
Abstract
Optical microresonators1, which confine light within a small cavity, are widely exploited for various applications ranging from the realization of lasers2 and nonlinear devices3,4,5 to biochemical and optomechanical sensing6,7,8,9,10,11. Here we use microresonators and suitable optical gain materials inside biological cells to demonstrate various optical functions in vitro including lasing. We explore two distinct types of microresonator—soft and hard—that support whispering-gallery modes. Soft droplets formed by injecting oil or using natural lipid droplets support intracellular laser action. The laser spectra from oil-droplet microlasers can chart cytoplasmic internal stress (∼500 pN μm–2) and its dynamic fluctuations at a sensitivity of 20 pN μm–2 (20 Pa). In a second form, whispering-gallery modes within phagocytized polystyrene beads of different sizes enable individual tagging of thousands of cells easily and, in principle, a much larger number by multiplexing with different dyes.
Similar content being viewed by others
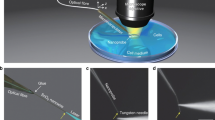
Main
Luminescent probes, including fluorescent dyes and proteins, quantum dots, bioluminescent molecules and plasmonic nanoparticles, have become indispensable tools in the fields of cell biology and medical sciences. Although these molecular probes are immensely useful, their relatively broad emission spectra, typically in the range of 30–100 nm, limit the number of probes that are simultaneously usable without ambiguity and often make their spectra indistinguishable from the broad background emission of endogenous molecules in tissues. It is fundamentally challenging to engineer molecules with much narrower spontaneous emission12. However, photonic principles such as optical resonance and stimulated emission allow spectral narrowing (filtering) via coherent loss or gain. The generation of narrowband resonant emission from biological cells has been demonstrated using cellular lasers with external cavities13,14 or photonic crystal needles15. Recently, we have sought to generate stand-alone cell lasers16, and we now describe effective approaches based on intracellular whispering-gallery mode (WGM) micro-resonators formed by soft and hard materials. WGMs are formed when light is circulating in a transparent spherical object as a result of being trapped due to total internal reflection at the interface. WGM cavities can have dimensions on the microscale or nanoscale17, although they are much larger than conventional luminescent probes.
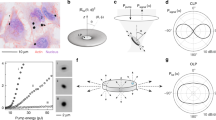
We first describe soft WGM cavities in the form of oil droplets in cells. Nile red dye-mixed polyphenyl ether (PPE), a chemically inert optical grade fluid with low viscosity (100 cP) and a refractive index of n = 1.69 (Fig. 1a), was injected into the cells. The size of the droplets was controlled18 to fall within the range of 4–20 µm, corresponding to a volume of 30–4,000 fl (Supplementary Movie 1 and Fig. 1b). Droplets larger than 7 µm demonstrate lasing upon pulsed excitation (λ = 535 nm, 5 ns, 10 Hz) with thresholds as low as a few nanojoules per pulse (Fig. 1c,d). This energy level is non-harmful for the cell13,14. The instantaneous heating of a droplet is calculated to be <1 °C, and the ambient temperature increase in the cytosol is negligible (Supplementary Section 3). When the droplet is under uniaxial stress, its shape deviates from a sphere and the deformation is manifested in the emission spectrum as a splitting of laser lines (Fig. 1e). For a small deformation the shape can be approximated as a spheroid, which supports laser oscillation in the equatorial plane (this has the lowest curvature and therefore the minimum optical loss). The modes were fitted to a model and the equatorial and polar semi-axes (a and b, respectively) were determined (Supplementary Section 1). From Laplace's law, the flattening stress Δσ is related to the local mean curvature of the droplet surface19, Δσ = 2γΔH, where γ is the surface tension and ΔH is the difference in the curvature (Supplementary Section 5). For small eccentricity e, that is, e2 ≪ 1, the stress is approximated as


a, Schematic of the injection of oil into the cytoplasm of a cell. b, Confocal fluorescence image of a cell with a PPE droplet doped with Nile red dye (red). The cell nucleus (blue) became kidney-shaped, giving space to the droplet. c, Bright-field (left) and laser-output (right) images of a cell with a droplet (arrows) above the lasing threshold. d, Output light intensity from a droplet as a function of pump pulse energy, showing a distinct laser threshold (arrow). Dashed line: linear fit to the fluorescence output below threshold. e, A typical output spectrum of the lasing modes. All modes are first radial modes, two modes with TE polarization and two with TM. Each mode is split into multiple submodes. From their splitting in these data, the spheroid is determined to be of oblate shape with equatorial and polar semi-axes measuring 8.3 and 8.5 µm, respectively. f, Time-lapse variation of the output spectrum for a live cell (left) and a dead cell fixed with formaldehyde (right). g, Standard deviation of the square of eccentricity and corresponding internal stress for live and fixed (dead) cells. Scale bars (b,c,f), 10 µm.
For the droplet in Fig. 1e we measured a = 8.5 and b = 8.3 µm, and, with γ ≈ 45 mN m−1 for the oil/water interface, we determined Δσ = 500 pN µm−2. Time-lapse traces of the output spectra revealed dynamic variations of the cellular stress in live cells (Fig. 1f). The mean fluctuation of the internal stress was measured to be ∼150 pN µm−2 (Fig. 1g). From the baseline fluctuation in dead cells (〈Δe2〉 = 0.19%), the force sensitivity is ∼20 pN µm–2 (20 Pa), which is an order of magnitude better than obtained from direct image-based analysis19.
We next investigated whether cells naturally containing lipid droplets (n = 1.47) can support laser oscillation (Fig. 2a). Adipocytes freshly extracted from porcine subcutaneous tissue contain a single lipid droplet with nearly perfect spherical geometry (Fig. 2b,c). After incubation with a lipophilic fluorescent dye and pumping with a pulsed laser, the cells exhibited lasing with WGM outputs (Fig. 2d) and a distinct threshold (Fig. 2e), thus demonstrating a completely natural intracellular optical cavity. Next, we examined the possibility of generating lasing from adipocytes in situ in tissues. Adipocytes in fat are closely packed and have random shapes (Fig. 2f), which have a lower cavity Q-factor and require a higher pump energy for lasing. To lower the threshold we injected a mixture of collagenase and lipophilic Nile red dye into the subcutaneous fat. The collagenase releases adipocytes from the tissue matrix, and they acquire spherical shapes20. An optical fibre was inserted through a needle puncture hole to excite the adipocytes with pulsed laser light and to collect the light emitted from the tissue (Fig. 2g). The adipocytes near the fibre tip readily showed lasing (Fig. 2h). In some cases, adipocytes at the periphery of the fat tissue (with a more round shape) also showed lasing (Supplementary Fig. 1), eliminating the need for collagenase.

a, Illustration of a typical mature subcutaneous adipocyte with a lipid droplet. b, Individual adipocytes extracted from subcutaneous porcine fat. c, Confocal image of an adipocyte containing a large lipid droplet (orange), which occupies the majority of the cell volume. The nucleus (blue) is visible next to the droplet. d, Spectrum from a 45 µm adipocyte above lasing threshold, showing typical WGM spectral peaks. Inset: fluorescence image of the cell above lasing threshold. e, Output energy as a function of pump energy. Dashed line: linear fit to the fluorescence output below laser threshold. f, Two-photon confocal image of adipocytes in situ in subcutaneous fat tissue, after intradermal injection of Nile red dye (yellow). g, Generation of cellular laser emission from within tissue. The pump laser is fibre-optically guided into the subcutaneous fat layer after injecting a mixture of collagenase and Nile red dye. h, Spectrum of light collected by the optical fibre from the tissue. Scale bars, 200 µm (b) and 20 µm (c,d,f).
Solid microspheres (such as polystyrene microspheres) offer a simple way to devise non-deformable, intracellular lasers (Fig. 3a). Polystyrene beads are readily internalized by endocytosis21,22 (Fig. 3b). We have observed that both macrophages and non-phagocytic cells (such as HeLa and NIH3T3) engulf beads up to 20 µm in diameter (d), large enough to exhibit lasing at low pump energy. The viability of HeLa cells 24 h after engulfing one or more polystyrene beads (>6 µm) was 98.4 ± 0.6% (compared to 99.4 ± 0.2% for cells without beads). WGM lasers offer multiple options for the position of the gain medium, including inside the resonator, outside the resonator and on the surface of the bead. We tested all three cases. First, fluorescent dye-embedded polystyrene beads provided gain within the beads. Beads larger than d = 11 µm supported lasing inside the cells at pump energy levels below a few nanojoules (Fig. 3c,d and Supplementary Fig. 2). The bead surface can also be functionalized with probe molecules such as antibodies or DNA for intracellular molecular sensing6,7,8,9,10,11. In the second case, the cell tracker dye 5-chloromethylfluorescein diacetate (CMFDA), which is retained in the cytosol, served as the gain medium for BaTiO3 beads. Lasing was observed in beads with dimensions as small as 8 µm (Fig. 3e). The gain medium interacts freely with the cell, generating amplified signals modulated by the cavity resonance. Third, the surfaces of high-index (n = 1.9) BaTiO3 beads were coated with fluorescent dye (Alexa Fluor 488). Although a monolayer of dye did not provide enough gain for lasing, its emission spectrum was strongly modulated by high-Q (700) resonance in beads with dimensions down to d = 3.5 µm, with relatively low fluorescence background (Fig. 3f). Any combinations of the three gain locations are of course possible. Furthermore, the gain and sensing media may be co-located or separated.

a, Schematic of a bead inside a cell. b, Confocal fluorescence image of a HeLa cell containing a polystyrene bead (green), nucleus (blue) and plasma membrane (red). c, Laser emission from a fluorescent polystyrene bead inside a cell. d, Emission spectra and images (insets) of a fluorescent polystyrene bead below (top) and above (bottom) lasing threshold (3.2 nJ). e, Laser output from a 8.7 µm non-fluorescent BaTiO3 bead embedded in a cell containing CMFDA dye in its cytoplasm. f, Spontaneous emission from a 3.5 µm BaTiO3 bead coated with Alexa 488 dye below laser threshold. Scale bars, 10 µm.
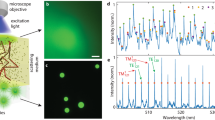
The precise wavelengths of the multiple spectral peaks from a bead above or below lasing threshold (Fig. 3) are uniquely determined by the size of the bead. We measured the output spectra from fluorescent polystyrene beads from each spatial location in a mid-plane of the cell (Fig. 4a) in a confocal hyperspectral imaging set-up with a numerical aperture of 1.25 and continuous-wave pumping with a 455 nm light-emitting diode (LED). A spatial map of the intensity of the resonance peaks has a ring shape (Fig. 4b), representing light circulating in the bead and leaking out in the tangential direction. By fitting the spectra with WGM theory23, the effective bead diameter d was calculated at each pixel. The diameter around the circumference varies by 1–2 nm, which may reflect the deviation of the actual bead from a perfect sphere (Fig. 4c). The precision of the mean diameter is ∼50 pm (Supplementary Fig. 3), which indicates a remarkable sensitivity of the WGM analysis (50 pm/7.7 µm = 6.5 × 10−6). The same type of analysis can be done for beads operated above threshold by pulsed pumping. However, when the number of lasing modes is less than three to four, the size measurement is frustrated or has an error of as much as one to two free spectral ranges (∼100 nm) (Supplementary Fig. 4).

a, Bright-field image of a HeLa cell containing a polystyrene fluorescent bead. b, False-colour image of the cell in a, showing the intensity of the oscillating WGMs. c, A bead diameter map calculated from confocal hyperspectral images of the WGM output. d,e, Multiple HeLa cells containing beads (d) and the corresponding bead diameter map (e). f, Time-lapse measurements of a resonant peak from a glass bead inside a HeLa cell. The addition of 2 g l–1 sodium chloride at t = 0 caused the peak wavelength to shift. Scale bars, 10 µm.
An intriguing application of this precision measurement is to use beads with different diameters to tag individual cells (Fig. 4d,e; Supplementary Fig. 5 and Supplementary Movie 2). The diameter interval between beads should be large enough to accommodate the typical wavelength variation of 2 nm due to refractive index changes arising from intracellular dynamics (Δλ/λ ≈ Δn/n + Δd/d), where n is the effective refractive index for the oscillating mode. With polydispersed beads with dimensions falling in the range 8–12 µm and a bin size of 2 nm, we can distinguish 2,000 individual beads. By using different fluorescent dyes with distinct emission bands this number can easily be increased several fold. Furthermore, each cell can engulf multiple particles (Supplementary Fig. 6b). Using three beads per cell and five different fluorescence dyes with non-overlapping spectra, it would be possible to individually tag 2,000×5C3 = 2 × 1011 cells, which is comparable to the number of cells in the human body and many orders of magnitude greater than achievable using state-of-the-art techniques such as stochastic brainbow recombination24 for in vivo cell tracking, or high-throughput on-chip cytometry and cell-based analysis.
To demonstrate intracellular sensing we measured the spectral changes of soda lime glass beads coated with Alexa Fluor 488 in HeLa cells, while the cell culture medium was provided with an additional 2 g l–1 of sodium chloride. Exposure of cells to hypertonic solution causes intracellular water to quickly diffuse out of the cell, reducing the cell volume. The shrinking is followed by a partial recovery via a regulatory volume increase25. The change in cell volume changes the concentration of the molecules in the cytoplasm, thereby also changing the refractive index. The time dependence of the shift in the position of a WGM (Fig. 4f) corresponds to Δn ≈ 1.0 × 10−2, which is close to the value of 7.6 × 10−3 calculated using the Boyle-van't Hoff law (Supplementary Section 6). As a control experiment, the same volume of medium was added without changing the osmolarity, and produced no response (Supplementary Fig. 7).
In this Letter we have demonstrated stand-alone cell lasers by using an intracellular WGM microresonator, a dye gain medium and far-field optical pumping with nanosecond pulses. A pulse duration of 100 ps could reduce the threshold pump energy (by ∼50 times) while supporting distinct WGM modes in the microresonators. The shape of the intracellular WGM cavities is not limited to spheres—cylinders, toruses and disks can also be used. The micrometre size may offer flexibility in cavity engineering for specific mode selection or direction-dependent radiation26. Biodegradable polymers27 may improve biocompatibility, and the use of smaller cavities causing minimal perturbations in cells would also be useful. High-refractive-index materials, such as semiconductors (n > 3)28 or the realization of metamaterials with extremely high refractive indices29 should enable the creation of sub-micrometre WGM lasers (Fig. 5). Plasmon-based spasers can have dimensions as small as tens of nanometres30, but the WGM analysis demonstrated here is most effective with microresonators accommodating multiple optical wavelengths.

Q-factors of WGMs are highly dependent on the refractive index and cavity size. The two dashed lines represent theoretical calculations of two Q-factors: 106 and 103, respectively. Circles indicate the measured minimum size of laser achieved inside a cell for a lipid droplet in an adipocyte, a polystyrene bead and a PPE droplet inside a HeLa cell, and BaTiO3 beads in 15 mM pyrromethene solution, as well as InGaP disk lasers in air28.
Methods
Optical set-up
For the pumping of the cell lasers and the collection of light, a ×40 1.25 NA or ×100 1.40 NA oil immersion objective was used. Pumping was achieved using an optical parametric oscillator with 5 ns pulse duration, tuned to 475 nm for green dyes or 535 nm for red dyes. The laser beam was slightly divergent at the objective entrance pupil, so the focus at the sample was located slightly further away from the objective focal plane, and the beam diameter at the objective focal plane was ∼20 µm wide. For measurements of polystyrene microsphere modes below the laser threshold (Fig. 4), a 455 nm LED was used as the excitation source. The collected light was sent through a dichroic mirror and split 50:50 into a charge-coupled device camera and an imaging spectrometer (300 mm focal length, 0.05 nm resolution). For all measurements except for the hyperspectral imaging, the spectrum was collected through the entrance slit of the spectrometer and therefore represents an integration along a line crossing the centre of a bead or droplet. For hyperspectral imaging the spectrometer slit was replaced by a 10 µm pinhole, and a two-dimensional raster scan was performed with an acquisition time of 0.1 s per pixel. The spectral peaks above a broad fluorescence background were integrated to obtain the laser intensity image shown in Fig. 4b. The individual spectral peaks after subtracting the background were fitted with a Lorentzian curve to obtain their central wavelengths, which were subsequently fitted to Supplementary equation (1), with a = b.
Cell culture
HeLa, NIH3T3 and RAW 264.7 cell lines were grown at 37 °C with 5% CO2 in full growth medium (Dulbecco's modified Eagle medium supplemented with 10% fetal bovine serum and 1% pen-strep). The cells were incubated in full growth medium supplied with beads for 12 h before laser experiments. The viability assay was performed 24 h after the cells were injected with PPE or were supplied with beads using ethidium homodimer-1.
Oil injection
Non-toxic, low-viscosity high-index polyphenyl ether oil (PPE, SL-5267, Santolubes) was doped with 5 mM Nile red (9-diethylamino-5- benzo[α]phenoxazinone) before injection into HeLa and NIH3T3 cells. The injection was performed using a microinjector (FemtoJet, Eppendorf) and a glass micropipette with a 1.0 µm outer diameter (Femtotip, Eppendorf). The size of the injected droplets was controlled by the injection time (ranging from 0.2 s to 1 s), with an injection pressure of 1,700 kPa.
Adipocytes
Fresh subcutaneous fat tissue was collected from the neck of a two-month-old pig, minced, and mixed with an equal volume of phosphate buffered saline (PBS) containing 2 mg ml–1 collagenase (type 1A). The mixture was incubated with frequent shaking at 37 °C for 30 min. The suspension was filtered through 250 μm nylon mesh to remove undigested tissue and centrifuged at 65g for 5 min to collect the top layer of fat. The adipocytes were stained by adding 1% 10 mM Nile red in acetone. In the tissue laser experiments, 1 ml PBS containing 1 mg ml–1 collagenase and 1% of 10 mM Nile red in acetone was injected into the subcutaneous porcine fat. The tissue was incubated at 37 °C for 15 min. For excitation and light collection a multimode fibre with a core diameter of 200 µm was used.
Fluorescent beads
Three different bead types were used: 8 µm mean diameter green fluorescent polystyrene spheres (Thermo Scientific, Fluoro-Max, 18% coefficient of variation), 15–19 µm soda lime glass beads (Cospheric) and polydisperse BaTiO3 beads with a broad size distribution of 1–40 µm (GL0175B, Mo-Sci). Polystyrene beads were incubated for 30 min at room temperature in 1 wt% poly-L-lysine hydrobromide (Mw = 30,000–70,000) in water and washed three times. Soda lime and BaTiO3 spheres were coated with Alexa Fluor 488 tetrafluorophenyl (TFP) ester dye (Life Technologies) as follows. Beads were washed with acetone and incubated in acetone containing 2% vol/vol 3-aminopropyltriethoxysilane for 15 min with mixing. The beads were washed twice with acetone, once with water, and dried at 120 °C for 30 min. The beads were redispersed in 0.1 M sodium bicarbonate buffer, pH 9.0. Alexa Fluor TFP ester (100 µg) dissolved in 10 µl dimethyl sulphoxide was added to a 200 µl bead dispersion in sodium bicarbonate buffer. After 2 h incubation, the beads were washed five times with water and transferred to PBS. All the above washing steps were performed by centrifugation at 5,000g for 5 min and with exchange of the medium.
References
Vahala, K. J. Optical microcavities. Nature 424, 839–846 (2003).
Qian, S. X., Snow, J. B., Tzeng, H. M. & Chang, R. K. Lasing droplets—highlighting the liquid–air interface by laser-emission. Science 231, 486–488 (1986).
Hill, M. T. et al. A fast low-power optical memory based on coupled micro-ring lasers. Nature 432, 206–209 (2004).
Kippenberg, T. J., Holzwarth, R. & Diddams, S. A. Microresonator-based optical frequency combs. Science 332, 555–559 (2011).
Spillane, S. M., Kippenberg, T. J. & Vahala, K. J. Ultralow-threshold Raman laser using a spherical dielectric microcavity. Nature 415, 621–623 (2002).
Armani, A. M., Kulkarni, R. P., Fraser, S. E., Flagan, R. C. & Vahala, K. J. Label-free, single-molecule detection with optical microcavities. Science 317, 783–787 (2007).
Vollmer, F. & Arnold, S. Whispering-gallery-mode biosensing: label-free detection down to single molecules. Nature Methods 5, 591–596 (2008).
Zhu, J. G. et al. On-chip single nanoparticle detection and sizing by mode splitting in an ultrahigh-Q microresonator. Nature Photon. 4, 46–49 (2010).
Fan, X. D. & White, I. M. Optofluidic microsystems for chemical and biological analysis. Nature Photon. 5, 591–597 (2011).
Baaske, M. D., Foreman, M. R. & Vollmer, F. Single-molecule nucleic acid interactions monitored on a label-free microcavity biosensor platform. Nature Nanotech. 9, 933–939 (2014).
Himmelhaus, M. & Francois, A. In-vitro sensing of biomechanical forces in live cells by a whispering gallery mode biosensor. Biosens. Bioelectron. 25, 418–427 (2009).
Zhang, J., Campbell, R. E., Ting, A. Y. & Tsien, R. Y. Creating new fluorescent probes for cell biology. Nature Rev. Mol. Cell Biol. 3, 906–918 (2002).
Gather, M. C. & Yun, S. H. Single-cell biological lasers. Nature Photon. 5, 406–410 (2011).
Jonáš, A. et al. In vitro and in vivo biolasing of fluorescent proteins suspended in liquid microdroplet cavities. Lab. Chip 14, 3093–3100 (2014).
Shambat, G. et al. Single-cell photonic nanocavity probes. Nano Lett. 13, 4999–5005 (2013).
Fan, X. & Yun, S.-H. The potential of optofluidic biolasers. Nature Methods 11, 141–147 (2014).
Hill, M. T. & Gather, M. C. Advances in small lasers. Nature Photon. 8, 908–918 (2014).
Zhang, Y. & Yu, L. C. Microinjection as a tool of mechanical delivery. Curr. Opin. Biotechnol. 19, 506–510 (2008).
Campàs, O. et al. Quantifying cell-generated mechanical forces within living embryonic tissues. Nature Methods 11, 183–189 (2014).
Gardan, D., Gondret, F. & Louveau, I. Lipid metabolism and secretory function of porcine intramuscular adipocytes compared with subcutaneous and perirenal adipocytes. Am. J. Physiol. Endocrinol. Metab. 291, E372–E380 (2006).
Cannon, G. J. & Swanson, J. A. The macrophage capacity for phagocytosis. J. Cell Sci. 101, 907–913 (1992).
Kobayashi, S. et al. Artificial induction of autophagy around polystyrene beads in nonphagocytic cells. Autophagy 6, 36–45 (2010).
Gorodetsky, M. L. & Fomin, A. E. Geometrical theory of whispering-gallery modes. IEEE J. Sel. Topics Quantum Electron. 12, 33–39 (2006).
Livet, J. et al. Transgenic strategies for combinatorial expression of fluorescent proteins in the nervous system. Nature 450, 56–62 (2007).
Schultz, S. G. Molecular Biology of Membrane Transport Disorders (Springer, 1996).
Wang, Q. J. et al. Whispering-gallery mode resonators for highly unidirectional laser action. Proc. Natl Acad. Sci. USA 107, 22407–22412 (2010).
Nizamoglu, S., Gather, M. C. & Yun, S. H. All-biomaterial laser using vitamin and biopolymers. Adv. Mater. 25, 5943–5947 (2013).
Zhang, Z. et al. Visible submicron microdisk lasers. Appl. Phys. Lett. 90, 111119 (2007).
Choi, M. et al. A terahertz metamaterial with unnaturally high refractive index. Nature 470, 369–373 (2011).
Stockman, M. I. Spasers explained. Nature Photon. 2, 327–329 (2008).
Acknowledgements
This research was supported in part by the US National Science Foundation (NSF; ECCS-1101947, EEC-1358296, ECCS-1505569) and the National Institutes of Health (P41 EB015903). M.H. was supported in part by the Marie Curie International Outgoing Fellowship no. 627274 within the 7th European Community Framework Programme. The authors thank J. Zhao and Wellman Centre Photopathology Core for technical support. Part of this work was performed at the Centre for Nanoscale Systems (CNS) at Harvard University, which is a member of the National Nanotechnology Infrastructure Network (NNIN) and supported by the NSF under award no. ECS-0335765.
Author information
Authors and Affiliations
Contributions
M.H. and S.H.Y. designed the study. M.H. carried out the experiments and analysed the data. M.H. and S.H.Y. wrote the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Supplementary information
Supplementary information (PDF 3501 kb)
Supplementary information
Supplementary Movie 1 (MOV 330 kb)
Supplementary information
Supplementary Movie 2 (MOV 406 kb)
Rights and permissions
About this article

Cite this article
Humar, M., Hyun Yun, S. Intracellular microlasers. Nature Photon 9, 572–576 (2015). https://doi.org/10.1038/nphoton.2015.129
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/nphoton.2015.129
This article is cited by
-
Transient stimulated Raman scattering spectroscopy and imaging
Light: Science & Applications (2024)
-
Plasmonic multi-wavelength random laser by gold nanoparticles doped into glass substrate
Journal of Optics (2024)
-
Lanthanide-based microlasers: Synthesis, structures, and biomedical applications
Nano Research (2024)
-
A biodegradable, flexible photonic patch for in vivo phototherapy
Nature Communications (2023)
-
Light distribution in fat cell layers at physiological temperatures
Scientific Reports (2023)
