
Abstract
The identification and functional characterization of microbial communities remains a prevailing topic in microbial oceanography as information on environmentally relevant pelagic prokaryotes is still limited. The Roseobacter group, an abundant lineage of marine Alphaproteobacteria, can constitute large proportions of the bacterioplankton. Roseobacters also occur associated with eukaryotic organisms and possess streamlined as well as larger genomes from 2.2 to >5 Mpb. Here, we show that one pelagic cluster of this group, CHAB-I-5, occurs globally from tropical to polar regions and accounts for up to 22% of the active North Sea bacterioplankton in the summer. The first sequenced genome of a CHAB-I-5 organism comprises 3.6 Mbp and exhibits features of an oligotrophic lifestyle. In a metatranscriptome of North Sea surface waters, 98% of the encoded genes were present, and genes encoding various ABC transporters, glutamate synthase and CO oxidation were particularly upregulated. Phylogenetic gene content analyses of 41 genomes of the Roseobacter group revealed a unique cluster of pelagic organisms distinct from other lineages of this group, highlighting the adaptation to life in nutrient-depleted environments.
Similar content being viewed by others


The Roseobacter group is, besides the SAR11 clade, one of the most abundant lineages of Alphaproteobacteria in ocean surface waters, and it is of particular importance during phytoplankton blooms1–4. Members of this group occur globally and show distinct distribution patterns with a preferred occurrence from temperate to polar regions5–7. They exhibit a wide range of different physiological traits, for example, aerobic anoxygenic photosynthesis (AAnP), degradation of dimethylsulfoniopropionate (DMSP) and oxidation of CO (ref. 8). Despite several well studied model strains9–11, little is known about the abundant pelagic clusters of the Roseobacter group12. Most information exists on the largest cluster, the Roseobacter clade affiliated (RCA) cluster6,13–15. A type species of this cluster, Planktomarina temperata RCA23, and its genome have been described16,17. Scarce information exists on cluster NAC11-7 (refs 13,18,19), for which an isolate has been obtained and its genome sequenced (strain HTCC2255). The isolate was lost but the genomic information provided a most valuable reference for, for example, environmental gene expression studies20.
The abundant CHAB-I-5 cluster, named after a sequence from the Mediterranean Sea21 and retrieved in the project CHABADA (CHAnges in BActerial Diversity and Activity), was composed exclusively of environmental sequences for a long time2,22. Only very recently have several isolates been obtained, but they have not been studied further23. Sequences related to this cluster have been found in various oceans22,24–27 and constitute 10 to 20% of small subunit (SSU) rRNA clone libraries in coastal waters28,29. However, detailed information on the occurrence and diversity of the CHAB-I-5 cluster, as well as its role in ocean biogeochemistry, is still missing. We thus aimed to investigate the biogeography of this cluster, obtaining genomic information from a CHAB-I-5 isolate and comparing its functional properties with other pelagic Roseobacter group organisms.
Results
Phylogenetic analyses of SSU rRNA gene sequences of the CHAB-I-5 cluster confirmed a distinct cluster within the Roseobacter group, supported by a bootstrap value of 73% and recovered reproducibly with maximum-likelihood calculations. This comprises two subclusters containing SSU rRNA gene sequences from uncultured organisms (Supplementary Fig. 1): subcluster A, with a sequence similarity of >99%, composed of sequences exclusively from free-living bacteria; subcluster B, exhibiting a sequence similarity of >96%, also including four sequences of >99% similarity from organisms associated with deep sea corals (Supplementary Table 1).
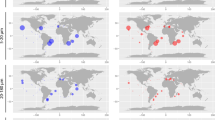
The application of a CHAB-I-5-specific primer set to DNA samples from the free-living and particle-associated fraction of bacterial communities from the North Sea (Supplementary Fig. 2) confirmed the presence of the CHAB-I-5 cluster almost exclusively in the free-living fraction. Furthermore, screening of marine surface waters from various oceans and geographic regions revealed that members of this cluster occur worldwide, from polar to tropical regions (Fig. 1 and Supplementary Table 2). These findings are in line with the biogeography of CHAB-I-5-related SSU rRNA gene sequences (>1,300 bp) from public databases (Fig. 1). The global distribution of CHAB-I-5 subclusters A and B did not exhibit significant differences (Supplementary Table 3).

For GenBank accession numbers see Supplementary Fig. 1 and Supplementary Tables 1 and 2. Water samples for PCR screening were obtained during eight cruises (Supplementary Table 3).
To quantify the occurrence of the CHAB-I-5 cluster, a transect from the southern to the northern North Sea from 54° to 60°N in July 2011 was analysed by quantitative PCR (qPCR) and 454 pyrosequencing of SSU rRNA amplicons (Fig. 2a and Supplementary Fig. 2). In addition, samples from in- and outside a phytoplankton bloom in the Skagerrak were analysed (Fig. 2b). The sea surface temperature ranged from 18.3 °C in the southern North Sea to 14.5 °C in the northern North Sea. Members of the CHAB-I-5 cluster were detected in every sample and constituted between 1 and 14% (mean = 8%) of the bacterioplankton as assessed by both methods. The proportion of active bacterioplankton, analysed by pyrosequencing of SSU rRNA transcripts, was systematically higher than that of total bacterioplankton and ranged between 3 and 22% (mean = 16%). There was no significant correlation of the relative abundance of the CHAB-I-5 cluster to chlorophyll a (Chl a) concentrations. These results emphasize that the CHAB-I-5 cluster is not only widely distributed, but also constitutes a major component of the total and notably the active bacterioplankton in the North Sea in the summer. This is consistent with previous studies that detected the CHAB-I-5 cluster in the North Sea27 and the Atlantic Ocean northwest of Spain, where it was more frequently found in the summer29. In contrast to the CHAB-I-5 distribution pattern, other pelagic clusters of the Roseobacter group are often positively correlated to phytoplankton blooms3,13,15,19,30. These dynamics suggest that the CHAB-I-5 cluster exhibits different substrate and growth requirements from other pelagic clusters of this clade.

a,b, Water samples were taken from the North Sea in July 2011 at 3–4 m depth and analysed by qPCR (error bars represent standard deviation from four technical replicates) and 454 pyrosequencing of the SSU rRNA gene of the total bacterial community and 454 pyrosequencing of the SSU rRNA gene of the total bacterial community and of the SSU rRNA of the active bacterial community. Samples were taken from a transect from 54° to 60°N (a) and stations inside (stations 8, 11, 17, 23) and outside (station 27) a phytoplankton bloom (b). For a map of the stations see Supplementary Fig. 2.
An organism affiliated to the CHAB-I-5 cluster, named strain SB2, was isolated from surface water of the North Sea. We were able to cultivate it and obtained enough biomass for genome sequencing, but, unfortunately, were unable to further maintain it as a pure culture. The draft genome encompasses 3.64 Mbp with a G+C content of 50.5%. Coding sequences constitute 88.85% of the genome (Table 1). The relatively small genome size and low G+C content compared with other members of the Roseobacter group is typical for roseobacters from pelagic habitats17,31. The G+C content of 50.5% is the fourth lowest of all roseobacters. Only strains HTCC2255, HTCC2150 and HIMB11 have lower values (Table 1). The genome of strain SB2 harbours one gene copy of the SSU rRNA and 3,569 genes, of which 98.6% encode proteins. Genes for all basic metabolic functions are encoded, as well as two complete flagella clusters, indicating possible motility.
To analyse the metabolic potential of strain SB2 and to put it into a functional and phylogenomic context, we compared its genome to those of other Roseobacter group members based on a multi locus sequence analysis (MLSA) of shared orthologous genes and on a distance matrix of the gene content. The MLSA analysis yielded a tree with five subclades, essentially in accordance with previous analyses17,32. Strain SB2 branches between subclades 1–3 and subclade 4 (Fig. 3). Its most closely related organism is Rhodobacteraceae bacterium HIMB11, isolated from Hawaiian coastal waters33. In contrast, in the gene content analysis, subclades 2–5 split up in various lineages, and the organisms show a different clustering (Fig. 3). Interestingly, strain SB2 and seven organisms described as exclusively pelagic form a distinct cluster in the gene content tree: the Pelagic Roseobacter Cluster (PRC). In the MLSA tree, these organisms encompass the three most deeply branching lineages and two lineages branching relatively deeply within or closely related to subclades 2 and 3 (Fig. 3).

a, Maximum-likelihood tree inferred with 500 bootstraps with RAxML. The alignment was created from 367 orthologous single-copy genes present in all genomes (MLSA) including the alphaproteobacterial strains Pelagibacter sp. HIMB122 and Nisaea sp. BAL199 as outgroups. b, Gene content tree of the same organisms based on an orthologues-content matrix representing the presence or absence of a gene in a certain genome, inferred with neighbour joining and 1,000 bootstraps. In both trees, bootstrap values >49% are indicated with a circle at the nodes (white, ≥50%; grey, ≥75%; black, 100%). Subclades of the Roseobacter group are colour-coded and the PRC is marked with a red dashed box. Scale bars, 10% sequence divergence.
To better understand this functional clustering, we compared the gene content of strain SB2 with the other PRC members as well as with the other genomes used for establishing the MLSA tree. Overall, strain SB2 shares 90% of its genes with members of the PRC and 95% with all other organisms used in the phylogenetic analysis (Fig. 4). A few genomic islands, encoding mainly hypothetical proteins, were identified based on divergent G+C content (Fig. 4). The SB2 genome encompasses many different energy acquisition modes. A complete cluster for AAnP encoded in this genome is also present in those of HIMB11, RCA23 and HTCC2083. Genes encoding CO oxidation, including both forms I and II of the coxL gene, are present in the genomes of SB2, HIMB11 and RCA23. Only roseobacters expressing both forms of the coxL gene are capable of oxidizing CO (ref. 8). Genes encoding the oxidation of inorganic sulfur compounds (sox) are present in all genomes of the PRC. This repertoire of energy-acquiring features provides strain SB2 with all potentially available modes present in the Roseobacter group, except photoheterotrophy via proteorhodopsin, which is present only in the genome of HTCC2255 and thus reflects the adaption of SB2 to an oligotrophic lifestyle.

The SB2 genome is not shown, but would cover all genomic features shown in the circular genomes including the genomic islands and features in the black wedges. The inner circle represents variations in G+C content of the SB2 genome. Selected features of the genome are marked. Central inset: Venn diagram (http://omics.pnl.gov/software/venn-diagram-plotter) illustrating the number of genes (without considering duplicates) shared among strain SB2, the PRC and the other genomes of the Roseobacter group used in this study. n, number of genomes.
Like the other PRC organisms, the genome of strain SB2 harbours the gene clusters for degrading DMSP by cleavage and demethylation pathways (Supplementary Table 4), as is common within the Roseobacter group10. The genome of all PRC members encodes the genes to acquire sulfur in the reduced form. Strains SB2, RCA23 and HTCC2255 lack the cysH gene, which is embedded in the gene cluster for assimilatory sulfate reduction in other roseobacters (Supplementary Table 5). Hence, it is unclear whether these strains are able to reduce oxidized sulfur compounds. They may, in fact, use exclusively reduced sulfur compounds, as is known from the SAR11 clade34.
The genomes of all PRC members encode glucose metabolism via the Entner–Doudoroff pathway, as is typical for many marine bacteria because of the enhanced supply of NADPH as compared to the Embden–Meyerhof–Parnas pathway and the possible protection against oxidative stress35. In contrast to the other PRC members, strain SB2 encodes a Na+/melibiose symporter (pfor_0c0141). A closely related orthologue is present in the single amplified genome AAA298-K06 of the Roseobacter group, obtained from the subtropical north Pacific gyre12. The genomes of all PRC members encode genes for nitrogen uptake in a reduced form as ammonium, amino acids, polyamines or urea. The SB2 genome, however, also encodes two unique nitrate/nitrite transporters (pfor_0c0726, _0c2842), but genes encoding enzymes of assimilatory nitrate or nitrite reduction were not detected. Therefore, the annotated functions of these genes remain ambiguous. With respect to vitamins, the PRC members are rather similar to other roseobacters encoding pathways for the synthesis of vitamin B12 (cobalamin), B6 (pyridoxine) and B2 (riboflavin, except HTCC2255) (Supplementary Table 4). They lack genes encoding the synthesis of vitamins B1 (thiamine) and B7 (biotin). All PRC members appear to be capable of taking up iron as Fe3+, and some also as Fe2+, for example, strains SB2 and HTCC2150 by a Fe2+/manganese transporter (sitABCD, Supplementary Table 4). PRC members lack siderophore uptake systems for iron, a feature quite common in other members of the Roseobacter group32. The presence of a gene cluster encoding only the type I and no other secretion system in the PRC members, except a putative gene cluster for type VI secretion systems in HTCC2083, reflects their adaptation to a free-living lifestyle.
Strain SB2 is unique within the PRC in carrying genes for phosphonate metabolism, even though many other members of the Roseobacter group encode this gene cluster32. Phosphonate constitutes ∼25% of the dissolved organic phosphorus pool in the ocean36. It is quite common as a P source among marine pelagic prokaryotes, including the SAR11 clade and cyanobacteria, in particular under oligotrophic conditions in subtropical and tropical oceans37,38. A distinct feature of strain SB2 when compared to the other PRC members is the high number of ABC transporter-related genes, in total 268, equivalent to 73.8 per Mbp (Table 1). A high proportion of ABC transporters, exhibiting high substrate affinities, is considered an adaptation of pelagic bacteria to a nutrient-poor environment39,40. In agreement with the energy acquisition features, these traits can be interpreted as adaptation of strain SB2 to oligotrophic conditions. These traits may also, at least partially, explain the relatively high abundance of the CHAB-I-5 cluster in the North Sea in the summer (this study) and near the Spanish coast in the northwest Atlantic29, that is, at elevated temperatures, and in stratified and nutrient-depleted conditions.
Based on the broad distribution pattern revealed by PCR screening, we analysed the presence of strain SB2 in metagenome and metatranscriptome data sets from the North Sea as well as from public data sets. At station 13 of the North Sea cruise, the genome was retrieved from the metagenome at 3 and 350 m depths to 94.8 and 91.8%, respectively, and from the corresponding metatranscriptome to 98.4 and 23.5%, respectively (Supplementary Table 6). These results underline the high activity of CHAB-I-5 members in surface waters of the North Sea as detected by pyrosequencing of SSU rRNA transcripts (Fig. 2). The SB2 genome accounted for 4.9% of the mapped metagenomic and 1.5% of the metatranscriptomic reads from 3 m and for 1.3 and 0.9% from 350 m, respectively. In a metagenome and metatranscriptome data set from the western English Channel, 0.63–2.72% and 0.02–2.34% of the reads were assigned to strain SB2, respectively (Supplementary Table 6 and Supplementary Fig. 3). In addition, the SB2 genome represented up to 0.69% of the reads from the Global Ocean Sampling (GOS) data set in the western Atlantic and eastern Pacific (Supplementary Table 6 and Supplementary Fig. 4). Small proportions (<0.1%) were even detected at stations near the equator, indicating a global distribution of SB2 populations at least in very low abundances.
The generally high metabolic activity of the SB2 population in the North Sea at station 13 was reflected by a three- to fivefold higher gene expression level of the DNA-directed RNA polymerase, the glutamate synthase and the anaplerotic malate dehydrogenase at both depths compared to the overall mean of all normalized reads (RPKM, reads per kilobase per million mapped reads; Supplementary Table 7). Several genes encoding transporter functions and transcribed at both depths exhibited much higher RPKM-normalized reads at 3 m relative to 350 m, such as the glutathione S-transferase and an ABC-Fe3+ transporter (Fig. 5a and Supplementary Table 7). At 350 m, the transcription of genes encoding one of two xylose/ribose and sulfate transporters and a haem exporter was upregulated relative to 3 m (Fig. 5a and Supplementary Table 7).

a, Ratio of RPKM values at depths of 3 m relative to 350 m (log2-transformed) of selected genes of SB2 and RCA23. Positive and negative values indicate higher gene expression at 3 and 350 m, respectively. b, Ratio of RPKM values of selected orthologous genes of SB2 relative to RCA23 (log2-transformed) at 3 and 350 m depths. Positive and negative values indicate higher gene expression in SB2 and RCA23, respectively. Further details on the selected genes are provided in Supplementary Table 7 (identified with colour coding).
We compared the gene expression patterns of strain SB2 with those of Planktomarina temperata RCA23, the only other abundant PRC member at station 13, constituting 7% of the total and active bacterioplankton at 3 m. Most striking for P. temperata was the highly upregulated transcription of genes encoding transposases, located on genomic islands and missing in the SB2 genome, at 3 m relative to 350 m (Fig. 5a and Supplementary Table 7). This phenomenon has already been reported for P. temperata in a phytoplankton bloom relative to non-bloom conditions41. Expression patterns of the photosynthetic gene cluster were rather similar for both organisms and depths (Fig. 5b). However, transcription of genes encoding a glutamate synthase, a glutamine synthetase and the cox gene cluster of strain SB2 was upregulated relative to P. temperata at both depths (Fig. 5b and Supplementary Table 7). Altogether, the gene expression patterns of both PRC organisms partly overlapped but were also distinct, particularly in the transcription of different transporter genes (Fig. 5b and Supplementary Table 7).
Discussion
Global biogeographic studies of relevant pelagic marine bacteria are important for a better functional understanding of their role in ocean biogeochemistry. Such studies are available for several major lineages of marine bacterioplankton, including selected lineages of the Roseobacter group7,42,43. Sequences of PRC members have been detected in many oceanic regions, including tropical, subtropical, temperate, subpolar and polar regions, and can constitute more than 10% of total bacterioplankton7,19. Detailed systematic biogeographic studies were conducted for the RCA/Planktomarina cluster, which occurs exclusively in upwelling and temperate to polar regions6,13–15. The CHAB-I-5 cluster, detected in previous studies in pelagic systems22,25,27–29 but not yet clearly defined, exhibits a much broader biogeography, ranging from tropical to polar regions. This indicates that members of this cluster are able to cope with higher temperatures and salinities than members of the RCA/Planktomarina cluster. The high abundance of the CHAB-I-5 cluster in the North Sea in the summer underscores its prominent role as a bacterioplankton component. However, it appears to have a different functional relationship to the occurrence of phytoplankton and the substrate spectrum from other clusters of the Roseobacter group.
Analysis of the draft genome of strain SB2 revealed that it is well adapted to an oligotrophic lifestyle, as reflected by various modes of energy acquisition and the very high number of ABC transporter-related proteins. Based on the gene content, SB2 clusters together with other organisms of the Roseobacter group with different phylogenetic affiliations and genome sizes ranging from 2.22 to 4.14 Mbp, but known to be exclusively pelagic and well adapted to oligotrophic nutrient conditions. Some, but not all of these organisms exhibit features of genome streamlining, such as a reduced genome size and a lack of plasmids, siderophores and secretion systems other than type I (Table 1 and Supplementary Table 4). As all of these organisms are deeply branching within the Roseobacter group (Fig. 3), they obviously underwent a different evolutionary history from other organisms of this group. By retaining a pelagic lifestyle under oligotrophic conditions, they appear to have lost a substantial number of gene families as compared to other members of the Roseobacter group44, even though this loss did not consistently lead to streamlined genomes. It would be interesting to know whether such gene content based clusters also exist in other lineages of marine bacteria as a result of their evolutionary adaptation to a pelagic and oligotrophic lifestyle.
The SB2 genome was present in different pelagic metagenome and metatranscriptome data sets. The transcription of gene clusters encoding flagella biosynthesis, AAnP and CO oxidation for acquiring complementary energy, of genes encoding the malate dehydrogenase for anaplerotic CO2-fixation and ABC transporters for various substrates revealed that this organism is highly active in the North Sea. Hence, this study sheds light on a so far neglected, but important, cluster of the Roseobacter group in global oceans and allows its quantitative and functional significance to be assessed in future studies. On the basis of the location of its isolation (island of Helgoland; old Germanic name, Forsetiland) and its pelagic lifestyle, we propose the name ‘Candidatus Planktomicrobium forsetii’ gen. nov., sp. nov., for CHAB-I-5 strain SB2.
Methods
Samples were collected during cruises with RV Heincke in the North Sea in July 2011 (Supplementary Fig. 2) and RV Polarstern in the Southern Ocean and across the Atlantic Ocean (Fig. 2 and Supplementary Table 3). Volumes of 0.5–1.0 l were filtered on 0.2 µm polycarbonate membranes (47 mm, Whatman) and stored in sterile 2 ml Eppendorf tubes at −80 °C until further processing. During the North Sea cruise, samples were prefiltered through 5.0 µm membranes, and additional samples were collected for bacterial community analysis by 454 pyrosequencing45 as well as for metagenome and metatranscriptome analysis30. Samples from other oceanic regions were collected as described in refs 46–48 (for further details see Supplementary Table 3).
Chl a concentrations and bacterial abundance were determined as described in ref. 17. DNA was extracted according to a modified standard protocol49 (Supplementary Methods).
CHAB-I-5-specific PCR detection
Specific primers targeting the SSU rRNA of the CHAB-I-5 cluster were designed using the ProbeDesign function of the ARB package50. Oligonucleotide sequences are CHAB-I-5_429f (5′-AAAGCTCTTTCGCCTGTGATG-3′) and CHAB-I-5_989r (5′-GCGACGACGAGTATGTC-3′). Conditions for the CHAB-I-5-specific PCR were optimized to 95 °C for 5 min; 30 cycles at 95 °C for 1 min, 58 °C for 1 min and 72 °C for 3 min and a final elongation step at 72 °C for 10 min. For details on PCR conditions and evaluation of the CHAB-I-5-specific primers, see Supplementary Methods.
qPCR assays
Primers targeting the bacterial SSU rRNA genes and the internal transcribed spacer (ITS) region of the CHAB-I-5 cluster were adapted from Buchan and co-authors25. SSU rRNA gene fragments were amplified and detected by qPCR performed with a 96-well LightCycler 480 Instrument II (Roche Diagnostics). The PCR reactions, using 15 µl SYBR Green I Master (Roche Diagnostics) including primers and a 5 µl template, were performed in quadruplicate. A plasmid containing the 16S–23S region of CHAB-I-5 strain SB2 was applied as standard. At a 1,000-fold dilution of DNA samples, both qPCR systems showed optimal results. The qPCR efficiency for Bacteria and CHAB-I-5-specific amplifications were 0.94 and 0.92, respectively, and the detection limit for CHAB-I-5-specific sequences was 15 gene copy numbers (0.074 fg DNA). Absolute cell numbers were calculated using the Fit Points method (LightCycler 480 II system, version 1.5) and abundances of the CHAB-I-5 cluster determined as percentage of total bacterial SSU rRNA genes. Our calculation is based on the existence of one copy of the SSU gene in the CHAB-I-5 cluster, as well as in total Bacteria as reference. As bacterial lineages may have more than one SSU gene copy51, our results yield conservative estimates of the relative CHAB-I-5 abundance. For details on the qPCR conditions see Supplementary Methods.
Sequencing and phylogeny based on the SSU rRNA
PCR products of the SSU rRNA gene were amplified as described above, purified using the peqGOLD Cycle-Pure kit (PEQLAB Biotechnologie) and sequenced by Sanger technology (GATC Biotech). Chimaeras, identified with Pintail52, were excluded from the analysis, and sequences were aligned with the ARB software package50. Phylogenetic trees were calculated with long sequences (>1,300 bp) using neighbour-joining and maximum-likelihood analyses.
Analysis of the active and total CHAB-I-5 cluster in the North Sea
DNA and RNA were extracted from the filters, and the bacterial community was investigated by amplification and sequencing of the V3–V5 region of the SSU rRNA gene as described in ref. 45. The resulting SSU rRNA gene and transcript sequences were processed and denoised using the QIIME 1.8.0 software package53. The remaining reverse primer sequences and chimaeras were removed, sequences of all samples were joined and clustered in operational taxonomic units at 1% genetic distance, and their taxonomy was determined by BLAST alignment against a modified version of the Silva database45.
Enrichment of strain SB2
A member of the CHAB-I-5 cluster, strain SB2, was enriched from a surface water sample collected near the island Helgoland in the North Sea (54.18 N, 7.89 E) on 24 September 2010. The strain was separated by dilution-to-extinction in glass test tubes with 4.5 ml of an artificial seawater medium54 (SWM). Supplements added after autoclaving were 1 ml l−1 of a sterile filtered trace element solution55, a multivitamin solution56 and 10 ml l−1 of 1 M NaHCO3 (autoclaved separately). For further details see Supplementary Methods. Finally, we obtained enough biomass for genome sequencing; however, in all further cultures, a contamination appeared that has not been removed so far.
Genome sequencing and annotation
Biomass of ∼20 ml of an SB2 culture grown in SWM at 15 °C was collected by centrifugation at 3,800g and 4 °C. Chromosomal DNA was isolated using the MasterPure complete DNA purification kit (Epicentre). The preparation of paired-end libraries with the Nextera XT library preparation kit and subsequent sequencing using the Genome Analyzer IIx were performed according to the protocol (Illumina). Sequencing resulted in 5,324,954 reads of 112 bp. De novo assembly with SPAdes version 2.5.1 (ref. 57) resulted in 38 contigs > 3 kbp with 111-fold coverage.
Protein-encoding genes were identified and annotated with the Prokka annotation pipeline using Prodigal v2.6 (ref. 58). The predicted coding DNA sequences were translated and annotated using the CDD, KEGG, UniProt, TIGRFam, Pfam and InterPro databases. Additional gene prediction analyses, functional annotation and comparisons were performed within the IMG-ER platform59. Contamination and completeness of the genome were tested (Supplementary Methods and Supplementary Tables 8 and 9).
Phylogenetic analysis
Sequenced genomes of roseobacters were selected as described in the Supplementary Methods. Total protein sequences from 43 genomes were extracted from the corresponding GenBank files and used for downstream analysis with an in-house pipeline at the Goettingen Genomics Laboratory. Briefly, clusters of orthologues were generated using proteinortho version 5 (ref. 60), inparalogues removed, the remaining sequences aligned with MUSCLE61 and poorly aligned positions automatically filtered from the alignments using Gblocks62. A maximum-likelihood tree from 367 orthologues was inferred with 500 bootstraps with RAxML63. The proteinortho clusters were also converted to an orthologues-content matrix representing the presence or absence of a gene in a certain genome. A phylogenetic tree was inferred with neighbour joining and 1,000 bootstraps. Both scripts, PO_2_MLSA.py and PO_2_GENECONTENT.py, are available at github (https://github.com/jvollme).
Metagenomics and metatranscriptomics
For metagenomic and metatranscriptomic analyses, DNA and cDNA were sequenced on an Illumina/Solexa GAIIx system. For sequence statistics see Supplementary Table 10. Sequences were processed as described in ref. 17 and mapped on the genomes of strain SB2 and P. temperata RCA23 using Bowtie2 as described in ref. 41. Paired-end reads were counted as single reads for each gene. Read numbers were normalized as RPKM and by multiplication with the unique RNA/DNA ratio for each species and sample, calculated by dividing the relative abundance in the metatranscriptome by the relative abundance in the metagenome41,64. Gene expression ratios were calculated by comparing normalized RPKM values from 3 and 350 m and SB2 and RCA23, respectively, and log2-transformed according to ref. 41 but not increased by one before log2-transformation.
Accession codes
The Whole Genome Shotgun project has been deposited at DDBJ/EMBL/GenBank under accession no. LGRT00000000. Here, version LGRT01000000 is described. Metagenome and metatranscriptome data have been deposited in the Sequence Read Archive of the National Center for Biotechnology Information (NCBI) under accession no. SRA082674.
References
Amin, S. A. et al. Interaction and signalling between a cosmopolitan phytoplankton and associated bacteria. Nature 552, 98–101 (2015).
Buchan, A., González, J. M. & Moran, M. A. Overview of the marine Roseobacter lineage. Appl. Environ. Microbiol. 71, 5665–5677 (2005).
Buchan, A., LeCleir, G. R., Gulvik, C. A. & González, J. M. Master recyclers: features and functions of bacteria associated with phytoplankton blooms. Nature Rev. Microbiol. 12, 686–698 (2014).
Giovannoni, S. J. & Stingl, U. Molecular diversity and ecology of microbial plankton. Nature 437, 343–348 (2005).
Pommier, T., Pinhassi, J. & Hagström, Å. Biogeographic analysis of ribosomal RNA clusters from marine bacterioplankton. Aquat. Microb. Ecol. 41, 79–89 (2005).
Selje, N., Simon, M. & Brinkhoff, T. A newly discovered Roseobacter cluster in temperate and polar oceans. Nature 427, 445–448 (2004).
Yooseph, S. et al. Genomic and functional adaptation in surface ocean planktonic prokaryotes. Nature 468, 60–66 (2010).
Cunliffe, M. Correlating carbon monoxide oxidation with cox genes in the abundant marine Roseobacter clade. ISME J. 5, 685–691 (2011).
Wagner-Döbler, I. et al. The complete genome sequence of the algal symbiont Dinoroseobacter shibae: a hitchhiker's guide to life in the sea. ISME J. 4, 61–77 (2010).
Moran, M. A., Reisch, C. R., Kiene, R. P. & Whitman, W. B. Genomic insights into bacterial DMSP transformations. Annu. Rev. Mar. Sci. 4, 523–542 (2012).
Thole, S. et al. Phaeobacter gallaeciensis genomes from globally opposite locations reveal high similarity of adaptation to surface life. ISME J. 6, 2229–2244 (2012).
Luo, H., Löytynoja, A. & Moran, M. A. Genome content of uncultivated marine roseobacters in the surface ocean. Environ. Microbiol. 14, 41–51 (2012).
West, N. J., Obernosterer, I., Zemb, O. & Lebaron, P. Major differences of bacterial diversity and activity inside and outside of a natural iron-fertilized phytoplankton bloom in the Southern Ocean. Environ. Microbiol. 10, 738–756 (2008).
Giebel, H. A., Brinkhoff, T., Zwisler, W., Selje, N. & Simon, M. Distribution of Roseobacter RCA and SAR11 lineages and distinct bacterial communities from the subtropics to the Southern Ocean. Environ. Microbiol. 11, 2164–2178 (2009).
Giebel, H.-A. et al. Distribution of Roseobacter RCA and SAR11 lineages in the North Sea and characteristics of an abundant RCA isolate. ISME J. 5, 8–19 (2011).
Giebel, H. A. et al. Planktomarina temperata gen. nov., sp. nov., belonging to the globally distributed RCA cluster of the marine Roseobacter clade, isolated from the German Wadden Sea. Int. J. Syst. Evol. Microbiol. 63, 4207–4217 (2013).
Voget, S. et al. Adaptation of an abundant Roseobacter RCA organism to pelagic systems revealed by genomic and transcriptomic analyses. ISME J. 9, 371–384 (2015).
González, J. M. et al. Bacterial community structure associated with a dimethylsulfoniopropionate-producing North Atlantic algal bloom. Appl. Environ. Microbiol. 66, 4237–4246 (2000).
Landa, M., Blain, S., Christaki, U., Monchy, S. & Obernosterer, I. Shifts in bacterial community composition associated with increased carbon cycling in a mosaic of phytoplankton blooms. ISME J. 10, 39–50 (2016). .
Varaljay, V. A. et al. Single-taxon field measurements of bacterial gene regulation controlling DMSP fate. ISME J. 9, 1677–1686 (2015).
Schäfer, H., Servais, P. & Muyzer, G. Successional changes in the genetic diversity of a marine bacterial assemblage during confinement. Arch. Microbiol. 173, 138–145 (2000).
Lekunberri, I. et al. The phylogenetic and ecological context of cultured and whole genome-sequenced planktonic bacteria from the coastal NW Mediterranean Sea. Syst. Appl. Microbiol. 37, 216–228 (2014).
Yang, S. J., Kang, I. & Cho, J. C. Expansion of cultured bacterial diversity by large-scale dilution-to-extinction culturing from a single seawater sample. Microb. Ecol. 71, 29–43 (2016).
Henriques, I. S., Almeida, A., Cunha, Â. & Correia, A. Molecular sequence analysis of prokaryotic diversity in the middle and outer sections of the Portuguese estuary Ria de Aveiro. FEMS Microbiol. Ecol. 49, 269–279 (2004).
Buchan, A., Hadden, M. & Suzuki, M. T. Development and application of quantitative-PCR tools for subgroups of the Roseobacter clade. Appl. Environ. Microbiol. 75, 7542–7547 (2009).
Rich, V. I., Pham, V. D., Eppley, J., Shi, Y. & DeLong, E. F. Time-series analyses of Monterey Bay coastal microbial picoplankton using a ‘genome proxy’ microarray. Environ. Microbiol. 13, 116–134 (2011).
Hahnke, S. et al. Physiological diversity of Roseobacter clade bacteria co-occurring during a phytoplankton bloom in the North Sea. Syst. Appl. Microbiol. 36, 39–48 (2013).
Suzuki, M. T. et al. Phylogenetic screening of ribosomal RNA gene-containing clones in Bacterial Artificial Chromosome (BAC) libraries from different depths in Monterey Bay. Microb. Ecol. 48, 473–488 (2004).
Alonso-Gutiérrez, J. et al. Bacterioplankton composition of the coastal upwelling system of ‘Ría de Vigo’, NW Spain. FEMS Microbiol. Ecol. 70, 493–505 (2009).
Wemheuer, B. et al. Impact of a phytoplankton bloom on the diversity of the active bacterial community in the southern North Sea as revealed by metatranscriptomic approaches. FEMS Microbiol. Ecol. 87, 378–389 (2014).
Luo, H., Swan, B. K., Stepanauskas, R., Hughes, A. L. & Moran, M. A. Evolutionary analysis of a streamlined lineage of surface ocean roseobacters. ISME J. 8, 1428–1439 (2014).
Newton, R. J. et al. Genome characteristics of a generalist marine bacterial lineage. ISME J. 4, 784–798 (2010).
Durham, B. P. et al. Draft genome sequence of marine alphaproteobacterial strain HIMB11, the first cultivated representative of a unique lineage within the Roseobacter clade possessing an unusually small genome. Stand Genomic Sci. 9, 632–645 (2014).
Tripp, H. J. et al. SAR11 marine bacteria require exogenous reduced sulphur for growth. Nature 452, 741–744 (2008).
Klingner, A. et al. Large-scale 13C flux profiling reveals conservation of the Entner–Doudoroff pathway as a glycolytic strategy among marine bacteria that use glucose. Appl. Environ. Microbiol. 81, 2408–2422 (2015).
Kolowith, L. C., Ingall, E. D. & Benner, R. Composition and cycling of marine organic phosphorus. Limnol. Oceanogr. 46, 309–320 (2001).
Sowell, S. M. et al. Transport functions dominate the SAR11 metaproteome at low-nutrient extremes in the Sargasso Sea. ISME J. 3, 93–105 (2009).
Feingersch, R. et al. Potential for phosphite and phosphonate utilization by Prochlorococcus. ISME J. 6, 827–834 (2012).
Giovannoni, S. J. et al. Genome streamlining in a cosmopolitan oceanic bacterium. Science 309, 1242–1245 (2005).
Lauro, F. M. et al. The genomic basis of trophic strategy in marine bacteria. Proc. Natl Acad. Sci. USA 106, 15527–15533 (2009).
Wemheuer, B. et al. The green impact: bacterioplankton response towards a phytoplankton spring bloom in the southern North Sea assessed by comparative metagenomic and metatranscriptomic approaches. Front. Microbiol. 6, 805 (2015).
Ghiglione, J.-F. et al. Pole-to-pole biogeography of surface and deep marine bacterial communities. Proc. Natl Acad. Sci. USA 109, 17633–17638 (2012).
Brown, M. V., Ostrowski, M., Grzymski, J. J. & Lauro, F. M. A trait based perspective on the biogeography of common and abundant marine bacterioplankton clades. Mar. Genom. 15, 17–28 (2014).
Luo, H., Csuros, M., Hughes, A. L. & Moran, M. A. Evolution of divergent life history strategies in marine alphaproteobacteria. MBio 4, e00373–13 (2013).
Osterholz, H. et al. Deciphering associations between dissolved organic molecules and bacterial communities in a pelagic marine system. ISME J. doi:10.1038/ismej.2015.231 (2016).
Baldwin, A. J. et al. Microbial diversity in a Pacific Ocean transect from the Arctic to Antarctic circles. Aquat. Microb. Ecol. 41, 91–102 (2005).
Gram, L., Melchiorsen, J. & Bruhn, J. B. Antibacterial activity of marine culturable bacteria collected from a global sampling of ocean surface waters and surface swabs of marine organisms. Mar. Biotechnol. 12, 439–451 (2010).
Wietz, M. et al. Wide distribution of closely related, antibiotic-producing Arthrobacter strains throughout the Arctic Ocean. Appl. Environ. Microbiol. 78, 2039–2042 (2012).
Zhou, J., Bruns, M. A. & Tiedje, J. M. DNA recovery from soils of diverse composition. Appl. Environ. Microbiol. 62, 316–322 (1996).
Ludwig, W. et al. ARB: a software environment for sequence data. Nucleic Acids Res. 32, 1363–1371 (2004).
Angly, F. E. et al. CopyRighter: a rapid tool for improving the accuracy of microbial community profiles through lineage-specific gene copy number correction. Microbiome 2, 11 (2014).
Ashelford, K. E., Chuzhanova, N. A., Fry, J. C., Jones, A. J. & Weightman, A. J. At least 1 in 20 16S rRNA sequence records currently held in public repositories is estimated to contain substantial anomalies. Appl. Environ. Microbiol. 71, 7724–7736 (2005).
Caporaso, J. G. et al. QIIME allows analysis of high-throughput community sequencing data. Nature Methods 7, 335–336 (2010).
Hahnke, S. et al. Planktotalea frisia gen. nov., sp. nov., isolated from the southern North Sea. Int. J. Syst. Evol. Microbiol. 62, 1619–1624 (2012).
Zech, H. et al. Growth phase-dependent global protein and metabolite profiles of Phaeobacter gallaeciensis strain DSM 17395, a member of the marine Roseobacter-clade. Proteomics 9, 3677–3697 (2009).
Martens, T. et al. Reclassification of Roseobacter gallaeciensis Ruiz-Ponte et al. 1998 as Phaeobacter gallaeciensis gen. nov., comb. nov., description of Phaeobacter inhibens sp. nov., reclassification of Ruegeria algicola (Lafay et al. 1995) Uchino et al. 1999 as Marinovum algicola gen. nov., comb. nov., and emended descriptions of the genera Roseobacter, Ruegeria and Leisingera. Int. J. Syst. Evol. Microbiol. 56, 1293–1304 (2006).
Bankevich, A. et al. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J. Comp. Biol. 19, 455–477 (2012).
Seemann, T. Prokka: rapid prokaryotic genome annotation. Bioinformatics 30, 2068–2069 (2014).
Markowitz, V. M. et al. IMG 4 version of the integrated microbial genomes comparative analysis system. Nucleic Acids Res. 42, D560–D567 (2014).
Lechner, M. et al. Proteinortho: detection of (co-)orthologs in large-scale analysis. BMC Bioinformatics 12, 124 (2011).
Edgar, R. C. & Edgar, R. C. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797 (2004).
Castresana, J. Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol. Biol. Evol. 17, 540–552 (2000).
Stamatakis, A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313 (2014).
Mortazavi, A., Williams, B. A., McCue, K., Schaeffer, L. & Wold, B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nature Methods 5, 621–628 (2008).
Acknowledgements
The authors thank J. Orchard, A. Neumann and O. Thomsen for their help in the laboratory, J. Lucas for the water sample from Helgoland and the crews of RV Heincke (grant no. AWI-HE361_00) and RV Polarstern (grant nos. AWI-PS ANT28-2_00, AWI-PS ANT28-4_00 and AWI-PS ANT28-5_00) for their support on board ship. The EAGER 2011 cruise was organized by the Continental Shelf Project of the Kingdom of Denmark and the Galathea 3 expedition was under the auspices of the Danish Expedition Foundation. This work was supported by the Deutsche Forschungsgemeinschaft (DFG) within the Transregional Collaborative Research Centre ‘Roseobacter’ (TRR 51).
Author information
Authors and Affiliations
Contributions
S.B., T.B. and M.S. designed the study. S.B. carried out the analyses of the phylogenetic cluster, biogeography and qPCR, and isolation of strain SB2. H.A.G. participated in sampling and provided data on chlorophyll and bacterial abundance in the North Sea. B.W., S.V., A.P. and R.D. carried out the genomic, metagenomic and metatranscriptomic analyses. L.G. and W.H.J. provided samples from various oceans. S.B. and M.S. wrote the major parts of the manuscript and all authors contributed to writing and revising it.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Supplementary information
Supplementary Text, Supplementary Figures 1-4, Supplementary Tables 1-6 and 8-10, Supplementary Table 7 Legend and Supplementary References. (PDF 11314 kb)
Supplementary Table 7
(XLS 1263 kb)
Rights and permissions
About this article

Cite this article
Billerbeck, S., Wemheuer, B., Voget, S. et al. Biogeography and environmental genomics of the Roseobacter-affiliated pelagic CHAB-I-5 lineage. Nat Microbiol 1, 16063 (2016). https://doi.org/10.1038/nmicrobiol.2016.63
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/nmicrobiol.2016.63
This article is cited by
-
Metagenome-assembled genomes reveal greatly expanded taxonomic and functional diversification of the abundant marine Roseobacter RCA cluster
Microbiome (2023)
-
Biogeographical and seasonal dynamics of the marine Roseobacter community and ecological links to DMSP-producing phytoplankton
ISME Communications (2022)
-
Availability of vitamin B12 and its lower ligand intermediate α-ribazole impact prokaryotic and protist communities in oceanic systems
The ISME Journal (2022)
-
TCA cycle enhancement and uptake of monomeric substrates support growth of marine Roseobacter at low temperature
Communications Biology (2022)
-
Mechanisms driving genome reduction of a novel Roseobacter lineage
The ISME Journal (2021)
