) PC12 cells were maintained with 10 μg/ml antimycin A, with (+) or without (−) 0.6 mM H2, for 24 h in a closed flask, and immunostained with antibodies to 8-OH-G or HNE. Fluorescence signals in response to 8-OH-G and HNE were quantified using 100 cells from each independent experiment (n = 4). *P < 0.05, **P < 0.01. (e) Phase-contrast pictures of PC12 cells 24 h after the exposure to antimycin A, with (+) or without (−) 0.6 mM H2. Arrows indicate dead cells. (f) Cell survival was assessed by manually counting the cells (Methods; n = 4). *P < 0.05, **P < 0.01 (compared with O μM H2). (g) PC12 cells were exposed to intracellular •OH produced by the Fenton reaction, with or without 0.6 mM H2. Cells were preincubated with 1 mM CuSO4, washed, and exposed for 1 h to 0.1 mM ascorbate (Vit. C) in order to reduce intracellular Cu2+ to Cu+ (Supplementary Methods). The cells were costained with propidium iodide (PI) (for dead cells) and Hoechst 33342 to visualize the nuclei. (h) Cell survival was assessed by manually counting the cells as in f (n = 5). *P < 0.05, **P < 0.01. Scale bars: 50 μm in a,c,e; 100 μm in g. Histograms represent mean ± s.d.
To verify that H2 protects against •OH, we pretreated cells with Cu2+ and then exposed them to ascorbate, in order to reduce intracellular Cu2+ to Cu+, which in turn catalyzes the production of •OH from cellular H2O2 that is endogenously produced. This treatment primarily induced •OH inside the cells (by the Fenton reaction), thus directly confirming that H2 protects cells against cellular •OH (Fig. 2g,h).
Spin-trapping identifies a free radical that is reduced by H2
To identify the free radical species that H2 reduces, we studied the effects of H2 on electron spin resonance (ESR) signals of spin-trapping reagents. We produced •OH by the cellular Fenton reaction, and semiquantified the cellular levels of •OH by spin-trapping using 5,5-dimethyl-1-pyrroline N-oxide (DMPO). Measurements of ESR indicated that H2 treatment did indeed decrease signals of •DMPO-OH derived from •OH (Fig. 3a–c).

(a) Standard electron spin resonance (ESR) signals of the •DMPO-OH radical were obtained by trapping •OH with a spin-trapping reagent (DMPO; details in Supplementary Methods). (b,c) PC12 cells were preincubated with 0.1 M DMPO and 2 mM CuSO4 for 30 min at 37 °C with or without 0.6 mM H2. After removal of this medium, the cells were treated with 0.2 mM ascorbate and 0.1 mM H2O2 for 5 min at 23 °C to produce •OH and then scraped into a flat cuvette for ESR measurement. Rectangle height reflects signal intensity. (d) The •DMPO-OH and •DMPO-H radicals18 and their corresponding ESR signals are illustrated. (e,f) PC12 cells were incubated in PBS containing 0.1 M DMPO and 30 μg/ml antimycin A for 7 min at 23 °C, with or without 0.6 mM H2, then scraped into a flat cuvette for ESR measurement. (g) A differential spectrum was obtained by subtracting the spectrum in f from that in e, in order to visualize the signals decreased by H2 treatment. * indicates •DMPO-OH signals derived from •OH. * and o indicate •DMPO-OH and •DMPO-H signals, respectively (a, b, d, e and g).
Moreover, when we induced O2−• production by treating cells with antimycin A in the presence of DMPO, we observed multiple ESR signals18. These signals seemed to consist of those from the •DMPO-OH and •DMPO-H radicals (Fig. 3d–f). The •DMPO-H radical is derived from the hydrogen radical (H•), which can be induced by porphyrins. To visualize the signals decreased by H2, we obtained the differential spectrum. We found that only •OH-derived signals were decreased by H2 treatment (Fig. 3e). These results strongly suggest the selective reduction of cellular •OH by H2 treatment.
H2 selectively reduces •OH and ONOO− in cell-free systems
Next, we confirmed in a pure solution that HPF fluorescence can be used to monitor the reduction of •OH by H2 during continuous •OH production by the Fenton reaction. In this condition, H2 suppressed increases in HPF signals in a dose-dependent manner (Fig. 4a–c). But when we mixed a solution containing H2 with HPF preoxidized with •OH, fluorescence signals from oxidized HPF did not decrease (data not shown), supporting the idea that H2 directly reacts with •OH.

(a,b) The Fenton reaction, which generates hydroxyl radicals, was initiated by adding H2O2 (to a final concentration of 5 μM) in a closed cuvette at 23 °C with gentle stirring (Supplementary Methods). Levels of •OH in the presence of various concentrations of H2 dissolved in the solution were assessed for HPF fluorescence. (a) Representative time course traces of HPF fluorescence at each concentration of H2. Baselines 1 and 2 show HPF fluorescence (in the presence of 0.8 mM H2) in the absence of H2O2 (baseline 1) and in the absence of ferrous perchlorate (baseline 2). (b) Mean ± s.d. of initial rates of increase in HPF fluorescence (four independent experiments). (c–g) Levels of •OH and two reactive nitrogen species (RNS: NO• and peroxynitrite (ONOO−)) remaining after incubation with 0.6 mM of H2 at 23 °C (details in the Supplementary Methods). Vitamin C (Vit. C) and superoxide dismutase (SOD) were used as positive controls. Signals generated in the absence of H2 (CTL) were set at 100%. Data represent mean ± s.d. (n = 6). *P < 0.05, ***P < 0.001. NBT-diformazan: oxidized form of nitroblue tetrazolium (NBT, a detector of O2−•). DAF-2: diaminofluorescein-2 (a detector of •NO).
Next, we examined the reactivity of H2 with other ROS or reactive nitrogen species (RNS). We prepared H2O2 and peroxynitrite (ONOO−) by dilution of the respective stock solutions, O2−• by the enzymatic reaction of xanthine oxidase with xanthine, and NO• by the spontaneous reaction of 1-hydroxy-2-oxo-3-(N-methyl-3-aminopropyl)-3-methyl-1-triazene (NOC7) in cell-free systems (Supplementary Methods online). H2 reduced ONOO− (Fig. 4d) somewhat, but did not reduce H2O2, NO• and O2−• (Fig. 4e–g). In cell-free experiments, we examined whether H2 reduced the oxidized forms of biomolecules involved in metabolic oxidation-reduction reactions. At room temperature and neutral pH, solutions saturated with H2 did not reduce the oxidized form of nicotinamide adenine dinucleotide (NAD+), the oxidized form of flavin adenine dinucleotide (FAD) or the oxidized form of cytochrome C (data not shown). Thus we infer that H2 does not affect the metabolism involved in oxidation-reduction reactions or the levels of O2−•, H2O2, and NO•, all of which play essential roles in signal transduction.
H2 protects neurons from in vitro ischemia and reperfusion
We also induced oxidative stress in a primary culture of neocortical cells19 under more physiological conditions. It is known that rapid transition from an ischemic condition to reperfusion results in oxidative stress damage20. To mimic ischemia, we subjected neocortical cells to oxygen glucose deprivation (OGD) under nitrogen or hydrogen gas for 60 min, followed by reperfusion with medium containing O2 and glucose.
HPF fluorescence showed that 10 min after the completion of OGD followed by reperfusion, •OH levels notably increased when H2 was absent, but diminished when H2 was present (Supplementary Fig. 4 online). At 24 h after OGD and reperfusion, H2 increased neuron survival and vitality (Supplementary Fig. 4), indicating that H2 protects neurons against oxidative stress–induced cell death.
Inhalation of H2 gas protects brain injury by reperfusion
To examine the therapeutic applicability of H2 as an antioxidant, we used a rat model of ischemia. ROS are generated during cerebral ischemia and reperfusion, and are one of the major causes of brain damage21,22. We produced focal ischemia in rats by occluding the middle cerebral artery (MCA) for 90 min, and then performed reperfusion for 30 min (ref. 23). In three of four conditions, rats inhaled H2 gas, mixed with nitrous oxide (N2O) for anesthesia, during the entire 120 min process (proportions of H2, O2 and N2O (vol/vol/vol) were 1%:30%:69%, 2%:30%:68%, and 4%:30%:66%); in the fourth condition, H2 was absent, (H2:O2:N2O (vol/vol/vol) was 0%:30%:70%). We carefully monitored physiological parameters during the experiments (Methods) and found no significant changes resulting from the inhalation of H2 (Supplementary Table 1 online). Additionally, there was no significant influence on cerebral blood flow, as measured by the Doppler effect (ref. 24 and Supplementary Fig. 5 online). H2 dissolved in arterial blood was increased by the inhalation of H2 in proportion to the concentration inhaled; the amount of H2 dissolved in venous blood was less than that in artery blood, suggesting that H2 had been incorporated into tissues (Fig. 5a).

(a) Rats inhaled H2 and 30% O2 for 1 h under the anesthetics N2O and halothane. Arterial (A) and venous (V) blood were collected, and the amount of H2 was examined by gas chromatography. (b) Rats underwent middle cerebral artery (MCA) occlusion. During the 120-min procedure, the indicated concentration of mixed gas was inhaled. One day after MCA occlusion, the forebrain was sliced into six coronal sequential sections and stained with the mitochondrial respiratory substrate TTC. Scale bar, 5 mm. (c) Infarct volumes of the brain were calculated in the brain slices. E and F, treatment with edaravone and FK506 (n = 6). *P < 0.05, **P < 0.01, ***P < 0.001, compared with 0% of H2. ##P < 0.01, ###P < 0.001 compared with 2% of H2. (d) Schematic of experiment with three different durations of hydrogen gas (2%) inhalation. (e) Infarct volumes of the brain for different durations of inhalation (calculated as in c) (n = 6). *P < 0.05, **P < 0.01, ***P < 0.001, compared with 0% of H2. #P < 0.05, ###P < 0.001 compared with 120 min of treatment. A, B and C represent the different durations of H2 gas inhalation (shown in d). Histograms represent mean ± s.d.
At 1 d after MCA occlusion, we sectioned and stained brains with 2,3,5-triphenyltetrazolium chloride (TTC), a substrate for mitochondrial respiration (Fig. 5b). We estimated infarct volumes by assessing the staining of brain areas (white indicates infarct, Fig. 5b,c), and found a clear H2-dependent decrease in infarct volume, with 2–4% of H2 providing the most substantial effect (Fig. 5c). We also noted that H2 exerted its effect only when it was inhaled during reperfusion; when H2 was inhaled during ischemia, infarct volume was not significantly decreased (Fig. 5d,e). For comparison, we tested two other compounds: edaravone (approved in Japan as an ROS scavenger for the treatment of cerebral infarction25) and FK506 (in clinical trials for cerebral infarction in the United States26). H2 was more effective than edaravone and as effective as FK506 in alleviating oxidative injury (Fig. 5c). These results indicate the potential of H2 for therapy.
Inhalation of H2 gas suppresses the progression of damage
At 1 week after MCA occlusion, the difference in infarct volume between untreated and H2-treated rats increased, compared to 1d after occlusion (Fig. 6a,b). The behavior of each rat, graded according to a neurological score27, revealed that the inhalation of H2 during ischemia and reperfusion improved movement (Fig. 6c). Moreover, although body weight and body temperature of H2-untreated rats gradually declined, those in H2-treated rats eventually recovered (Fig. 6d,e). Thus H2 suppressed not only the initial brain injury, but also its progressive damage.

Rats inhaled 2% of hydrogen gas during the 120-min ischemia and reperfusion procedure and were maintained for 1, 3 or 7 d. (a) One week after MCA occlusion, the brains were sliced and stained with hematoxylin and eosin. Three representative slices are shown. Scale bar, 5 mm. (b) Infarct volumes (light-pink regions in a) were calculated (n = 6). ***P < 0.001. (c) Neurological scores were graded on a scale of 0 to 5, as described previously27: score 0, no neurological deficit; 1, failure to fully extend the right forepaw; 2, circling to the right; 3, falling to the right; 4, unable to walk spontaneously; and 5, dead. When a rat's neurological score was judged to be between 1 and 2, 2 and 3, or 3 and 4, the score was set at 1.5, 2.5 and 3.5, respectively. Closed and open circles represent treatment with or without H2 (n = 6). ***P < 0.001. (d,e) Body weights and temperature were monitored with (closed circles) or without (open circles) inhalation of 2% hydrogen gas (n = 6). *P < 0.05, **P < 0.01, ***P < 0.001. (f) On days 3 or 7 after MCA occlusion, coronal 6-μm sections from the ischemic core area in the temporal cortex were stained with antibody to Iba1 (a microglial marker). Scale bar, 200 μm (100 μm in the inset). (g) Cells positive for the Iba1 antibody28, per field of view (FOV), were counted in the ischemic core area, as indicated in f (n = 6). *P < 0.05. Data represent mean ± s.d.
We examined H2-mediated molecular changes at 12 h, 3 d or 7 d after occlusion, by staining brain sections with antibodies to 8-OH-G in order to assess the extent of nucleic acid oxidation (Supplementary Fig. 6 online), and with antibodies to HNE to assess lipid peroxidation (Supplementary Fig. 6). For both of these oxidative markers, staining was substantially reduced in H2-treated rats as compared to untreated rats. We also stained identical regions of the brain with antibodies to Iba1 (ref. 28) and antibodies to GFAP, which are specific to activated microglia and to astrocytes, respectively (Fig. 6f,g and Supplementary Fig. 6). We found a distinct H2-dependent decrease in the accumulation of microglia, indicative of inflammation and remodeling. Taken together, these results indicate that H2 can markedly decrease oxidative stress and suppress brain injury.
Discussion
This study shows that molecular hydrogen can selectively reduce ROS in vitro. As •OH and ONOO− are much more reactive than other ROS (ref. 14), it stands to reason that H2 will react with only the strongest oxidants. This is advantageous for medical procedures, as it means that the use of H2 should not have serious unwanted side effects. It is likely that H2 is mild enough not to disturb metabolic oxidation-reduction reactions or to disrupt ROS involved in cell signaling—unlike some antioxidant supplements with strong reductive reactivity, which increase mortality, possibly by affecting essential defensive mechanisms29.
H2 has a number of advantages as a potential antioxidant: it effectively neutralizes •OH in living cells, and, unlike most known antioxidants, which are unable to successfully target organelles30, it has favorable distribution characteristics: it can penetrate biomembranes and diffuse into the cytosol, mitochondria and nucleus. Despite the moderate reduction activity of H2, its rapid gaseous diffusion might make it highly effective for reducing cytotoxic radicals. Its ability to protect nuclear DNA and mitochondria suggests that it could reduce the risk of life style–related diseases and cancer.
H2 markedly decreased oxidative stress and suppressed brain injury caused by ischemia and reperfusion. Inhalation of H2 gas was more efficacious than a treatment currently approved for cerebral infarction and, furthermore, mitigated hepatic injury caused by ischemia and reperfusion (K. Fukuda, S.A., M.I., Y. Yamamoto, I.O. and S.O., unpublished data). This finding indicates that the beneficial effects of H2 are not specific to cerebral injury but can be used for injuries in other organs.
This study suggests that H2 protects cells and tissues against strong oxidative stress by scavenging •OH. However, it remains possible that H2 also protects from stress by directly or indirectly reducing other strong oxidant species in living cells. For instance, H2 may induce cytoprotective factors; however, we found no H2-induced change in the expression of several genes involved in cytoprotection or reduction (K.N., M.I., I.O. and S.O., unpublished data). Further studies will reveal the mechanisms by which H2 protects cells and tissues against oxidative stress.
Acute oxidative stress may be caused by several factors, including inflammation, intense exercise, cardiac infarction, cessation of blood flow and organ transplantation. For treatment, H2 dissolved in saline could easily be delivered intravascularly. For prevention, H2 saturated in water could be administered. Inhalation of H2 has already been used in the prevention of decompression sickness in divers and has shown a good safety profile31. Notably, H2 has no risk of flammability or explosion at a concentration of less than 4.7% in air. We propose that H2, one of the most well-known molecules, could be widely used in medical applications as a safe and effective antioxidant with minimal side effects.
Methods
Hydrogen and oxygen measurements.
We measured molecular hydrogen (H2) and oxygen (dioxygen, O2) dissolved in solution by using a hydrogen electrode (ABLE) and an oxygen electrode (Strathkelvin Instruments), respectively. We determined hydrogen gas concentration by gas chromatography (Teramecs). For measuring H2 levels in blood, we pretreated rats with heparin to avoid blood clotting, collected arterial and venous blood (5 ml each) in test tubes, and then immediately injected the blood samples into closed aluminum bags containing 30 ml of air. After complete transfer of the H2 gas from the blood to the air in the closed bag, we subjected 20 ml of the air to gas chromatography using standard H2 gas, in order to quantify the amount of H2.
Hydrogen treatment of cultured cells.
Over a 2-h period, we dissolved H2 beyond saturation levels into DMEM medium under 0.4 MPa pressure. We dissolved O2 into a second medium by bubbling O2 gas at the saturated level (42.5 mg/l), and CO2 into a third medium by bubbling CO2 gas. All three media were maintained at atmospheric pressure. We then combined the three media (H2 medium:O2 medium:CO2 medium) in the proportion 75%:20%:5% (vol/vol/vol) and added fetal bovine serum (FBS) to achieve a final concentration of 1%. For culture, we put the combined medium into a culture flask and immediately examined H2 or O2 concentration with an H2 or O2 electrode. Then we filled the culture flask with mixed gas consisting of 75% H2, 20% O2 and 5% CO2 (vol/vol/vol) and cultured cells in the closed culture flask. We prepared degassed medium lacking H2 by stirring the medium, which had been saturated with H2, in an open vessel for 4 h, and checked the concentration of H2 with a hydrogen electrode. In the experiments on the dose dependence of H2 (results shown in Fig. 2f), we diluted the combined medium with a fourth medium containing 1% FBS equilibrated with air containing 5% CO2, in order to obtain the desired concentration of H2; we then filled the culture flasks with the mixed gas diluted with air containing 5% CO2.
Induction of oxidative stress by antimycin A and menadione.
We maintained PC12 cells at 37 °C in DMEM medium containing 1% FBS with or without 0.6 mM H2 in a closed flask filled with mixed gases as described above. We treated the cells with menadione or antimycin A, which inhibit complex I or complex III, respectively, of the mitochondrial electron transport chain, and thus produce O2−• (by accelerating the leakage of electrons). After exposure to antimycin A for 24 h, we assessed cell survival by manually counting the cells double-stained with 1 μM propidium iodide (dead cells labeled pink) and 5 μM Hoechst 33342 (dead and living cells labeled blue) under a fluorescent microscope. To examine the protective effect by H2 on mitochondria, we pretreated cells with 4.5 g/l 2-deoxy-D-glucose (an inhibitor of glycolysis) and 1 mM pyruvate (a substrate of oxidative phosphorylation) for 30 min, exposed them to antimycin A with or without 0.6 mM H2 and then quantified cellular ATP levels using a cellular ATP measurement kit (TOYO B-Net.).
Cerebral infarction model.
Animal protocols were approved by the Animal Care and Use Committee of Nippon Medical School. We anesthetized male Sprague-Dawley rats (body weight: 250–300 g) with halothane (4% for induction, 1% for maintenance) in a mixture of nitrous oxide and oxygen (70%:30%, vol/vol). We maintained temperature (37.5 ± 0.5 °C) using a thermostatically controlled heating blanket connected to a thermometer probe in the rectum, and, at the same time, monitored physiological parameters (using a cannula in the tail artery), including blood gases (pCO2 and pO2), pH, glucose level and blood pressure. We attempted to maintain constant levels of pH and pO2 by regulating the amount of halothane and the N2O:O2 ratio. We produced focal ischemia by performing intraluminal occlusion of the left middle cerebral artery (MCA), using a nylon monofilament with a rounded tip and a distal silicon rubber cylinder as previously described23. The rats underwent MCA occlusion for 90 min and then reperfusion for 30 min; they inhaled H2 gas during the entire process except in the experiments corresponding to Figure 5d,e. We treated rats with edaravone and FK506 using the most effective concentrations (refs. 25, 23 and Fig. 5c). After the rats recovered from anesthesia, they were maintained at 23 °C.
At 24 h after MCA occlusion, we removed brains under anesthesia and sliced them into six coronal sequential sections (2 mm thick). We stained the sections with 2,3,5-triphenyltetrazolium chloride (TTC) (3%), and then measured infarct and noninfarct areas using an optical dissector image analysis system (Mac SCOPE, Mitsuya Shoji). We outlined the border between infarct and noninfarct tissues, and obtained the area of infarction by subtracting the nonlesioned area of the ipsilateral hemisphere from that of the contralateral side. We calculated the volume of infarction as infarct area × thickness. At 12 h, 3 d or 7 d after MCA occlusion, we quickly removed brains under anesthesia, and fixed them with 10% formalin. We sliced paraffin-embedded brains into a series of 6-μm sections, and stained sections with hematoxylin and eosin (H&E). We then quantified the pink areas with a graphic analyzer system (Mac Scope). For immunostaining, we stained the sections with antibodies by using VECTASTAIN ABC reagents according to the supplier's instructions.
Statistical analysis.
We used StatView software (SAS Institute) for the statistical analyses. For single comparisons, we performed an unpaired two-tailed Student's t-test; for multiple comparisons, we used an analysis of variance (ANOVA) followed by Fisher's exact test. We performed experiments for quantification in a blinded fashion.
Note: Supplementary information is available on the Nature Medicine website.
References
Wallace, D.C. A mitochondrial paradigm of metabolic and degenerative diseases, aging, and cancer: a dawn for evolutionary medicine. Annu. Rev. Genet. 39, 359–407 (2005).
Reddy, P.H. Amyloid precursor protein-mediated free radicals and oxidative damage: implications for the development and progression of Alzheimer's disease. J. Neurochem. 96, 1–13 (2006).
Ohta, S. A multi-functional organelle mitochondrion is involved in cell death, proliferation and disease. Curr. Med. Chem. 10, 2485–2494 (2003).
Wright, E., Jr ., Scism-Bacon, J.L. & Glass, L.C. Oxidative stress in type 2 diabetes: the role of fasting and postprandial glycaemia. Int. J. Clin. Pract. 60, 308–314 (2006).
Winterbourn, C.C. Biological reactivity and biomarkers of the neutrophil oxidant, hypochlorous acid. Toxicology 181, 223–227 (2002).
Chinopoulos, C. & Adam-Vizi, V. Calcium, mitochondria and oxidative stress in neuronal pathology. Novel aspects of an enduring theme. FEBS J. 273, 433–450 (2006).
Sauer, H., Wartenberg, M. & Hescheler, J. Reactive oxygen species as intracellular messengers during cell growth and differentiation. Cell. Physiol. Biochem. 11, 173–186 (2001).
Turrens, J.F. Mitochondrial formation of reactive oxygen species. J. Physiol. (Lond.) 552, 335–344 (2003).
Sheu, S.S., Nauduri, D. & Anders, M.W. Targeting antioxidants to mitochondria: a new therapeutic direction. Biochim. Biophys. Acta 1762, 256–265 (2006).
Liu, H., Colavitti, R., Rovira, I.I. & Finkel, T. Redox-dependent transcriptional regulation. Circ. Res. 97, 967–974 (2005).
Murad, F. Discovery of some of the biological effects of nitric oxide and its role in cell signaling. Biosci. Rep. 24, 452–474 (2004).
Buxton, G.V., Greenstock, C.L., Helman, W.P. & Ross, A.B. Critical review of rate constants for reactions of hydrated electrons, hydrogen atoms and hydroxyl radicals (•OH/•O-) in aqueous solution. J. Phys. Chem. Ref. Data 17, 513–886 (1988).
Ohsawa, I., Nishimaki, K., Yasuda, C., Kamino, K. & Ohta, S. Deficiency in a mitochondrial aldehyde dehydrogenase increases vulnerability to oxidative stress in PC12 cells. J. Neurochem. 84, 1110–1117 (2003).
Setsukinai, K., Urano, Y., Kakinuma, K., Majima, H.J. & Nagano, T. Development of novel fluorescence probes that can reliably detect reactive oxygen species and distinguish specific species. J. Biol. Chem. 278, 3170–3175 (2003).
Tomizawa, S. et al. The detection and quantification of highly reactive oxygen species using the novel HPF fluorescence probe in a rat model of focal cerebral ischemia. Neurosci. Res. 53, 304–313 (2005).
Kamiya, H. Mutagenicities of 8-hydroxyguanine and 2-hydroxyadenine produced by reactive oxygen species. Biol. Pharm. Bull. 27, 475–479 (2004).
Petersen, D.R. & Doorn, J.A. Reactions of 4-hydroxynonenal with proteins and cellular targets. Free Radic. Biol. Med. 37, 937–945 (2004).
Falick, A.M., Mahan, B.H. & Myers, R.J. Paramagnetic resonance spectrum of the 1Δg oxygen molecule. J. Chem. Phys. 42, 1837–1838 (1965).
Asoh, S. et al. Protection against ischemic brain injury by protein therapeutics. Proc. Natl. Acad. Sci. USA 99, 17107–17112 (2002).
Halestrap, A.P. Calcium, mitochondria and reperfusion injury: a pore way to die. Biochem. Soc. Trans. 34, 232–237 (2006).
Lipton, P. Ischemic cell death in brain neurons. Physiol. Rev. 79, 1431–1568 (1999).
Ferrari, R. et al. Oxidative stress during myocardial ischaemia and heart failure. Curr. Pharm. Des. 10, 1699–1711 (2004).
Nito, C., Kamiya, T., Ueda, M., Arii, T. & Katayama, Y. Mild hypothermia enhances the neuroprotective effects of FK506 and expands its therapeutic window following transient focal ischemia in rats. Brain Res. 1008, 179–185 (2004).
Takada, J. et al. Adenovirus-mediated gene transfer to ischemic brain is augmented in aged rats. Exp. Gerontol. 38, 423–429 (2003).
Zhang, N. et al. Edaravone reduces early accumulation of oxidative products and sequential inflammatory responses after transient focal ischemia in mice brain. Stroke 36, 2220–2225 (2005).
Labiche, L.A. & Grotta, J.C. Clinical trials for cytoprotection in stroke. NeuroRx 1, 46–70 (2004).
Murakami, K. et al. Mitochondrial susceptibility to oxidative stress exacerbates cerebral infarction that follows permanent focal cerebral ischemia in mutant mice with manganese superoxide dismutase deficiency. J. Neurosci. 18, 205–213 (1998).
Ito, D. et al. Microglia-specific localisation of a novel calcium binding protein, Iba1. Brain Res. Mol. Brain Res. 57, 1–9 (1998).
Bjelakovic, G., Nikolova, D., Gluud, L.L., Simonetti, R.G. & Gluud, C. Mortality in randomized trials of antioxidant supplements for primary and secondary prevention: systematic review and meta-analysis. J. Am. Med. Assoc. 297, 842–857 (2007).
James, A.M., Cocheme, H.M. & Murphy, M.P. Mitochondria-targeted redox probes as tools in the study of oxidative damage and ageing. Mech. Ageing Dev. 126, 982–986 (2005).
Fontanari, P. et al. Changes in maximal performance of inspiratory and skeletal muscles during and after the 7.1-MPa Hydra 10 record human dive. Eur. J. Appl. Physiol. 81, 325–328 (2000).
Acknowledgements
This work was supported by grants to S.O. from the Ministry of Health, Labor and Welfare (H17-Chouju-009, longevity science; and 17A-10, nervous and mental disorders) and the Ministry of Education, Culture, Sports, Science and Technology (16390257).
Author information
Authors and Affiliations
Contributions
S.O. conceived the experiments. S.O., I.O., K.K. and Y.K. designed the experiments. I.O., S.A. and S.O. performed data analysis. I.O., M.I., K.T., M.W., K.N, K.Y., S.A. and S.O. performed the experiments. S.O. and I.O. wrote the paper.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Supplementary Fig. 1
Molecular hydrogen dissolved in culture medium does not reduce cellular hydrogen peroxide and nitric oxide. (PDF 351 kb)
Supplementary Fig. 2
pH, H2 and O2 maintain constant in culture medium in a closed flask filled with a mixed gas. (PDF 75 kb)
Supplementary Fig. 3
Several methods confirm protection of cells by H2 against oxidative stress. (PDF 470 kb)
Supplementary Fig. 4
Molecular hydrogen protects cultured neurons from ischemia and reperfusion in vitro. (PDF 553 kb)
Supplementary Fig. 5
Cerebral blood flow is not influenced by H2 inhalation. (PDF 417 kb)
Supplementary Fig. 6
The brain after induction of ischemia reperfusion injury with or without H2 treatment was immunostained. (PDF 305 kb)
Supplementary Table 1
Physiological parameters during cerebral ischemia reperfusion (PDF 255 kb)
Rights and permissions
About this article
Cite this article
Ohsawa, I., Ishikawa, M., Takahashi, K. et al. Hydrogen acts as a therapeutic antioxidant by selectively reducing cytotoxic oxygen radicals. Nat Med 13, 688–694 (2007). https://doi.org/10.1038/nm1577
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/nm1577
This article is cited by
-
Effects of dietary Clostridium butyricum and rumen protected fat on meat quality, oxidative stability, and chemical composition of finishing goats
Journal of Animal Science and Biotechnology (2024)
-
Si-based agent alleviated small bowel ischemia–reperfusion injury through antioxidant effects
Scientific Reports (2024)
-
Hydrogen gas can ameliorate seizure burden during therapeutic hypothermia in asphyxiated newborn piglets
Pediatric Research (2024)
