
Abstract
Despite progress in mass spectrometry (MS)-based phosphoproteomics, large-scale in vivo analyses remain challenging. Here we report a 'spike-in' stable-isotope labeling with amino acids in cell culture (SILAC) methodology using standards derived from labeled mouse liver cell lines, using which we analyzed insulin signaling. With this approach we identified 15,000 phosphosites and quantitatively compared 10,000 sites in response to insulin treatment, creating a very large, accurately quantified in vivo phosphoproteome dataset.
Similar content being viewed by others

Main
Mass spectrometry–based phosphoproteomics has evolved into a powerful technology for system-wide phosphoproteomic studies1,2,3,4. It has been used to characterize the phosphoproteome of different systems, ranging in complexity from bacteria to yeast and mammalian cells lines. To characterize physiological or pathological responses of different organs in mammals, it is essential to extend these studies to measure phosphorylation changes in vivo. However, identification and quantification of phosphorylated peptides (phosphopeptides) from mammalian tissues has not been practical because of the complexities of tissue proteomes and the extremely challenging nature of the required proteomic analysis. The largest tissue phosphoproteomic study to date has mapped ∼7,000 phosphorylation sites (phosphosites) in the mouse liver and ∼15,000 sites in mouse brain5. That study and others6,7, however, did not use accurate quantification and did not address the regulation of the phosphoproteome in these tissues.
Quantitative phosphoproteomic analyses of tissues are in principle possible using mice labeled via stable-isotope labeling with amino acids in cell culture (SILAC)8. We initially sought to investigate the mouse liver phosphoproteome using liver from mice labeled by SILAC as an internal standard. We had combined this with the standard workflow in our laboratory, including collision-induced dissociation for phosphopeptide fragmentation on a linear ion trap Orbitrap mass spectrometer9. However, the complexity of the tissue material as well as technical limitations posed by the longer peptides generated by LysC digestion of tissue from mouse labeled with lysine via SILAC limited accurate quantification and site localization to less than 1,000 phosphosites from 80 liquid chromatography–tandem mass spectrometry (LC-MS/MS) runs. This prompted us to develop an alternative but still highly accurate in vivo approach. Motivated by our recently described super-SILAC technique10, we developed a methodology that relies on SILAC-based quantitation but uses phosphopeptides derived from a mouse liver cell line (Hepa1-6) as an internal standard for in vivo measurements. As a proof of principle, we applied the method to study insulin signaling in vivo. Insulin is an important regulator of metabolic and mitogenic processes, and alterations of this pathway cause pathologies such as insulin resistance and type-2 diabetes11. Investigating insulin signaling in animals provides valuable information that complements cell-line studies because it allows consideration of the contribution of different tissues and environmental influences such as diet and physical activity.
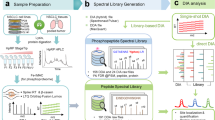
To generate a phosphopeptide standard for these measurements, we used the general principle of 'spike-in' SILAC12,13 and metabolically labeled mouse hepatoma Hepa1-6 cells with heavy forms of lysine (4,4,5,6-D4-L-lysine) and arginine (13C614N4-L-arginine). We divided the cells into two populations: one left untreated (control) and the other stimulated with insulin to achieve an adequate representation of insulin-dependent and insulin-independent phosphosites in the standard (Fig. 1a). The choice of standard mixture depends on the complexity of the analyzed samples, and here we were interested in representing signaling in hepatocytes, by far the most prominent cell type in the liver. Hepa1-6 cells, in which insulin signaling is intact, turned out to adequately reflect the phosphoproteome of interest. Compared to chemically synthesizing labeled phosphopeptides, our 'phospho-SILAC' standard has several advantages. First, it leads to the economical generation of tens of thousands of standard peptides present at roughly the desired amounts for accurate relative quantification of phosphosites across different conditions. Second, it allows mixing the standard with the sample early in processing, avoiding quantification errors introduced during the proteomics workflow. Furthermore, in contrast to labeling tissue via SILAC, this technology can be extended to human material.

(a) Experimental design for the analyses of control (PBS-treated) and insulin-treated mice. A mixture of insulin-treated and untreated Hepa1-6 cells labeled via SILAC serves as an internal standard. The mixtures are digested, and the resulting peptides are analyzed by high-resolution LC-MS/MS. Ratios from control and insulin-treated samples are then compared ('ratio of ratios'). SCX, strong cation exchange. (b,c) Distribution of the ratios between the phosphopeptides of the control livers and of the Hepa1-6 cells (b) and between phosphopeptides of insulin-treated livers and of Hepa1-6 cells (c) (labeled liver/Hepa1-6). (d,e) 'Ratio of ratios' histogram (d) and boxplot of the phosphosite ratios between insulin-treated and control (labeled insulin/PBS) liver samples (e) (representative example of one technical replicate; triplicate distribution is even narrower).
To analyze the samples, we used the sodium dodecyl sulfate (SDS)-based filter-aided sample preparation (FASP) method14 for complete solubilization and digestion, and a linear ion trap Orbitrap Velos instrument with very high sequencing speed15. The FASP method enables unbiased and clean sample preparation without the need to add inhibitors of proteases or phosphatases because 4% SDS acts as a potent phosphatase and protease inhibitor. The high ion current transmission of LTQ Orbitrap Velos instrument combined with better transmission of the fragment ions from the collision cell adjacent to the C-trap enabled routine use of higher-energy collision dissociation fragmentation16,17. By acquiring full scan and fragmentation scan (denoted by MS and MS/MS scan) measurements in the Orbitrap analyzer, we obtained parts-per-million–range mass accuracy for both precursor peptides and their fragments, which enabled identification of peptides and sites that would otherwise be lost in low-accuracy spectra. Analysis of the mass spectrometric data, including determination of quantitative ratios was done in the MaxQuant environment (Online Methods). All mass spectrometric data and annotated spectra of all phosphosites are available in Tranche (Supplementary Note).
To test our methodology and workflow, we injected two mice each with 1 unit kg−1 body weight of insulin or phosphate-buffered saline (PBS) as a control. We collected livers of these mice 15 min after injection and snap-froze them in liquid nitrogen, which preserves the phosphoproteome. Before processing the samples, we spiked our phospho-SILAC standard into the liver tissue lysates from control and insulin injected animals. To determine whether our standard mixture contained most of the phosphopeptides of the liver sample at the appropriate abundance to yield quantifiable ratios, we analyzed the histogram of the ratios between SILAC heavy and light amino acid–containing peptide pairs. The ratio distribution of phosphopeptides was unimodal, indicating that there is a good match between liver and cell-line phosphoproteomes, and that the great majority of the light phosphopeptides from the sample of interest had counterparts in the heavy amino acid–labeled internal standard (Fig. 1b,c). However the distribution was not centered at one, as peptides in tissue and cell-line phosphoproteomes were not quantitatively identical. This also has been observed in the super-SILAC study10 but is inconsequential for 'ratio of ratio' determination as described below.
To test the reproducibility of our workflow, we separately analyzed three technical replicates derived from both insulin-treated and control liver samples, and found a high correlation between measurements with less than 0.4% of the ratios were regulated in opposite direction across different technical replicates (Supplementary Fig. 1). We therefore concluded that our methodology is robust and reproducible.
Using the described method, we mapped 20,491 phosphosites (average localization probability of 0.99 for the 14,857 class I phosphosites, whereas the remaining phosphosites had less than 0.75 localization probability but were still identified with 99% confidence), providing to our knowledge the largest in vivo phosphoproteome dataset not only for liver but for any other mammalian tissue analyzed so far. The approach allowed accurate quantitative comparison of 10,000 phosphosites, on average, in response to insulin treatment. A summary of the identification and quantification of phosphosites in the individual experiments is available in Supplementary Table 1.
Comparison of liver samples with the standard generates a heavy-to-light ratio between the two forms of the peptide. Because the internal standard is the same for all the samples, the 'ratio of ratios' between the insulin- and PBS-injected liver samples allows direct comparison of phosphopeptide abundances. The distribution of these 'ratios of ratios' between insulin-treated and PBS-treated mice was remarkably narrow, demonstrating high accuracy of quantitation (Fig. 1d,e). The narrow distribution also implies that the large majority of phosphosites were not regulated in response to insulin. In fact, only about 1,000 sites responded to stimulation by insulin.
To define a cut-off value for phosphorylation changes resulting from insulin stimulation, we performed box-plot statistics on the ratios of the medians of the three insulin-stimulated and the three control phosphoproteome measurements. This showed that ratio changes of less than 0.5 or more than 1.5 were significant (lying outside the quartiles by 1.5 times the interquartile distance; Supplementary Fig. 2). This demonstrated that both the depth and the quantitative accuracy of the in vivo experiment were at least on par with that achieved in cell-line experiments. This is particularly important for studying insulin signaling, which involves only moderate changes in phosphorylation.
For our analysis, we chose 15 min of insulin treatment because we wanted to characterize intermediate to late stages of signaling. At this time point, we did not expect to detect tyrosine phosphorylation of the initial mediators of the pathway, for example, Irs1 and Irs2, but rather expected to measure the increased serine phosphorylation of later mediators, such as Akt or mTOR substrates. To confirm the activation efficiency of the insulin signaling pathway in the liver by the in vivo treatment, we assessed the Ser473 phosphorylation of Akt by western blot. As expected, livers from insulin-treated, but not from PBS-injected control mice, showed strong upregulation of Akt phosphorylation (Supplementary Fig. 3).
Much is already known about the insulin-signaling pathway in liver cells, but a global view of the insulin phosphoproteome in vivo has been lacking. Insulin interacts with its receptor, a tyrosine kinase, initiating a signaling cascade that culminates in alterations of metabolism and activation of the mitogenic program. Our dataset revealed phosphosites on many of the known targets of the insulin cascade (Fig. 2 and Table 1). Examples of known targets include the key signaling mediators Irs1 and Irs2, for which we quantified more than twofold changes in the phosphorylation of several serine sites after insulin stimulation. In addition, the dataset contains hundreds of regulated sites that to our knowledge previously had not been described for insulin signaling (Supplementary Tables 2 and 3).

After interacting with the insulin receptor tyrosine kinase, insulin activates mediators that regulate the metabolic and mitogenic effects of insulin. Phosphorylation changes after insulin treatment of mice are shown. The pathway in the figure summarizes the literature11, and the phosphosites identified as regulated in this study are indicated by colored circles.
In summary, our results demonstrate that our strategy of using SILAC-prepared phosphorylated standard mixtures is well suited to identify and especially to quantify protein phosphorylation in complex tissue samples to a depth previously only achievable in cell-culture experiments. The method is generic and can, in principle, be applied to any organism and tissue. In more heterogeneous systems such as human tumor biopsies, a super-SILAC strategy involving a mixture of several cell lines might be more appropriate for phosphoproteomics experiments than the mixture of a single nonstimulated and stimulated cell line we used here. We illustrated the utility of this approach by quantifying changes in the tissue phosphoproteome in vivo, after systemic administration of insulin. Our strategy is applicable to any type of signaling analysis, ranging from responses to hormones, to inflammatory responses and pharmacological treatments in model organisms or in humans. Our phosphopeptide standard technology is thus a generic means of studying phosphorylation responses in vivo. We hope that it will aid the understanding of physiological responses and help support the development of therapeutics for pathological conditions.
Methods
Data information.
All mass spectrometric raw files, annotated spectra and MaxQuant output tables have been deposited in the open-access public repository Tranche (http://proteomecommons.org/tranche/), as listed in the Supplementary Note.
Insulin treatment of mice and preparation of liver samples for mass-spectrometric analysis.
PBS or insulin from bovine pancreas (Sigma) (1 U kg−1 body weight) was administered to 10-week-old C57BL/6 male mice (two mice for each condition) intraperitoneally after overnight fasting. The mice were housed in a 12 h light–12 h dark cycle pathogen-free facility and fed a chow diet in the animal house of the Max Planck Institute of Biochemistry, which has a license for breeding and housing of laboratory animals (5.1.-568 rural districts office). They were killed by cervical dislocation 15 min after the treatment. Livers were homogenized with an IKA Ultra Turbax blender (speed setting 5) in a lysis buffer (4% SDS, 100 mM Tris-HCl (pH 7.6) and 0.1 M DTT). Lysis buffer was added to the liver tissue before the homogenization (roughly 1 mg tissue:5 μl buffer). Depending on the amount of tissue, samples were homogenized for 30 s to a few minutes at room temperature (25 °C) to avoid SDS precipitation. The homogenates were incubated for 3 min at 95 °C, sonicated with Branson sonifier 250 for 4–5 min at output control 1 and duty cycle 10%, centrifuged at 16,000g at 20 °C for 5 min, supernatant was collected for the analyses, and the biological replicates were pooled.
SILAC labeling of Hepa1-6 cells and preparation of the internal standard.
To generate phospho-SILAC standards, we cultured Hepa1-6 cells in SILAC medium (Invitrogen) containing heavy isotope–labeled 13C614N4-L-arginine and 4,4,5,6-D4-L-lysine (Cambridge Isotope Laboratories) supplemented with 10% dialyzed fetal bovine serum (FBS) (Invitrogen). Labeled Hepa1-6 cells were starved for 24 h and treated with 100 nM insulin for 15 min. The cells were washed three times with PBS and collected in lysis buffer. After 3 min of incubation at 95 °C and sonication, the samples were centrifuged at 16,000g at 20 °C for 5 min. For the internal standard, equal amounts of protein from Hepa1-6 cells that were untreated and treated with insulin were mixed.
Protein digestion by filter-aided sample preparation.
For protein digestion, 10 mg of proteins from control or insulin treated liver homogenates and 10 mg of proteins from SILAC-labeled Hepa1-6 internal standard lysates were mixed and diluted in buffer containing 8 M urea and 100 mM Tris-HCl (pH 8.5). The protein concentration was determined by the Bradford method using 5 μl of samples diluted 1:20 in 1 ml of measurement solution, compared to a BSA standard curve. This dilution reduced interference of SDS with the assay. The samples were loaded onto 15 ml AmiconUltra Ultracel-30K filters (Millipore) (5 mg protein per filter) and processed according to the FASP protocol14. Briefly, SDS from the samples was removed by urea exchange followed by alkylation with iodoacetamide (Sigma). Urea was then replaced with 20 mM NH4HCO3, and trypsin (Promega) was added (1 μg trypsin per 100 μg protein) for overnight digestion at 37 °C. Peptides were collected from the filter by centrifugation followed by a wash with water. Recovery from the filter was around 50%, and the peptide concentration of the resulting solution (pooled from four filters) was ∼1.45 mg ml−1.
Peptide fractionation by strong cation exchange chromatography.
Eluted peptides were adjusted to pH 2.7 and 30% acetonitrile (the final volume of the solution was 10 ml) and loaded onto a 1 ml Resource S column (GE Healthcare) equilibrated with 30% acetonitrile containing 5 mM KH2PO4 (pH 2.7). Multiply phosphorylated peptides did not bind to the column and were collected in the flow-through. Bound peptides were eluted in a 30-min increasing salt gradient with a buffer containing 5 mM KH2PO4 and 150 mM KCl in six fractions.
Enrichment of phosphopeptides by TiO2 beads.
The fractions and the flow through were incubated one and four times, respectively, with 5 mg of TiO2 beads (MZ-Analysentechnik) prepared in 30 mg ml−1 solution of dihydrobenzoic acid (Sigma). After incubation, the beads were washed with a solution of 30% acetonitrile with 0.5% (v/v) trifluoroacetic acid (TFA) in water followed by a second wash with 80% acetonitrile with 0.1% TFA. The phosphopeptides were then eluted from the beads with 15% NH3 and loaded on C18 StageTips.
Reverse phase chromatography and mass spectrometry.
Peptides were separated on a 15-cm column with 75 μm inner diameter packed in-house with reversed-phase material ReproSil-Pur 120 C18-AQ 3 μm resin (Dr. Maisch GmbH). Peptides were separated on a binary buffer system of 0.5% acetic acid (solution A) and 80% acetonitrile in 0.5% acetic acid (solution B) in a Proxeon Easy nLC system (Proxeon Biosystems, now Thermo Fisher Scientific). For each sample, 4.5 μl of peptides were loaded on the column at a flow rate of 400 nl min−1. Peptides were separated by a linear gradient of buffer B from 5% to 35% in 90 min.
Liquid chromatography was interfaced to an LTQ Orbitrap Velos instrument via a Proxeon nanoelectrospray ionization source. The mass spectrometer was operated in a data-dependent mode, with full scan acquisition in the Orbitrap at 30,000 resolution (at 400 m/z) after filling to a target value of 106 ions in the C-trap. Lock mass of 445.120025 Da was used but without dedicated injection. For the data-dependent scans, the ten most intense ions were selected with assigned charge state >1 to a target value of 30,000 ions within a maximum injection time of 150 ms and fragmented by higher energy collision dissociation fragmentation (HCD) in the collision cell with a normalized collision energy of 40. The fragment ions were then detected in the Orbitrap at 7,500 resolution.
Data processing.
The data were processed with MaxQuant18 version 1.1.1.21, and the peptides were identified from the MS/MS spectra searched against the International Protein Index (IPI) mouse database (version 3.68) using the Andromeda search engine19. Cysteine carbamidomethylation was used as a fixed modification and methionine oxidation, protein N-terminal acetylation and serine, threonine and tyrosine phosphorylation as variable modifications. For the identification, the false discovery rate was set to 0.01 for peptides, proteins and sites and the minimum peptide length allowed was six amino acids.
References
Witze, E.S., Old, W.M., Resing, K.A. & Ahn, N.G. Nat. Methods 4, 798–806 (2007).
Bodenmiller, B., Mueller, L.N., Mueller, M., Domon, B. & Aebersold, R. Nat. Methods 4, 231–237 (2007).
Boersema, P.J., Mohammed, S. & Heck, A.J. J. Mass Spectrom. 44, 861–878 (2009).
Choudhary, C. & Mann, M. Nat. Rev. Mol. Cell Biol. 11, 427–439 (2010).
Huttlin, E.L. et al. Cell 143, 1174–1189 (2010).
Villen, J., Beausoleil, S.A., Gerber, S.A. & Gygi, S.P. Proc. Natl. Acad. Sci. USA 104, 1488–1493 (2007).
Wisniewski, J.R., Nagaraj, N., Zougman, A., Gnad, F. & Mann, M. J. Proteome Res. 9, 3280–3289 (2010).
Kruger, M. et al. Cell 134, 353–364 (2008).
Olsen, J.V. et al. Cell 127, 635–648 (2006).
Geiger, T., Cox, J., Ostasiewicz, P., Wisniewski, J.R. & Mann, M. Nat. Methods 7, 383–385 (2010).
Taniguchi, C.M., Emanuelli, B. & Kahn, C.R. Nat. Rev. Mol. Cell Biol. 7, 85–96 (2006).
Geiger, T. et al. Nat. Protoc. 6, 147–157 (2011).
Ishihama, Y. et al. Nat. Biotechnol. 23, 617–621 (2005).
Wisniewski, J.R., Zougman, A., Nagaraj, N. & Mann, M. Nat. Methods 6, 359–362 (2009).
Olsen, J.V. et al. Mol. Cell. Proteomics 8, 2759–2769 (2009).
Olsen, J.V. et al. Nat. Methods 4, 709–712 (2007).
Nagaraj, N., D'Souza, R.C., Cox, J., Olsen, J.V. & Mann, M. J. Proteome Res. 9, 6786–6794 (2010).
Cox, J. & Mann, M. Nat. Biotechnol. 26, 1367–1372 (2008).
Cox, J. et al. J. Proteome Res. 10, 1794–1805 (2011).
Acknowledgements
We thank the members of our laboratory for fruitful discussions and help. T. Walther, R. Farese, B. Blagoev and I. Kratchmarova critically read the manuscript. This project was supported by HepatoSys and by Diabetes Genome Anatomy Project (US National Institutes of Health grant DK60837).
Author information
Authors and Affiliations
Contributions
M. Monetti designed and performed the experiments, analyzed the data and wrote the paper. N.N. contributed to experiments, data analysis and writing the manuscript. K.S. assisted with data analysis and writing the manuscript. M. Mann supervised the work and wrote the paper.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Supplementary Text and Figures
Supplementary Figures 1–3, Supplementary Table 1 and Supplementary Note (PDF 2084 kb)
Supplementary Table 2
List of all class I sites quantified in at least two experiments with details including the coefficient of variation and directional variability of the ratios. (XLS 14899 kb)
Supplementary Table 3
List of regulated class I sites and sites that are exclusively quantified either in insulin- or PBS- treated samples. (XLS 2075 kb)
Rights and permissions
About this article
Cite this article
Monetti, M., Nagaraj, N., Sharma, K. et al. Large-scale phosphosite quantification in tissues by a spike-in SILAC method. Nat Methods 8, 655–658 (2011). https://doi.org/10.1038/nmeth.1647
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/nmeth.1647
This article is cited by
-
Cell-type-specific metabolic labeling of nascent proteomes in vivo
Nature Biotechnology (2017)
-
Fatty acid synthesis configures the plasma membrane for inflammation in diabetes
Nature (2016)
-
Identification of KasA as the cellular target of an anti-tubercular scaffold
Nature Communications (2016)
-
High-throughput phosphoproteomics reveals in vivo insulin signaling dynamics
Nature Biotechnology (2015)
-
Phosphoproteomics reveals malaria parasite Protein Kinase G as a signalling hub regulating egress and invasion
Nature Communications (2015)
